

Abstract
Voltage-gated sodium channels composed of a pore-forming α subunit and auxiliary β subunits are responsible for the upstroke of the action potential in cardiac muscle. However, their localization and expression patterns in human myocardium have not yet been clearly defined. We used immunohistochemical methods to define the level of expression and the subcellular localization of sodium channel α and β subunits in human atrial myocytes. Nav1.2 channels are located in highest density at intercalated disks where β1 and β3 subunits are also expressed. Nav1.4 and the predominant Nav1.5 channels are located in a striated pattern on the cell surface at the z-lines together with β2 subunits. Nav1.1, Nav1.3, and Nav1.6 channels are located in scattered puncta on the cell surface in a pattern similar to β3 and β4 subunits. Nav1.5 comprised approximately 88% of the total sodium channel staining, as assessed by quantitative immunohistochemistry. Functional studies using whole cell patch-clamp recording and measurements of contractility in human atrial cells and tissue showed that TTX-sensitive (non-Nav1.5) α subunit isoforms account for up to 27% of total sodium current in human atrium and are required for maximal contractility. Overall, our results show that multiple sodium channel α and β subunits are differentially localized in subcellular compartments in human atrial myocytes, suggesting that they play distinct roles in initiation and conduction of the action potential and in excitation–contraction coupling. TTX-sensitive sodium channel isoforms, even though expressed at low levels relative to TTX-sensitive Nav1.5, contribute substantially to total cardiac sodium current and are required for normal contractility. This article is part of a Special Issue entitled “Na+ Regulation in Cardiac Myocytes”.
Keywords: Sodium channels, Myocardium, Immunocytochemistry, Contractility
1. Introduction
Cardiac action potentials are generated and propagated through the coordinated activity of multiple types of ion channels. Voltage-gated sodium channels generate the upstroke of the action potential, and their activation and inactivation set the conduction velocity through cardiac tissue and the refractory period between conducted action potentials. Mutations in genes encoding voltage-gated sodium channels are known to cause arrhythmias [1] and to be involved in cardiomyopathy [2–4]. Differential expression and localization of sodium channel subunits are likely to be important determinants of electric excitability of cardiac myocytes. This study defines the subcellular localization of sodium channel subunits in human atrial myocardium.
Voltage-gated sodium channels are composed of a pore-forming α subunit with one or two auxiliary β subunits [5]. Ten different genes encoding sodium channel α subunits have been identified and nine have been functionally expressed [5,6]. The different α subunit isoforms have distinct patterns of development and localization in the nervous system, skeletal and cardiac muscle, and they have different pharmacological properties. Isoforms preferentially expressed in the central nervous system (Nav1.1, 1.2, 1.3, 1.6) are inhibited by nanomolar concentrations of the puffer fish toxin, tetrodotoxin (TTX), a highly specific sodium channel blocker. The isoform present in adult skeletal muscle (Nav1.4) is also blocked by nanomolar TTX-concentrations. In contrast, the primary cardiac isoform (Nav1.5) requires micromolar concentrations of TTX for inhibition due to substitution of a cysteine for an aromatic residue in the pore region [6,7].
Four genes encoding different β-subunits—β1, β2, β3 and β4—have been identified [8,9]. β1 and β3 are noncovalently associated with α subunits, whereas β2 and β4 are disulfide-linked to α. The β subunits modulate channel gating, interact with extracellular matrix, cytoskeleton, and cell adhesion molecules, play a role in adhesive interactions, and influence cell surface localization of sodium channels [10]. Our previous work showed that sodium channel β subunits are differentially localized in the transverse tubules, surface membrane, and intercalated disks of mouse ventricular myocytes [11].
Nav1.5 has often been termed the “cardiac” sodium channel. However, we showed previously that the “brain” sodium channels Nav1.1 and Nav1.3 are also expressed in mouse heart and have distinct distributions and functions from Nav1.5 [11–13], and other groups have extended these findings [14–18]. Multiple sodium channel isoforms are also expressed in human atrial myocytes [19].
2. Materials and methods
2.1. Tissue samples
Samples of human atrial tissue were obtained from patients undergoing elective cardiac surgery for multiple indications. Tissue from patients with congestive heart failure or atrial rhythm disorders including atrial fibrillation was excluded to avoid structural and/or electrophysiological alterations in the right atrial myocardium. A detailed description of tissue isolation and preparation is provided in Supplemental Material. All procedures conformed to the principles outlined in the Declaration of Helsinki and were in agreement with the policies of the Ethics Committee of the University of Würzburg.
2.2. Immunohistochemistry of human atrial tissue
A full description of the antibodies and immunohistochemical methods is given in Supplemental Material. Patches of atrial muscle were removed during surgery, immediately frozen in liquid nitrogen and sectioned (10 μm) on a cryostat. After mounting, fixation and antibody treatment, sections were viewed using Bio-Rad MRC 600 or Leica TCS SL confocal microscopes. More than 20 tissue sections from 14 patients were analyzed for each antibody.
2.3. Quantification of immunohistochemistry
For these studies we used saturating concentrations of antibodies recognizing each sodium channel α subunit [20]. All images in a single experiment were collected using the same laser gain and offset settings so that the intensity of staining of different antibodies could be compared. Staining intensity for each antibody was normalized to staining of Nav1.5 from the same experiment. For quantification, three to five images from 5 different atrial samples of the same patients as mentioned above were analyzed for each antibody.
2.4. Electrophysiology
Single human cardiac myocytes were prepared from right atrial tissue according to the modified protocol of Bustamante et al. [21,22]. Whole-cell sodium current (INa) was recorded at room temperature from rod-shaped, striated and Ca2+-tolerant cells within 8 h of isolation.
2.5. Contractility measurements in human atrial muscle
Human atrial muscle fibers were harvested during open-heart surgery from patients who underwent elective coronary bypass surgery and used for contractility studies as previously described [23,24]. In brief, fibers were mounted between a force transducer and a micrometer screw and constantly superfused with oxygenated Krebs–Henseleit (KHS) buffer at 37 °C with a pO2 of >500 mm Hg. The fibers were electrically stimulated at 1 Hz, 25% above threshold, and were initially stretched to optimal length (Lmax), which was indicated by maximum isometric force amplitude; length was not adjusted further during the experiment. Fibers were superfused with oxygenated KHS buffer containing different TTX concentrations. These experiments were carried out in the presence of atropine (100 nM) and propranolol (1 μM) to prevent effects of neurotransmitters that might be released from autonomic nerve endings.
3. Results
3.1. Localization of sodium channel α subunits in human right atrium
Human atrial tissue samples from 14 patients were stained with antibodies recognizing individual α subunits of voltage-gated sodium channels to determine their localization. Nav1.1, Nav1.3 and Nav1.6 were observed in puncta on the surface of myocytes (Figs. 1A-E). The number of puncta and the intensity of staining were greatest for Nav1.1 and Nav1.6; puncta were fewer and less intensely stained for Nav1.3. None of these channels was located at the z-lines which were marked by staining for α-actinin (Figs. 1B, C). In contrast, antibodies recognizing Nav1.2 channels stained the cell surface most intensely near the intercalated disks (Fig. 1G); pale striated staining for Nav1.2 was also seen (Figs. 1G, I). Double labeling with connexin 43 to mark intercalated disks confirmed the intercalated disk localization of the high density Nav1.2 staining (Figs. 1G-J). Staining was abolished when primary antibodies were omitted (Fig. 1F).
Fig. 1.

Immunostaining of sodium channel α subunits Nav1.1, 1.3, 1.6, 1.2, 1.4 and 1.5 in human atrium. Tissue section double-labelled with anti-Nav1.1 (A) and α actinin (B) demonstrating punctate surface labelling of Nav1.1 channels that does not overlap with the z-lines when the images are merged (C). Human atrial tissue labelled with anti-Nav1.3 (D) or anti-Nav1.6 (E) illustrating punctate surface staining of these channels, similar to anti-Nav1.1. (F) Control section in which the primary antibody was omitted illustrating specificity of staining. Tissue section double labelled with anti-Nav1.2 (G) and anti-connexin 43 (H) illustrating a high density of Nav1.2 channels at the intercalated disk region (I, merged) and relatively low density of staining in a banded pattern along the length of the myocyte. (J) Higher magnification of a tissue section double labelled with anti-Nav1.2 (green) and anti-connexin 43 (red) illustrating the staining of these two proteins at the intercalated disk region. (K) Staining of anti-Nav1.4 on atrial tissue illustrating staining in a banded pattern along the myocyte. (L) Atrial tissue double-labelled with anti-Nav1.5 (green) and anti-connexin 43 (red). (M, N) High magnification image of tissue double-labelled with anti-Nav1.5 (M) and α-actinin (N) showing that the banded pattern of Nav1.5 staining is in register with the staining of α-actinin at the z-lines (O). Scale bars = 5 μm.
Nav1.4 and Nav1.5 channels also were observed at the myocyte surface, but in a striated pattern (Figs. 1K, L). The striated Nav1.5 staining was in register with α-actinin marking the z-lines (Figs. 1M-O). This pattern suggests that Nav1.5 channels cluster at a cell surface specialization anchored to the proteins of the z-line. In contrast to Nav1.2, increased staining near intercalated disks was not observed for Nav1.4 and Nav1.5.
Surface staining by Nav1.5 was confirmed with an optical z-series (Figs. 2A–C, green). Nav1.5 staining was observed across the entire width of the myocyte in sections that included the cell surface (Fig. 2A). In progressively deeper sections (Figs. 2B, C), staining was seen at the periphery of the myocyte where the cross section intersected the surface membrane, but was reduced or absent from the cell interior in the center of the cross-section. Connexin 43 is also seen in a band traversing the entire myocyte cross-section in cell surface sections (Fig. 2A, red) but is reduced in intensity and restricted to the edges of the myocyte in the deeper sections (Figs. 2B, C, red).
Fig. 2.

Localization of Nav1.5 relative to SERCA ATPase and CaV1.2. (A–C) Images from a z-series through atrial tissue stained with anti-Nav1.5 (green) and anti-connexin 43 at the cell surface (A) and progressively deeper sections (B and C) to confirm surface localization of sodium channel proteins. Arrows and arrowheads emphasize locations where depth-dependent changes in staining are particularly evident. (D–F) Optical plane showing both cell surface and intracellular staining for Nav1.5 (green, D and F) and SERCA ATPase (red). (G–I) Distribution of Nav1.5 (green, G, I) relative to SERCA ATPase (red, H, I). J–L, Localization of Nav1.5 (green, J, L) relative to CaV1.2 (red, K, L). Scale bars A–F = 5 μm; G–L = 2 μm.
We further assessed the cell-surface localization of the Nav1.5 sodium channel by co-labeling with an antibody recognizing the SERCA ATPase in the sarcoplasmic reticulum (Figs. 2D–I). Optical sections including the surface or through the center of the cell show Nav1.5 at the surface (Figs. 2D, F) but SERCA ATPase throughout the cross-section of the myocyte in the deeper sections (Figs. 2E, F). The most intense bands of SERCA ATPase staining fall between the bands of Nav1.5 staining that mark the z-lines (Figs. 2G–I), consistent with primary localization of the SERCA ATPase in the sarcoplasmic reticulum between the z-lines. Conversely, the CaV1.2 channel was found in a striated pattern in the cell interior but in register with Nav1.5 on the myocyte surface (Figs. 2K, L). Staining for CaV1.2 occurred at each z-line in doublets flanking the staining for Nav1.5 (Figs. 2J, L) and for α-actinin (not shown).
Collectively, these data indicate that Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.5, and Nav1.6 are all expressed in human atrial myocardium, but are localized in distinct patterns. In Fig. 1 staining for each α subunit isoform is shown at similar brightness to clearly illustrate the pattern of staining for each channel subtype. When imaged quantitatively, staining by anti-Nav1.5 was much more intense than that of the other antibodies, suggesting that this channel is present in highest density (see Section 3.3 below for quantification).
3.2. Localization of sodium channel β subunits in human right atrium
Voltage-gated sodium channel β subunits were also differentially localized on the surface of human atrial myocytes. The β1 subunit was observed only at intercalated disks, as indicated by double-labeling for connexin 43 (Figs. 3A–C). In contrast, sodium channel β2 subunits were observed in a banded pattern in register with z-lines as defined by α-actinin (Figs. 3D–F). A third pattern of staining was observed for sodium channel β3 subunits, which were observed in cell surface puncta, similar to Nav1.1, Nav1.3 and Nav1.6 α subunits (Figs. 3G–I). β3 was sometimes observed near intercalated disks (Figs. 3J–L), like β1. β4 subunits were also found in puncta, similar to β3, but were not concentrated at intercalated disks (Figs. 3M–O). Thus, the related β1 and β3 subunits, which are non-covalently associated with α subunits, are found at the intercalated disks, like Nav1.2 α subunits. The β3 subunit is also found in puncta, like Nav1.1, Nav1.3 and Nav1.6 α subunits. In contrast, the disulfide-linked β2 subunit is found at the z-line, like the Nav1.2, Nav1.4 and Nav1.5 α subunits. Finally, the β2-like disulfide-linked β4 subunit is found in puncta, similar to the Nav1.1, Nav1.3 and Nav1.6 α subunits.
Fig. 3.

Double immunostaining of sodium channel β subunits in human atrium with connexin 43 and α-actinin. Tissue section double labelled with anti-β1 (A) and anti-connexin 43 (B) antibodies illustrating dense labelling at the region of the intercalated disk of atrial tissue. (C) Merged picture of images in (A) and (B). Section double labelled with anti-β2 (D) and α-actinin (E) illustrating labelling of the β2 subunit of sodium channels in a banded pattern in register with the z-lines when the images are merged (F). (G) Atrial tissue double labelled with anti-β3 (G) and α-actinin (H) showing non-uniform, punctate staining in muscle fiber when the images are merged (I). Tissue section labelled with anti-β3 (J) and anti-connexin 43 (K) illustrating that β3 subunits are present at the intercalated disk region as shown in the merged image (L). Double labelling of atrial section with anti-β4 (M) and anti-α actinin (N) illustrating a punctate pattern of staining that is in register with the z-lines as shown in the merged image (O). Scale bars = 2 μm.
3.3. Levels of sodium channel α subunit expression in human right atrium
The images of Figs. 1 and 3 were obtained using gain and offset settings that optimized visualization of each antibody and emphasized protein localization. To estimate the relative levels of the α subunit proteins, we measured the relative intensity of staining of each α subunit antibody when applied at saturating concentration [20]. We measured mean pixel intensity for each antibody and determined the percentage of total mean pixel intensity represented by each antibody (Fig. 4). Using this approach Nav1.1 accounted for 2.4% of total sodium channel staining, Nav1.2 for 2.5%, Nav1.3 for 0.4%, Nav1.4 for 2.6% and Nav1.6 for 4.4%. When combined, these TTX-sensitive sodium channels accounted for 12.3% of total sodium channel staining. The majority of staining (87.7% of the total mean pixel density) was due to TTX-resistant Nav1.5. These quantitative immunohistochemical results suggest that the TTX-resistant Nav1.5 channels are the predominant channels in human atrial tissue but that TTX-sensitive sodium channels are also expressed. If each TTX-resistant and TTX-sensitive channel molecule contributed equally to total sodium current, approximately 12% of the total current would be sensitive to nanomolar concentrations of TTX, while the remainder, due to Nav1.5, would be blocked by micromolar TTX concentrations.
Fig. 4.
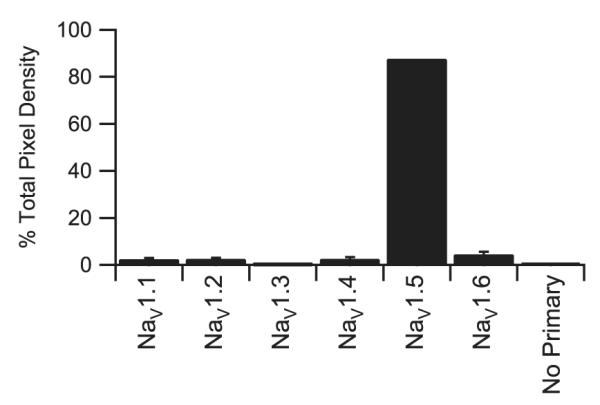
Immunocytochemical quantification of TTX-sensitive sodium channel α subunit isoforms. Quantification of staining by antibodies recognizing TTX-sensitive sodium by channel α subunits Nav1.1, Nav1.2, Nav1.3, Nav1.4 or Nav1.6 as well as the TTX-resistant Nav1.5 sodium channel expressed as normalized pixel density.
3.4. TTX-sensitivity of sodium current in human atrial myocytes
To determine the TTX-sensitivity of functional sodium channels in human atrium, total sodium current, INa, was recorded from single human atrial cardiomyocytes using whole-cell voltage-clamp, and concentration–response curves for block of sodium current by TTX were plotted (Fig. 5). We initially used a holding potential of −100 mV and a test potential of 0 mV to activate all sodium channel isoforms. Under these conditions, 20 nM TTX blocked 14 ± 2.7% (n = 19) of the current and 100 nM TTX blocked 25.0 ± 7.0% (n = 10) of the peak current (Figs. 5A, B). Since 20 nM TTX is only expected to block ≈1% of TTX-resistant Nav1.5, the substantial block at this concentration is consistent with the presence of sodium current conducted by TTX-sensitive sodium channel isoforms. Higher concentrations of TTX blocked the remaining sodium current with a concentration dependence expected for Nav1.5. Block occurred without alteration in sodium current kinetics (data not shown). A fit of the concentration–response curves with a two-component binding isotherm indicated that 27% of current was blocked by TTX with an IC50 of 12.7 nM and the remaining 73% of current was blocked with and IC50 of 2.7 μM (Fig. 5B). This result is consistent with the conclusion that approximately 73 ± 2% of the sodium current at a holding potential of −100 mV is conducted by TTX-resistant Nav1.5 channels and the remaining 27% of the current is conducted by TTX-sensitive Nav1.1, Nav1.2, Nav1.3, Nav1.4 and Nav1.6 channels.
Fig. 5.

Concentration–response relationship for the effect of TTX on total cardiac peak sodium current in human atrial myocytes. (A) Examples of current traces during treatment with TTX in two different human atrial myocytes. Depolarizations to 0 mV from a holding potential of −100 mV were applied every 5 s, and peak sodium current was measured and compared in the absence and presence of the indicated TTX concentrations. (B) Concentration–response relationship from experiments like those in A. After exposure to TTX, toxin was washed out of the bath to insure that rundown had not occurred. Reversal values are plotted as the open point at 0 concentration. Error bars represent SEM. The fit curve is a two component binding isotherm with IC50 values of 12.7 nM and 2.7 μM. The higher affinity component represents 27% of the total current. (C) Effect of holding potential and test potential on TTX inhibition. Bars display normalized currents ± SEM. Filled squares, TTX effect at a holding potential of −100 mV and a test pulse to −60 mV; open circles, TTX effect at a holding potential of −70 mV and test pulse to 0 mV. Control conditions were recorded at −100 mV holding and 0 mV test potential. All displayed data were normalized to control conditions. Fit of the two-component binding isotherm with IC50 values fixed to the values determined in panel B indicating that 67% of the channels were TTX-sensitive. Recordings were from 12 different patients with 3–30 myocytes studied per TTX concentration.
Different sodium channel α-subunit isoforms have different voltage-dependent activation and inactivation properties, and we used these differences to alter the fraction of total current contributed by Nav1.5. TTX-resistant Nav1.5 sodium channels inactivate at more negative potentials than the TTX-sensitive isoforms [25]. Therefore, we used a holding potential of −70 mV to increase the fraction of recorded current conducted by TTX-sensitive channel isoforms. This holding potential inactivates 38% of the total sodium current (data not shown). When sodium current was elicited from a holding potential of −70 mV, the percentage blocked by 20 nM TTX (n = 19) increased to 46% from the 14% observed with a holding potential of −100 mV (Fig. 5C), which was consistent with 67 ± 18% of sodium current being conducted by TTX-sensitive sodium channels at a holding potential of −70 mV based on curve fitting as in Fig. 5B. Thus, reducing the fraction of total sodium current contributed by Nav1.5 by using a more depolarized holding potential substantially increased the fraction of TTX-sensitive sodium current.
Conversely, the fraction of total current contributed by Nav1.5 can be enhanced by using a test depolarization of −60 mV and a holding potential of −100 mV. Since Nav1.5 channels activate at more negative potentials than the TTX-sensitive isoforms, the −60 mV test pulse is expected to partially activate Nav1.5, but not the TTX-sensitive channels [26]. Using this protocol, no detectable block of sodium current was observed at 20 nM, consistent with all of the current being due to Nav1.5 (Fig. 5C). Thus, isolating current conducted by the Nav1.5 channel in this way makes the entire population of channels TTX-resistant. The sum of these experiments indicates that human atrial myocytes express functional TTX-sensitive sodium channels, in addition to TTX-resistant Nav1.5, and these contribute approximately 27% of total sodium current.
3.5. Changes in contractility of atrial muscle strips due to blockade of TTX-sensitive sodium channel α subunits
TTX was also used to determine the functional relevance of TTX-sensitive sodium channels for atrial contractility (Fig. 6). Concentrations of TTX that are expected to block primarily TTX-sensitive sodium channels, 20 nM (n = 5), 50 nM (n = 7), and 100 nM (n = 7), reduced contraction amplitude by 11.1 ± 5.6%, 17.6 ± 4.8%, and 30 ± 6.8%, respectively (Fig. 6). From the parameter values from the concentration–response curves of Fig. 5B, we can estimate the fraction of total sodium current blocked at each concentration and the amount of block that is due to TTX-sensitive (Fig. 6, green) and TTX-resistant sodium channels (Fig. 6, blue), respectively. Over this concentration range, the reduction in peak sodium current is largely attributable to selective block of TTX-sensitive sodium channels (Fig. 6). In parallel with block of the TTX-sensitive channels, both the amplitude and the velocity of contraction of the atrial muscle bundles were reduced (Fig. 6, red and black). Thus, block of TTX-sensitive sodium channels results in substantial effects on contraction of atrial muscle strips, approximately consistent with their effects on total sodium current amplitude.
Fig. 6.
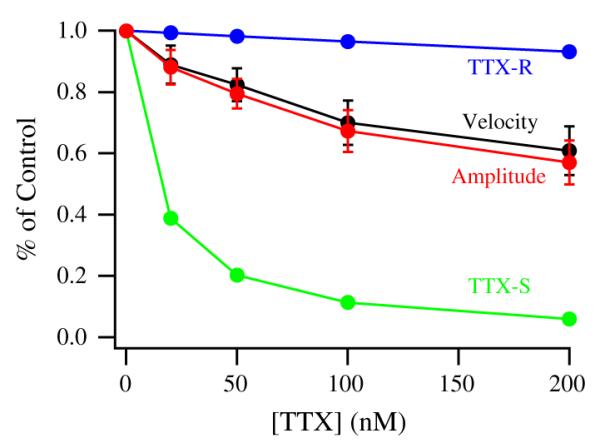
Concentration dependent effects of specific sodium channel blockade with TTX on human atrial contractility showing contraction amplitude (red) and velocity (black). The fractions of sodium current due to TTX-resistant (blue) and TTX-sensitive (green) channels remaining at each TTX concentration. The values were calculated from the best fit data from Fig. 5B.
4. Discussion
4.1. Expression and differential localization of sodium channel subtypes in human right atrium
Our immunocytochemical results show that there is detectable expression of five TTX-sensitive sodium channel α subunit proteins (Nav1.1, Nav1.2, Nav1.3, and Nav1.4, and Nav1.6) in atrial muscle, in addition to the predominant TTX-resistant isoform Nav1.5. This remarkable diversity of sodium channel expression in the human atrium is reminiscent of the diversity of expression of voltage-gated potassium channel isoforms in the heart [27–29]. Quantitative determination of RNA levels indicated that each TTX-sensitive isoform is expressed at far lower levels than Nav1.5 [19,30]. This is consistent with the lower protein expression for TTX-sensitive isoforms in mouse ventricle [20].
Diversity of expression of these sodium channel isoforms is accompanied by diversity of localization. The quantitatively dominant Nav1.5 channels are localized on the cell surface in register with the z-lines, suggesting a cell surface specialization. Of the TTX-sensitive isoforms, only Nav1.4 exhibits a similar distribution. The β2 and β4 subunits that are disulfide linked to α subunits are also found at z-lines. Conversely, Nav1.1 and Nav1.6 channels have a diffuse punctate distribution, as does a fraction of the β3 and β4 subunits. Nav1.2 channels have a strikingly different distribution, being localized at the intercalated disks along with the β1 subunits and a fraction of the β3 subunits. The markedly different localization of these sodium channel subtypes suggests that they have distinct physiological functions in these subcellular compartments of atrial myocytes.
4.2. Sodium channel complexes in human atrium
Sodium channel complexes are thought to contain a single α subunit in association with one or two β subunits [5,9]. β1 and β3 subunits are closely related and both are noncovalently associated with α subunits. Similarly, β2 and β4 subunits are also closely related and are disulfide-linked to α subunits. It is likely that native sodium channel complexes are composed of an α subunit in association with one β1/β3 subunit and/or one β2/β4 subunit. Although our results do not show direct physical interaction or strict colocalization between specific sodium channel α and β subunits, the co-localization of specific subsets of sodium channel subunits in distinct subcellular compartments suggests that different subunit combinations may form sodium channel complexes in these compartments of atrial myocytes. Nav1.5 α subunits and β2/β4 subunits are found in a striated pattern on the cell surface and may form complexes. Likewise Nav1.2 α subunits and β1/β3 subunits are found at intercalated disks where they could form complexes. Nav1.1 and Nav1.6 α subunits and β3 and β4 subunits are found in punctate clusters at the cell surface where they could form complexes. As sodium channel β subunits have differential modulatory effects on α subunits and interact with distinct sets of extracellular matrix, cell adhesion, cytoskeletal, and signal transduction proteins [9], the association of α subunits with different β subunits suggested by these co-localization data may provide specialization of function of sodium channels in distinct subcellular locations.
4.3. TTX-sensitive and TTX-resistant sodium currents in human atrium
Our results show that the predominant Nav1.5 α subunit accounts for 88% of the sodium channel protein as estimated by immunostaining, and 73% of the functional sodium current as estimated by whole-cell voltage clamp and TTX inhibition. Considering the differences in sodium channel gating parameters between TTX-sensitive and TTX-resistant sodium channels [31], these results are in reasonable agreement and indicate that TTX-sensitive sodium channels comprise a significant fraction of the sodium channel protein and contribute a significant component of sodium current in the human atrium. An earlier study in human atrial myocytes reported that TTX-insensitive sodium current accounted for 92% of total sodium current [32]. These studies agree with ours in showing that there is substantial TTX-sensitive sodium current in atrial myocytes. The quantitative differences in the estimates of TTX-sensitive sodium currents may reflect differences in cell dissociation, which can damage cell-surface sodium channels during proteolytic treatments, but may also reflect differences in recording technique using different sodium concentrations and membrane potentials.
In addition to these results on human atrium, other studies have provided functional evidence for the importance of TTX-sensitive sodium current in the heart. Already in 1979, Coraboeuf et al. described that TTX-sensitive sodium current contributes to the action potential duration of Purkinje fibers in dog heart, since low concentrations of TTX led to a reduction of the action potential duration [33]. Our previous data demonstrated TTX-sensitive sodium channel expression and TTX-sensitive sodium current in mouse ventricular myocytes and sino-atrial node [12]. More recent studies indicate that TTX-sensitive sodium channels generate 10% and 22% of the peak sodium current in canine ventricle and Purkinje fibers, respectively [34], approximately 20% in rat ventricular myocytes [17], and approximately 8% in mouse ventricular myocytes [35].
4.4. Physiological function of sodium channel subtypes in the heart
Our studies of atrial contractility demonstrate the functional importance of TTX-sensitive sodium current in human atrial myocytes since blocking them selectively with a low concentration of TTX results in a concomitant loss of contractile function (Fig. 6). The reduced contractile function may be due to the differential localization of the TTX-sensitive isoforms, particularly the localization of Nav1.2 at intercalated disks. The importance of the TTX-sensitive sodium channel α subunit isoforms may also reside in differences between their voltage dependence of gating and that of the Nav1.5 TTX-resistant isoform. The TTX-sensitive subtypes have faster activation and positively shifted voltage dependence of inactivation relative to Nav1.5, making them better suited to trigger action potentials rapidly in response to initial depolarization by incoming action potentials. The positive voltage dependence may also provide a margin of safety for triggering action potentials and maintaining rapid conduction in more depolarized cardiac cells. This margin of safety may become particularly important in supporting physiological function of cardiac cells depolarized by pathophysiological challenges such as ischemia. This role may be further enhanced in chronic pathologic conditions since relatively large TTX-sensitive currents were observed in ventricular tissue and myocytes from post-infarction remodeled myocardium [36] and surviving Purkinje fibers [37].
Localization of sodium channel subtypes in human atrial myocytes differs markedly from that in mouse ventricular myocytes. The predominant Nav1.5 channels are localized at the cell surface in a striated pattern near each z-line, rather than at the intercalated disks. Only the Nav1.2 channel is localized at intercalated disks in atrial myocytes. As this channel is quantitatively minor, it is likely that conduction is continuous rather than saltatory in human atrium. Thus, the action potential likely is initiated by Nav1.2 channels at the intercalated disk in concert with nearby Nav1.5 channels and progressively activates other cell surface Nav1.5 channels and TTX-sensitive channels to spread smoothly along the atrial myofiber. The localization of the predominant Nav1.5 channels and the skeletal muscle isoform TTX-sensitive Nav1.4 at the z-lines is similar to the location of CaV1.2 calcium channels in atrial tissue (Fig. 2). Such close co-localization may ensure rapid activation of the calcium channels and triggering of contraction. Thus, the Nav1.5 and Nav1.4 channels in human atrial myocytes are likely to serve an analogous function to the t-tubular TTX-sensitive sodium channels in mouse ventricle, which aid in depolarization of the t-tubules and initiation of contraction [12].
Due to the lack of availability, we could not use atrial tissue from healthy human donors. Therefore, concomitant cardiovascular disease might have influenced ion channel expression and function in the investigated specimens. This concern is greatest for Nav1.3 because it is expressed during embryonic and early postnatal stages. Conditions such as heart failure are known to provoke the activation of a fetal gene program [38]. On the other hand, Nav1.3 and the other TTX-sensitive sodium channel subtypes were observed in our studies of healthy mouse cardiac myocytes [11,20]. We did not include cells harvested from patients with atrial fibrillation, because there is evidence for an up-regulation of the sodium channel isoform Nav1.1 in this rhythm disorder [39]. Therefore, although disease-related remodeling might have had an unknown impact on the results presented here, the correlations among several different species in expression of TTX-sensitive sodium channel subtypes in the heart suggest that this sodium channel diversity is indeed characteristic of the normal heart.
In conclusion, we show isoform-specific subcellular localizations of sodium channel α and β subunits and provide evidence for the composition of sodium channel complexes in human heart. The presence of TTX-sensitive sodium channels with the positive voltage dependence of gating and more rapid activation and inactivation [31] suggests additional roles for TTX sensitive channels. In addition to their function in conducting the action potential within the cell and from cell to cell, they may play a safety function in diseased myocardium where cells have a less hyperpolarized resting membrane potential with the consequence that the predominant Nav1.5 channels may be unavailable for activation. The exact physiological function of the different sodium channel subunits in human heart remains to be fully determined and is an important focus for future research.
Supplementary Material
Acknowledgments
We thank Marco Abesser for excellent technical assistance.
Sources of funding This work was primarily supported by the Deutsche Forschungsgemeinschaft, DFG, (Ma2252) and the Interdisziplinäres Zentrum für klinische Forschung der Universität Würzburg (IZKF) to S.K.G. Maier. The authors gratefully acknowledge the financial support provided by the National Institutes of Health (P01 HL44948 and R01 HL085372) to W.A. Catterall.
Footnotes
Disclosures None.
Appendix A. Supplementary data Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.yjmcc.2013.05.006.
References
- [1].Clancy CE, Kass RS. Inherited and acquired vulnerability to ventricular arrhythmias: cardiac Na+ and K+ channels. Physiol Rev. 2005;85:33–47. doi: 10.1152/physrev.00005.2004. [DOI] [PubMed] [Google Scholar]
- [2].McNair WP, Ku L, Taylor MR, Fain PR, Dao D, Wolfel E, et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation. 2004;110:2163–7. doi: 10.1161/01.CIR.0000144458.58660.BB. [DOI] [PubMed] [Google Scholar]
- [3].George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–9. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. SCN4B-encoded sodium channel β4 subunit in congenital long-QT syndrome. Circulation. 2007;116:134–42. doi: 10.1161/CIRCULATIONAHA.106.659086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- [6].Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol. 2001;63:871–94. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- [7].Satin J, Kyle JW, Chen M, Bell P, Cribbs LL, Fozzard HA, et al. A mutant of TTX-resistant cardiac sodium channels with TTX- sensitive properties. Science. 1992;256:1202–5. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- [8].Yu FH, Westenbroek RE, Silos-Santiago I, McCormick KA, Lawson D, Ge P, et al. Sodium channel β4, a new disulfide-linked auxiliary subunit with similarity to β2. J Neurosci. 2003;23:7577–85. doi: 10.1523/JNEUROSCI.23-20-07577.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brackenbury WJ, Isom LL. Na channel beta subunits: overachievers of the ion channel family. Front Pharmacol. 2011;2:53. doi: 10.3389/fphar.2011.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Isom LL. Sodium channel β subunits: anything but auxiliary. Neuroscientist. 2001;7:42–54. doi: 10.1177/107385840100700108. [DOI] [PubMed] [Google Scholar]
- [11].Maier SK, Westenbroek RE, McCormick KA, Curtis R, Scheuer T, Catterall WA. Distinct subcellular localization of different sodium channel α and β subunits in single ventricular myocytes from mouse heart. Circulation. 2004;109:1421–7. doi: 10.1161/01.CIR.0000121421.61896.24. [DOI] [PubMed] [Google Scholar]
- [12].Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–8. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Maier SK, Westenbroek RE, Yamanushi TT, Dobrzynski H, Boyett MR, Catterall WA, et al. An unexpected requirement for brain-type sodium channels for control of heart rate in the mouse sinoatrial node. Proc Natl Acad Sci U S A. 2003;100:3507–12. doi: 10.1073/pnas.2627986100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lei M, Jones SA, Liu J, Lancaster MK, Fung SS, Dobrzynski H, et al. Requirement of neuronal- and cardiac-type sodium channels for murine sinoatrial node pacemaking. J Physiol. 2004;559:835–48. doi: 10.1113/jphysiol.2004.068643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brette F, Orchard CH. Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem Biophys Res Commun. 2006;348:1163–6. doi: 10.1016/j.bbrc.2006.07.189. [DOI] [PubMed] [Google Scholar]
- [16].Brette F, Orchard CH. No apparent requirement for neuronal sodium channels in excitation–contraction coupling in rat ventricular myocytes. Circ Res. 2006;98:667–74. doi: 10.1161/01.RES.0000209963.02720.70. [DOI] [PubMed] [Google Scholar]
- [17].Duclohier H. Neuronal sodium channels in ventricular heart cells are localized near T-tubules openings. Biochem Biophys Res Commun. 2005;334:1135–40. doi: 10.1016/j.bbrc.2005.06.203. [DOI] [PubMed] [Google Scholar]
- [18].Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, et al. Characterization of sodium channel α- and β-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303–10. doi: 10.1161/01.cir.103.9.1303. [DOI] [PubMed] [Google Scholar]
- [19].Greener ID, Monfredi O, Inada S, Chandler NJ, Tellez JO, Atkinson A, et al. Molecular architecture of the human specialised atrioventricular conduction axis. J Mol Cell Cardiol. 2011;50:642–51. doi: 10.1016/j.yjmcc.2010.12.017. [DOI] [PubMed] [Google Scholar]
- [20].Westenbroek RE, Bischoff S, Catterall WA, Scheuer T, Maier SKG. In depth analysis of sodium channel expression patterns in mouse ventricular myocytes using isoform-specific antibodies and quantitative immunocytochemistry. J Mol Cell Cardiol. 2013 doi: 10.1016/j.yjmcc.2013.08.004. [submitted for publication] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bustamante JO, Watanabe T, Murphy DA, McDonald TF. Isolation of single atrial and ventricular cells from the human heart. Can Med Assoc J. 1982;126:791–3. [PMC free article] [PubMed] [Google Scholar]
- [22].Maier S, Aulbach F, Simm A, Lange V, Langenfeld H, Behre H, et al. Stimulation of L-type Ca2+ current in human atrial myocytes by insulin. Cardiovasc Res. 1999;44:390–7. doi: 10.1016/s0008-6363(99)00229-1. [DOI] [PubMed] [Google Scholar]
- [23].Vahl CF, Bonz A, Timek T, Hagl S. Intracellular calcium transient of working human myocardium of seven patients transplanted for congestive heart failure. Circ Res. 1994;74:952–8. doi: 10.1161/01.res.74.5.952. [DOI] [PubMed] [Google Scholar]
- [24].Bonz A, Vahl CF, Hagl S. Contractile behaviour and intracellular calcium during afterloaded contraction in mitral valve disease. Thorac Cardiovasc Surg. 1997;45:280–6. doi: 10.1055/s-2007-1013750. [DOI] [PubMed] [Google Scholar]
- [25].Kirsch GE, Brown AM. Kinetic properties of single sodium channels in rat heart and rat brain. J Gen Physiol. 1989;93:85–99. doi: 10.1085/jgp.93.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Smith RD, Goldin AL. Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci. 1998;18:811–20. doi: 10.1523/JNEUROSCI.18-03-00811.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Patel SP, Campbell DL. Transient outward potassium current, ‘Ito’, phenotypes in the mammalian left ventricle: underlying molecular, cellular and biophysical mechanisms. J Physiol. 2005;569:7–39. doi: 10.1113/jphysiol.2005.086223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Charpentier F, Merot J, Loussouarn G, Baro I. Delayed rectifier K+ currents and cardiac repolarization. J Mol Cell Cardiol. 2010;48:37–44. doi: 10.1016/j.yjmcc.2009.08.005. [DOI] [PubMed] [Google Scholar]
- [30].Chandler NJ, Greener ID, Tellez JO, Inada S, Musa H, Molenaar P, et al. Molecular architecture of the human sinus node. Circulation. 2009;119:1562–75. doi: 10.1161/CIRCULATIONAHA.108.804369. [DOI] [PubMed] [Google Scholar]
- [31].Fozzard HA, Hanck DA. Structure and function of voltage-dependent sodium channels: comparison of brain II and cardiac isoforms. Physiol Rev. 1996;76:887–926. doi: 10.1152/physrev.1996.76.3.887. [DOI] [PubMed] [Google Scholar]
- [32].Sakakibara Y, Wasserstrom JA, Furukawa T, Jia H, Arentzen CE, Hartz RS, et al. Characterization of the sodium current in single human atrial myocytes. Circ Res. 1992;71:535–46. doi: 10.1161/01.res.71.3.535. [DOI] [PubMed] [Google Scholar]
- [33].Coraboeuf E, Deroubaix E, Coulombe A. Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. Am J Physiol. 1979;236:H561–7. doi: 10.1152/ajpheart.1979.236.4.H561. [DOI] [PubMed] [Google Scholar]
- [34].Haufe V, Cordeiro JM, Zimmer T, Wu YS, Schiccitano S, Benndorf K, et al. Contribution of neuronal sodium channels to the cardiac fast sodium current INa is greater in dog heart Purkinje fibers than in ventricles. Cardiovasc Res. 2005;65:117–27. doi: 10.1016/j.cardiores.2004.08.017. [DOI] [PubMed] [Google Scholar]
- [35].Haufe V, Camacho JA, Dumaine R, Gunther B, Bollensdorff C, von Banchet GS, et al. Expression pattern of neuronal and skeletal muscle voltage-gated Na+ channels in the developing mouse heart. J Physiol. 2005;564:683–96. doi: 10.1113/jphysiol.2004.079681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Huang B, El-Sherif T, Gidh-Jain M, Qin D, El-Sherif N. Alterations of sodium channel kinetics and gene expression in the postinfarction remodeled myocardium. J Cardiovasc Electrophysiol. 2001;12:218–25. doi: 10.1046/j.1540-8167.2001.00218.x. [DOI] [PubMed] [Google Scholar]
- [37].Bril A, Kinnaird AA, Man RY. Comparison of the sodium currents in normal Purkinje fibres and Purkinje fibres surviving infarction—a pharmacological study. Br J Pharmacol. 1989;97:999–1006. doi: 10.1111/j.1476-5381.1989.tb12554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, et al. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–67. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- [39].Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, et al. Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol. 2010;55:2330–42. doi: 10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.