

Abstract
Osteoarthritis (OA) is a progressive disorder with high incidence in the ageing human population that still has no treatment currently. This disorder induces the breakdown of articular cartilage, leading to the exposure and damage of bone surfaces. For a global understanding of OA development, the systematic integration of known OA-related proteins with protein–protein interaction (PPI) networks is required. In this work, the OA-related interactome was reconstructed using multiple data sources to have the most up-to-date information on OA-related proteins and their interactions. We then combined emergent concepts in network medicine to detect new unclassified OA-related proteins. The mapping of known OA-related proteins with PPI networks showed that these proteins are locally connected to each other and agglomerated in a large component. To expand this module, we applied a diffusion-based algorithm that probabilistically induces more searches in the vicinity of the seed OA-related proteins. As a result, the 10 topmost ranked proteins were connected to the OA disease module, supporting the local hypothesis. We computed structural modules and selected those that had the highest enrichment of OA-related proteins. The identified molecules show a link between structural topology and disease dysfunctionality. Interestingly, the protein Q6EEV6 was highlighted for OA association by both methods, reinforcing the potential involvement of this protein. These results suggest that similar disease-connected modules may exist in different human disorders, which could lead to systematic identification of genes or proteins that have a joint role in specific disease phenotypes.
Keywords: osteoarthritis disorder, network medicine, chondrocytes
1. Introduction
Osteoarthritis (OA) is a progressive disorder mainly characterized by the breakdown of articular cartilage, leading to the exposure and damage of bone surfaces [1]. OA is a widespread cause of joint degeneration, mostly affecting synovial joints, such as those found in the knees, hips and fingers, and is one of the leading disabling human conditions worldwide. Despite its high incidence, no treatment currently exists that can replace damaged cartilage or prevent cartilage degeneration [2]. A combination of various anti-inflammatory drugs that reduce joint inflammation is currently the best method to treat OA patients, although the most effective treatment appears to be weight loss to reduce the stress on joint cartilage. This lack of appropriate pharmaceutical intervention suggests that new approaches are needed to understand the functional perturbations linked to OA and to identify key proteins that can be targeted for therapeutic use.
Cartilage is a type of connective tissue found in the nose and ears, as well as in joints, such as the knees and fingers. Three major types are found in the human body, including articular (hyaline), elastic and fibrocartilage. Articular cartilage is the most prevalent and is found at the surface of synovial joints, primarily functioning to facilitate joint motion and distribute the forces of weight bearing [3]. Chondrocytes are the only cell population found within mature articular cartilage, constituting approximately 2–5% of the total tissue, with the rest consisting of a tough and flexible extracellular matrix (ECM). Chondrocytes control matrix turnover through the production and release of collagen, proteoglycans and enzymes involved in cartilage metabolism [4]. Chondrocytes have been the most studied component of the disease system to determine how changes in their function may be linked to OA. Whereas healthy articular cartilage is believed to be permanent, consisting of chondrocytes with low metabolic activity, in conditions, for example OA, they enter a stage of maturation becoming proliferative and then hypertrophic chondrocytes [5]. In this state, chondrocytes increase ECM synthesis but cannot compensate for the increased matrix degradation by proteolytic matrix enzymes. This leads to an overall decrease in matrix synthesis resulting in the breakdown of articular cartilage.
Biomedical knowledge combined with network science tools aim to identify genes or proteins that can be probably associated with specific disease phenotypes [6,7]. Several computation-based methods have been proposed to uncover disease-associated genes. Disease candidate molecules were prioritized using linkage methods [8–10], where macromolecules that interact directly with a disease protein were assumed to be associated with the same disorder. Another approach seeks to exploit the fact that molecules which belong to the same structural and functional module may have a joint role. The extension of this concept to disease phenotypes means that identifying an unclassified protein that belongs to a disease module with high enrichment over a random selection may indicate that the protein is likely to be involved in the same disorder [11,12].
Diffusion-based methods were also used to identify proteins that are related to specific disorders. The random walk particles navigate through the complex interactome structure, starting from those protein products originated by known disease genes. The nodes most often visited by the random walkers are considered to be more related to the known disorder genes. Several algorithmic versions of this concept have been applied to human diseases, from prostate cancer to Alzheimer's disease [13,14]. One of the leading network-based diffusion algorithms is the PageRank, which is not only well known to rank web content but can also potentially classify any type of nodes given a suitable network structure [15,16]. The algorithm does not rank nodes using their intrinsic features, rather it uses the collective wisdom of the network, considering each link as a node that has as a direct vote to increase the node's ranking. Recently, the PageRank algorithm has been successfully used to identify disease-causing genes based on protein interactions, to detect cancer genes in protein networks and to classify the metabolic network of the tuberculosis bacterium [17–19]. It is expected that the advent of full-genome sequencing and genome-wide association studies can help to provide more accurate disease gene (seeds) as well as better validation methods.
Network and systems approaches have not been widely applied to complex skeletal diseases as arthritis so far. A molecular interaction map was presented for processes ongoing in patients affected by rheumatoid arthritis (RA) based on the results of functional genomic analyses and pathways available in the literature [20], which identified a new potential drug target for the treatment of RA [21]. Gene expression data from pooled joint tissue were overlaid with a protein interaction network to identify components that were significantly upregulated during the development of OA [22]. However, for a more global understanding of OA development, the systematic integration of known OA-related proteins with protein interaction networks is required.
In this study, we combine the recently introduced concepts in network medicine (structural proximity, modularity and diffusion algorithmic approach) to detect new unclassified OA-related proteins using protein interaction networks [6]. We first constructed a manually curated interactome network composed of all proteins linked to at least one chondrocyte protein. We then overlaid OA-related proteins onto this protein interaction network and showed that the non-random placement of the OA-related proteins in the interactome offers a new avenue for the prediction of OA relation. Next, the fully connected component composed of only OA-related proteins was extracted and the PageRank-based diffusion algorithm, with a personalized vector constructed using the known OA-related proteins, was applied to the interactome network. The personalized PageRank algorithm has already been used for biological applications in the context of protein interaction networks [18].
The results show that the 10 topmost ranked proteins, excluding natural overlaps with personalized vector proteins, are neighbours of the main OA-related connected component, which leads to an expansion of the OA disease module. Moreover, the computation of the modularity applied to the full network leads to the identification of a set of modules, in which the enrichment of OA-related proteins over a random selection was significantly higher. Among them, one module shared a protein already identified in the 10 topmost ranked proteins, leading to the same prediction using two independent methods.
2. Material and methods
(a). Datasets
To construct a suitable network for the investigation of OA, we first used the complete list of protein–protein interactions from the Human Protein Reference Database (HPRD) [23] as a baseline protein interaction network. We then used several large-scale proteomic studies to identify proteins that were detected in healthy and osteoarthritic chondrocytes. Seventeen studies were used in total (table 1), among which nine focused on healthy chondrocyte proteomics, whereas the remaining eight contained data about chondrocyte proteins that had their expression altered in OA. All 17 studies were used to build the chondrocyte protein interaction network, as it was assumed that proteins detected in OA cells were still present in healthy cells.
Table 1.
List of proteomic studies used to construct the chondrocyte protein interactome.
| healthy chondrocyte proteome | altered proteins in OA |
|---|---|
| Akiyama et al. [24] | Gobezie et al. [25] |
| Cillero-Pasto et al. [26] | Guo et al. [27] |
| Guo et al. [28] | Lambrecht et al. [29] |
| Jmeian & El Rassi [30] | Ling et al. [31] |
| Pecora et al. [32] | Ma et al. [33] |
| Ruiz-Romero et al. [34] | Rosenthal et al. [35] |
| Ruiz-Romero et al. [36] | Ruiz-Romero et al. [37] |
| Ruiz-Romero et al. [38] | Wu et al. [39] |
| Vincourt et al. [40] |
Each protein identified in these proteomic studies was queried within the HPRD interaction network to extract all proteins interacting with the query protein, assuming that if one interaction partner is present in chondrocytes, the second partner must be present too for the interaction to occur, even though the second partner may not necessarily have been detected by the proteomic analysis. This process resulted in a protein interaction network specific to chondrocyte cells. All proteins names were converted to UniProt accession identifiers in order to eliminate any duplication owing to different naming conventions. Proteins that have altered levels in OA were extracted from the eight OA studies and labelled by a specific attribute. The complete network comprised 2955 nodes, among which 168 were related to OA (figure 1). Background data for the full network is provided in the electronic supplementary material.
Figure 1.
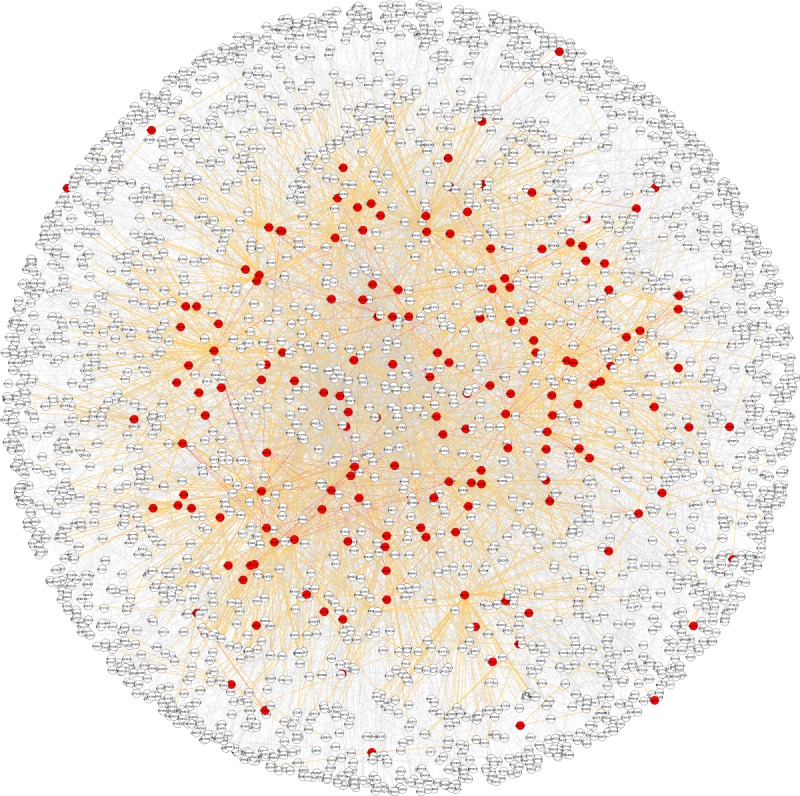
The chondrocyte protein interactome network is composed of 2955 nodes, 168 out of them being OA-related proteins (labelled in red). Links between OA-related proteins are denoted in red. Interactions between OA-related and non-OA-related proteins are indicated by orange links.
(b). PageRank algorithm with personalized vector
The PageRank algorithm describes a diffusion process, where a particle moves by following the links of the network and jumps to randomly selected nodes with given probabilities. These probabilities are parameters of the model. As a result, the frequency of visitation of a node by the particle at the stationary state of this diffusion process is the PageRank of the node and it is used to determine its ranking relative to other nodes of the network [15,16].
The fundamental PageRank algorithm can be described using the following equation:
| 2.1 |
where 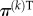 is the PageRank row vector at the kth iteration. Each component of the vector is associated with a node, therefore
is the PageRank row vector at the kth iteration. Each component of the vector is associated with a node, therefore 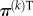 gives the probability that the particle is at each node of the network at time step k. The matrix G can be written as
gives the probability that the particle is at each node of the network at time step k. The matrix G can be written as
| 2.2 |
where 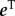 is the row vector of all 1 s and α is a parameter that indicates the proportion of time the random surfer follows a random teleportation process, or conversely 1 – α is the proportion of time it is guided through the links on the network structure. The stochastic matrix S reads as
is the row vector of all 1 s and α is a parameter that indicates the proportion of time the random surfer follows a random teleportation process, or conversely 1 – α is the proportion of time it is guided through the links on the network structure. The stochastic matrix S reads as 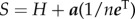 , which is constructed using the row normalized probability transition matrix H and the dangling node vector
, which is constructed using the row normalized probability transition matrix H and the dangling node vector 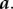 This column vector has 1 s in components associated with nodes with no-outgoing links and 0 s for the rest of the components.
This column vector has 1 s in components associated with nodes with no-outgoing links and 0 s for the rest of the components.
Here, we can distinguish between random teleportation αF and personalized teleportation αP probabilities [18]. Each i component of the personalized vector takes the value 1/W if the node i is a known OA-related protein, and 0 otherwise. W is a normalizing weighted factor that represents the total number of OA-related proteins.
Then, equation (2.2) can read as
| 2.3 |
By substituting the expression of matrix S and by defining a personalized vector as 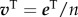 , then equation (2.1) reads as
, then equation (2.1) reads as
| 2.4 |
Our computations were performed using αF = αP = 0.15 and a value of ɛ = 0.0001 for the error of convergence of the algorithm at the stationary state.
(c). Statistical significance of the observed size of the osteoarthritis-related connected component
The OA-related network was randomized using a shuffling algorithm that exchanges edges in the network but preserves the degree distribution. For each of the 100 network samples, we generated with the same size as the observed OA-related network, we performed 200 shuffling edges steps. Then, the mean value and standard deviation of the observed giant connected component were computed. The statistical significance of the analysis was examined using the two-tailed p-value, as shown in the Results section.
(d). Modularity algorithm
To identify topological modules, we used a multi-scale method in which modularity is optimized by means of a greedy local algorithm. The nodes that form a discovered partition are then merged and a supernode for each new partition is created. The processed is repeated in the re-scaled network [41].
The algorithm starts with as many different modules as there are nodes. For each node i, the gain of modularity is evaluated when removing node i from its module and by assigning it to another module j. The node i will be finally assigned to the community that offers the highest gain and is positive. The process is applied for all nodes until no new gains can be achieved. The modularity of the network will increase in each iteration and tends to maximize the following modularity functional form:
where m is the number of edges, 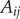 is an element of the adjacency matrix of the network, ki denotes the degree of node i and ci is the community to which node i is assigned. The δ(x, y) function is 1 if x = y and 0, otherwise.
is an element of the adjacency matrix of the network, ki denotes the degree of node i and ci is the community to which node i is assigned. The δ(x, y) function is 1 if x = y and 0, otherwise.
(e). Community analysis
The detection of communities in networks is a common method to identify groups of closely related entities or functional modules. We applied a community detection algorithm by Blondel et al. [41] to detect groups of highly connected OA-related proteins in the protein–protein interaction (PPI) network. This algorithm constructs a hierarchical community structure for the network, where modularity is maximized at each level. We focused on the first level and tested the detected communities for OA enrichment. The probability of detecting a number n of OA-related proteins in a community of size N, considering that the ratio of OA-related proteins in the full network was 0.0569 (168/2955), was calculated using the hypergeometric distribution. A community was deemed to be significantly enriched in OA-related proteins if this probability was lower than 0.05. As there is generally no unique solution to decompose the network into communities maximizing modularity, the exact composition of communities may vary when the algorithm is run several times. We therefore repeated the analysis 10 times and retained for each significant community the composition observed the largest number of times.
3. Results
(a). The osteoarthritis-related proteins are locally connected to each other and agglomerated in a large component
The assembled interactome network is composed of 2955 nodes, of which 168 are known OA-related proteins. The visualization of the giant connected component of the network can be seen in figure 1. Note that 37 nodes (including six OA-related proteins) and 21 edges did not belong to the main component so they are absent in figure 1. The total number of links is 5477 with 151 links connecting two OA-related proteins (red), 3310 connecting OA-related to non-OA-related proteins (orange) and 2016 connecting non-OA-related proteins to each other.
Evidence of the non-random localizations of disease genes is necessary in our network-based approach to identify unclassified disease-related molecules. To investigate whether the OA-related proteins were distributed randomly in the network, we extracted all the OA-related proteins by keeping only edges that were connecting two OA-related proteins. The resulting network is shown in figure 2. Out of 168 proteins, 162 are connected to the main component of the interactome and 89 nodes form an OA-related connected component. These results show that the OA-related proteins tend to be locally connected to each other and a large fraction of them are agglomerated in a large component (table 2).
Figure 2.
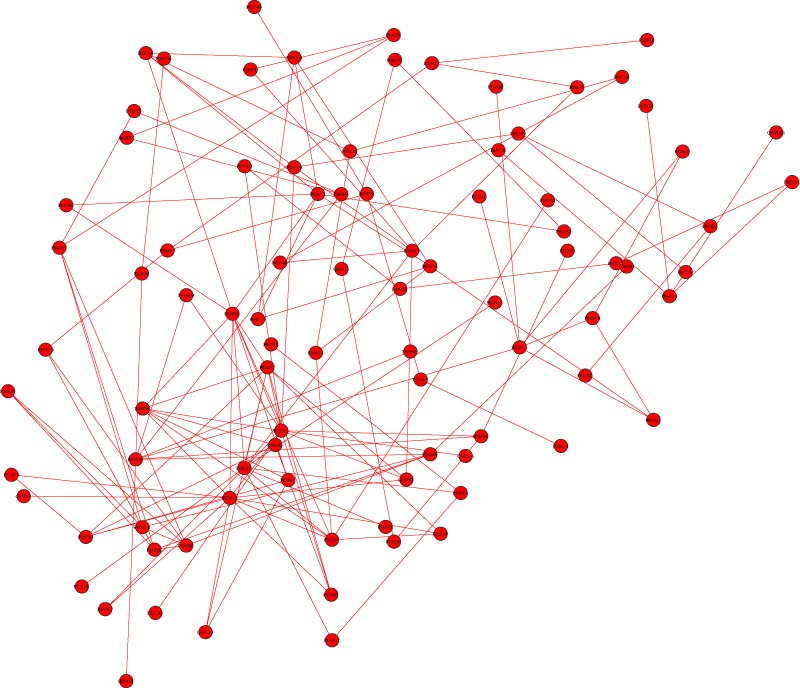
Extracted giant connected component composed of OA-related proteins. Links between OA-related and non-OA-related proteins were removed. The layout of proteins is identical to figure 1.
Table 2.
Main statistical network features of the analysed networks.
| full protein network (giant connected component) | OA-related extracted network | OA-related giant connected component | |
|---|---|---|---|
| nodes | 2955 (2918) | 168 (89) | 89 |
| average degree | 3.7 | 1.79 | 3.2 |
| clustering degree | 0.069 | 0.092 | 0.174 |
| average path length | 4.3 | 4.8 | 4.8 |
The analysis of the degree distribution of the full interactome network reveals a non-random structure. The network follows a power law, a characteristic pattern of scale-free networks. On the other hand, the OA-related network, which is of smaller size, decays faster than a power law. The cumulative degree distributions of the networks are shown in figure 3. The fact that the full interactome network follows a scale-free distribution also allows the use of diffusion-based algorithms to analyse the network.
Figure 3.

(a) The cumulative degree distributions for both the fully connected network and the extracted OA-related connected network.
(b). The osteoarthritis-related connected component statistically emerges as disease module seed to be expanded
To evaluate the significance of the OA-related connected network, we performed two statistical analyses (see Material and methods for details). The network was randomized with 2000 shuffling edges for each of the 100 network samples. The component size distribution for randomized networks is shown to be normal distribution (figure 4, light blue curve), which has a mean value of 97 and a standard deviation of 2.45. The results show that the observed giant component (89) is over 3 s.d. from the mean of a randomized network sample (97). The statistical significance analysis leads to a two-tailed p-value of 8.70 × 10–3. This indicates that the observed size of the OA-related connected component would be highly unlikely in a random network.
Figure 4.
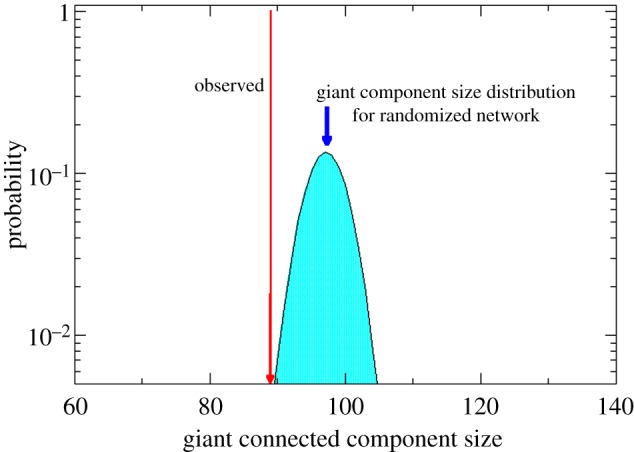
Statistical significance analysis of the giant connected component. The expected value obtained in a randomized network (blue curve) is over 3 s.d. from the observed value. The statistical significance analysis gives a two-tailed p-value of 8.70 × 10–3 (blue curve).
(c). Expansion of the osteoarthritis-related connected component
It has been hypothesized that molecular routes can overlap with the shortest paths between known disease-related molecules. This network parsimony principle has supported the idea of using diffusion-based algorithms. We therefore applied the PageRank algorithm with personalized vector to the interactome network in order to predict new disease candidate proteins that are likely to be associated with OA. As shown in a recent work, the success of the PageRank algorithm in ranking nodes in large networks is rooted in the fact that the degree distribution follows a power law, which makes the top-ranked node easier to find and the ranking more reliable when the networks grow in size [42]. Here, the personalized vector is simply a vector that includes the known OA-related proteins. This vector takes a non-zero weighted probability for each protein that is associated with OA, and a zero score for the remaining proteins, where the sum of all scores is normalized to one. The PageRank results showed that several already known OA-related proteins appeared in the top ranks. Therefore, we selected the 10 top-ranked proteins excluding known OA-related proteins (table 3). These proteins were added to the OA-related network and are shown in figure 5 (green nodes). Interestingly, the results show that the identified proteins are in the neighbourhood of the OA-related connected component and always adjacent to at least one OA-related protein. This fact supports the idea of a disease module that can be expanded using the collective wisdom of the full interactome network, unveiling new potential disease-protein candidates.
Table 3.
List of top 10 candidate proteins predicted to be OA-related by the PageRank algorithm, excluding known OA-related proteins.
| UnitProt | standard name | main biological activity |
|---|---|---|
| P26641 | elongation factor 1-γ (EEF1G) | protein biosynthesis |
| Q6EEV6 | small ubiquitin-related modifier 4 (SUMO-4) | ubiquitin conjugation pathway |
| P17252 | protein kinase C-α (PKCA) | angiogenesis, regulation of apoptosis |
| P61981 | 14-3-3 protein gamma (YWHAG) | cellular membrane organization |
| P68104 | elongation factor 1-α 1 (EEF1A1) | protein biosynthesis |
| P28482 | mitogen-activated protein kinase 1 (MAPK1) | transcription regulation |
| P63104 | 14-3-3 protein zeta/delta (YWHAZ) | cellular membrane organization |
| P35222 | catenin β-1 (CTNNB1) | cell adhesion, transcription regulation |
| P31946 | 14-3-3 protein β/α (YWHAB) | cellular membrane organization |
| P62736 | actin, aortic smooth muscle (ACTA2) | cell motility |
Figure 5.
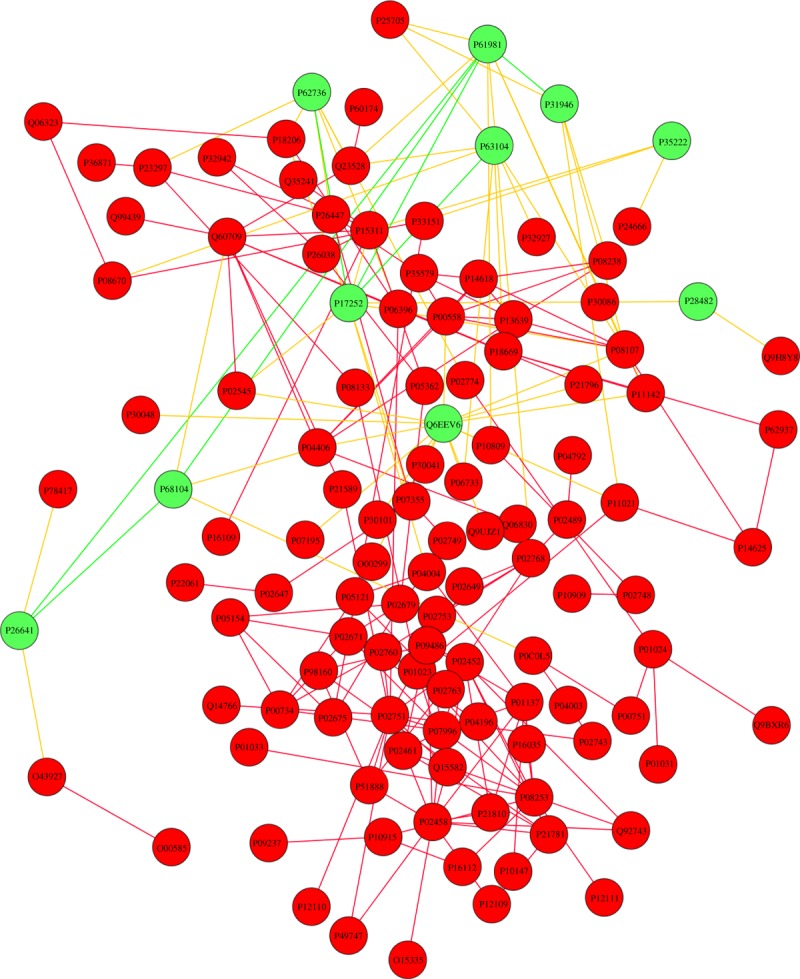
Expansion of the OA-related giant component by adding the 10 top-ranked proteins by PageRank analysis excluding known OA-related proteins (green nodes). Links between newly identified proteins are shown in green, whereas links between new proteins and known OA-related proteins are shown in yellow.
(d). Module enrichment analysis
In network medicine, the disease module hypothesis assumes that the molecules related to a specific disorder tend to be located in the same network neighbourhood, forming a structural disease module. To support our analysis done on new identified OA-related proteins using a diffusion algorithmic method, we used a different approach based on maximizing modularity in complex networks that detects modules with the highest enrichment of OA-related proteins.
The detection of communities in networks is a common method to identify groups of closely related entities or functional modules. We applied a community detection algorithm by Blondel et al. [41] to detect groups of highly connected OA-related proteins in the PPI network. This method optimizes modularity using a greedy local algorithm, where nodes are iteratively exchanged between modules as long as the modularity can be increased and is positive. This method is widely used owing to its efficiency to deal with large networks as well as the high quality of results it provides. It scored the highest rank when evaluated in computer-generated benchmark problems [43,44].
We focused on those modules that showed highest enrichment of OA-related proteins. Overall, four significant communities enriched in OA-related proteins were detected (figure 6). Ranked from the highest to lowest significance, the first community contains three OA-related proteins out of seven (p = 0.005) (figure 6b), the next contains three OA-related proteins out of 11 (p = 0.021) (figure 6c), the next contains two OA-related proteins out of five (p = 0.029) (figure 6d) and the last contains two OA-related proteins out of six (p = 0.041) (figure 6e). All non-OA-related proteins belonging to these communities are therefore likely to be subjected to disease perturbations owing to their high interconnectivity with disease-associated proteins.
Figure 6.
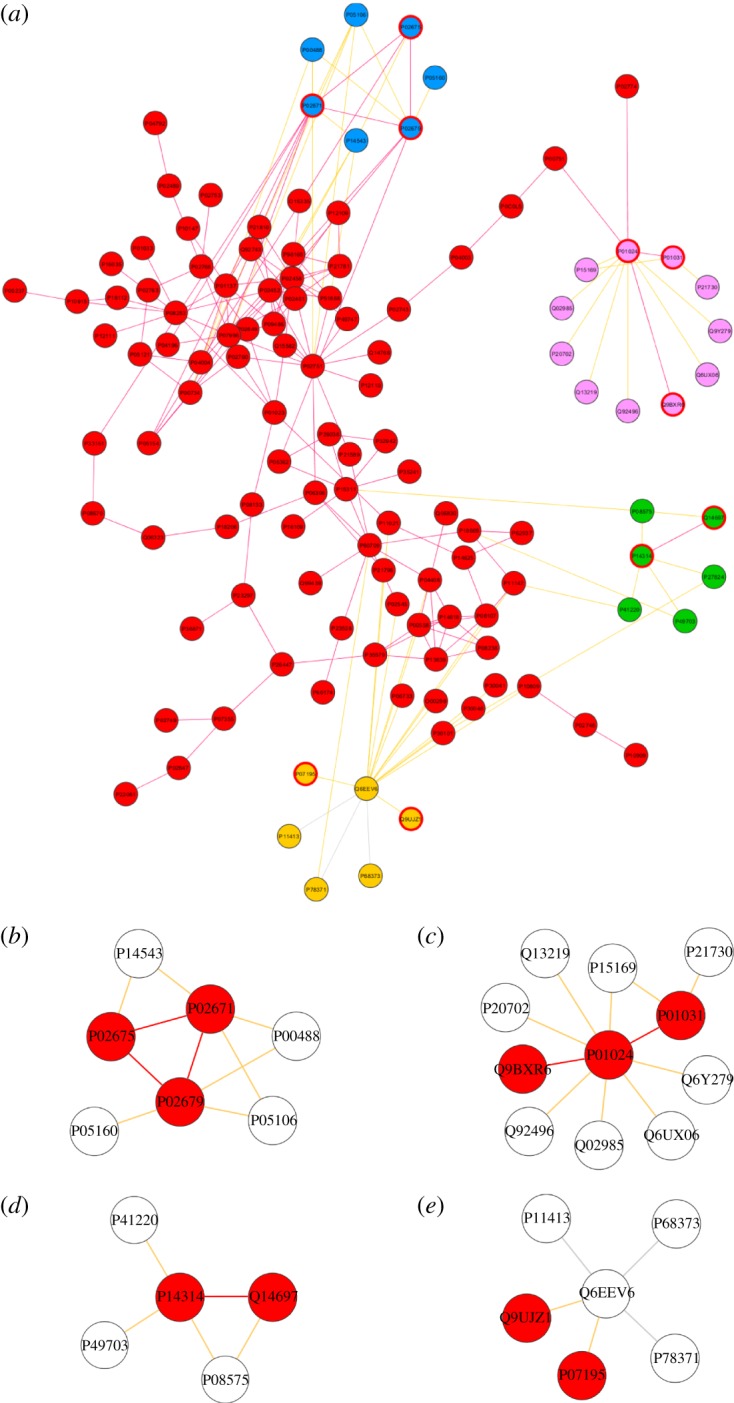
(a) Identified modules with significant OA enrichment connected to the main OA-related connected component. Nodes of each module are denoted by a specific colour. (b–e) In each module, the OA-related proteins are highlighted in red.
We then filtered the proteins found in these modules to investigate whether there is an overlap between them and the 10 top-ranked proteins detected by the diffusion algorithm (table 3). Interestingly, one protein was predicted by these two independent methods, namely Q6EEV6 (figure 7).
Figure 7.
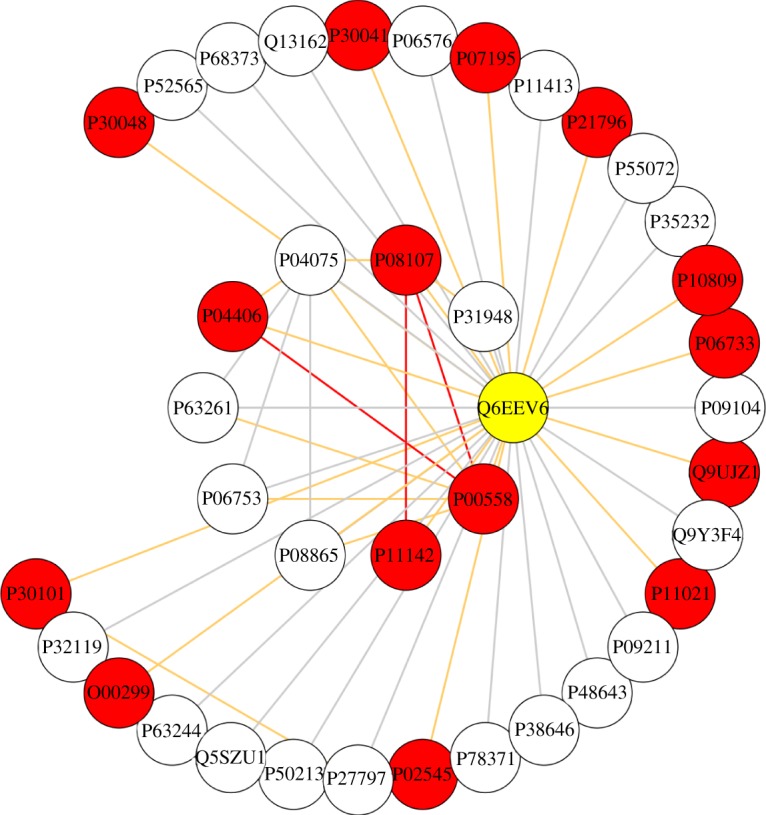
The protein Q6EEV6 was identified among the top 10 candidates for OA association by the PageRank analysis and also by the modularity analysis. Note the complex interconnectivity in the protein's neighbourhood.
The probability for this simultaneous detection to occur randomly is only 2 × 10−5. Q6EEV6 is the small ubiquitin-related modifier 4 (SUMO-4). SUMO-4 was previously associated with susceptibility to type I diabetes and cellular response to oxidative stress, but no explicit link to cartilage degradation or skeletal diseases was found. In total, SUMO-4 was found to interact with 15 OA-related proteins, including several proteins involved in glycolysis and redox regulation (table 4). The potential involvement of this protein in OA is hereby reinforced.
Table 4.
List of OA-related interacting partners of the Q6EEV6 (SUMO-4) protein.
| UnitProt | standard name | main biological activity |
|---|---|---|
| P00558 | phosphoglycerate kinase 1 (PGK1) | glycolysis |
| P11142 | heat shock cognate 71 kDa protein (HSP8) | transcription regulation stress response |
| P04406 | glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | glycolysis translation regulation |
| P08107 | heat shock 70 kDa protein 1A/1B (HSPA1A/1B) | protein folding stress response |
| P30048 | thioredoxin-dependent peroxide reductase, mitochondrial (PRDX3) | redox regulation |
| P30041 | peroxiredoxin-6 (PRDX6) | redox regulation |
| P07195 | l-lactate dehydrogenase B chain (LDHB) | glycolysis |
| P21796 | voltage-dependent anion-selective channel protein 1 (VDAC1) | ion transport |
| P10809 | 60 kDa heat shock protein, mitochondrial (HSPD1) | protein folding |
| P06733 | α-enolase (ENO1) | glycolysis transcription regulation |
| Q9UJZ1 | stomatin-like protein 2, mitochondrial (STOML2) | mitochondrial regulation |
| P11021 | 78 kDa glucose-regulated protein (HSPA5) | protein complex assembly |
| P02545 | prelamin-A/C (LMNA) | nuclear assembly |
| O00299 | chloride intracellular channel protein 1 (CLIC1) | ion transport |
| P30101 | protein disulfide-isomerase A3 (PDIA3) | redox regulation |
4. Discussion and conclusion
Network medicine is based on several widely used principles related to network structure and functionality. The local hypothesis or structural proximity suggests that proteins, which belong to the same disorder, are assumed to interact with each other. Next, the disease module hypothesis assumes that the molecules related to a specific disorder tend to be located in the same network neighbourhood, leading to a structural disease module. Moreover, molecular routes often overlap with the shortest paths between known disease-related molecules. This network parsimony principle has encouraged the usage of diffusion-based algorithms. While the structural proximity has been very well experimentally supported, the existence of disease modules overlapping with structural modules and the parsimony principles have been less well quantified or verified.
In this work, we have combined these three organizing principles to analyse the OA-related network. The interactome was reconstructed using multiple data sources and manual curation to have the most up-to-date information on OA-related proteins and their interactions. First, the integration of known OA-related proteins with protein interaction networks has shown that the OA-related proteins are locally connected to each other and are furthermore agglomerated in a large component. This component may represent the seed of the OA disease module, which can be expanded by identifying OA-related proteins in the neighbourhood. To this end, we have applied a diffusion-based algorithm with a personalized vector that probabilistically induces more searches in the structural vicinity of the seed OA-related proteins. As shown in Ivan & Grolmusz [18], this feature is essential to successfully identify new candidate disease proteins in protein–protein interaction networks. In addition, we also allowed random teleportation to explore distant locations on the network with the same probability. As a result, the 10 topmost ranked proteins were connected to the OA disease module, supporting the local hypothesis.
To investigate the overlap between the structural and disease module, we computed structural modules and selected those ones that had the highest enrichment of OA-related proteins. The identified molecules show a link between structural topology and disease dysfunctionality. In addition, they confirm the local hypothesis because they are also connected to the main OA-related connected component. Interestingly, the protein Q6EEV6 (SUMO-4) was identified among the top 10 candidates for OA association by the PageRank analysis and was also detected in the modular analysis. SUMO-4 mutations were previously associated with other diseases. A study by Guo et al. [45] suggested that a substitution in the SUMO-4 gene is associated with increased risk of type I diabetes mellitus. While subsequent studies confirmed these findings in Asian populations [46,47], these observations were not consistent in European Caucasians [48], suggesting that further study is required to fully elucidate the relationship between SUMO-4 mutations and type I diabetes mellitus. The effect of SUMO-4 mutations in RA and systemic lupus erythematosus has been investigated, showing no consistent sign of predisposition to these diseases [49–51].
Several glycolytic enzymes were found among the top candidate proteins predicted by our analysis to be involved in OA. The importance of glycolysis in the prevention of cartilage breakdown and responses to apoptotic processes has already been suggested [29,37]. Chondrocytes require a large amount of energy to regulate the cartilage ECM through the release of proteins, such as collagens and proteoglycans. Perturbation of this cellular function may lead to a decrease in ATP production, thereby preventing the normal control of matrix turnover and contributing to the gradual breakdown of cartilage, as the decreased ECM is no longer as effective in distributing the forces of weight bearing.
Other proteins are involved in the stress response and regulation of apoptosis. The link between cartilage degeneration and chondrocyte apoptosis has already been described and is thought to play an important role in the pathogenesis of OA [52]. In particular, the level of protein kinase C-α (P17252) was found to be significantly increased in human knee OA cartilage by a recent study [53] in correlation with an increase in chondrocyte apoptosis. This observation confirms that our network analysis is a valid approach to identify candidate proteins for disease association, as the link between P17252 and OA is too recent to have been included in databases and did not appear in our original dataset.
The third main group of proteins is related to cellular adhesion and mechanotransduction. Actins are ubiquitously expressed in eukaryotic cells and changes in the cytoskeleton are believed to be associated with the onset of the disease [54]. Protein kinase C-α itself is known to be involved in the regulation of cell adhesion and motility [55]. Catenin β-1 (P35222) is another detected protein that is involved in the regulation of cell adhesion through the Wnt signalling pathway. Although a direct link between catenin β-1 and OA was not established, the activation of catenin β-1 signalling was observed to lead to premature chondrocyte differentiation in mice, which is a symptom of OA [56,57].
To summarize, we have reconstructed a large-scale OA-related interactome, in which the module of OA-related proteins emerged as potential disease seed. The presence of a main connected component of OA-related proteins enables the discovery of newly identified proteins using network diffusion algorithms. The promising results shown here suggest that similar disease-connected modules may also exist in different human disorders. The curation of a high volume of data for other disorders and its overlapping with the background interactome network could also lead to highly connected disorder modules (seeds) that could be expanded using a rich variety of computational methods [43]. This network medicine approach could lead to systematic identification of genes or proteins that can have a joint role in a specific disease phenotype.
Acknowledgements
We thank Raymond Boot-Handford and Timothy Hardingham for helpful feedback on this manuscript.
Funding statement
This work was financed in part by the European Commission through the Sybil project (602300).
References
- 1.Altman R, et al. 1986. Development of criteria for the classification and reporting of osteoarthritis: Classification of osteoarthritis of the knee. Arthritis Rheum. 29, 1039–1049 (doi:10.1002/art.1780290816) [DOI] [PubMed] [Google Scholar]
- 2.Messier SP. 2009. Obesity and osteoarthritis: disease genesis and nonpharmacologic weight management. Med. Clin. North Am. 93, 145–159 (doi:10.1016/j.mcna.2008.09.011) [DOI] [PubMed] [Google Scholar]
- 3.Koopman WJ, Moreland LW. 2005. Arthritis and allied conditions: a textbook of rheumatology. Philadelphia, PA: Lippincott Williams & Wilkins [Google Scholar]
- 4.Muir H. 1995. The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioessays 17, 1039–1048 (doi:10.1002/bies.950171208) [DOI] [PubMed] [Google Scholar]
- 5.Sandell L, Aigner T. 2001. Articular cartilage and changes in arthritis: cell biology of osteoarthritis. Arthritis Res. 3, 107–113 (doi:10.1186/ar148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabasi A-L, Gulbahce N, Loscalzo J. 2011. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 (doi:10.1038/nrg2918) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Csermely P, Korcsmáros T, Kiss HJM, London G, Nussinov R. 2013. Structure and dynamics of biological networks: a novel paradigm of drug discovery. A comprehensive review. Pharmacol. Ther. 138, 333–408 (doi:10.1016/j.pharmthera.2013.01.016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oti M, Snel B, Huynen MA, Brunner HG. 2006. Predicting disease genes using protein-protein interactions. J. Med. Genet. 43, 691–698 (doi:10.1136/jmg.2006.041376) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krauthammer M, Kaufmann CA, Gilliam TC, Rzhetsky A. 2004. Molecular triangulation: bridging linkage and molecular-network information for identifying candidates genes in Alzheimer's disease. Proc. Natl Acad. Sci. USA 101, 15 148–15 153 (doi:10.1073/pnas.0404315101) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Franke L, van Bakel H, Fokkens L, de Jong ED, Egmont-Petersen M, Wijmenga C. 2006. Reconstruction of a functional human gene network, with an application for prioritizing positional candidates genes. Am. J. Hum. Genet. 78, 1011–1025 (doi:10.1086/504300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Navlakha S, Kingsford C. 2010. The power of protein interaction networks for associating genes with diseases. Bioinformatics 26, 1057–1063 (doi:10.1093/bioinformatics/btq076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lage K, et al. 2007. A human phenome-interaction network of protein complexes implicated in genetic disorders. Nat. Biotechnol. 25, 309–316 (doi:10.1038/nbt1295) [DOI] [PubMed] [Google Scholar]
- 13.Kohler S, Bauer S, Horn D, Robinson PN. 2008. Walking the interactome for prioritization of candidate disease genes. Am. J. Hum. Genet. 82, 949–958 (doi:10.1016/j.ajhg.2008.02.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. 2010. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 6, e1000641 (doi:10.1371/journal.pcbi.1000641) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brin S, Page L. 1998. The anatomy of a large-scale hypertextual web search engine. Comput. Newt. ISDN Syst. 30, 107–117 (doi:10.1016/S0169-7552(98)00110-X) [Google Scholar]
- 16.Langville AN, Meyer CD. 2006. Google's PageRank and beyond: the science of search engine rankings. Princeton, NJ: Princeton University Press [Google Scholar]
- 17.Chen P, Aronow BJ, Jegga AG. 2009. Disease candidate gene identification and priorization using protein interaction networks. BMC Bioinform. 10, 73–83 (doi:10.1186/1471-2105-10-73) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ivan G, Grolmusz V. 2011. When the web meets the cell: using PageRank for analyzing protein interaction networks. Bioinformatics 27, 405–407 (doi:10.1093/bioinformatics/btq680) [DOI] [PubMed] [Google Scholar]
- 19.Banky D, Ivan G, Grosmusz V. 2013. Equal opportunity for low-degree network nodes: a PageRank-based method for protein target identification in metabolic graphs. PLoS ONE 8, e54204 (doi:10.1371/journal.pone.0054204) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu G, Zhu L, Dent JE, Nardini C. 2010. A comprehensive molecular interaction map for rheumatoid arthritis. PLoS ONE 5, e10137 (doi:10.1371/journal.pone.0010137) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dent J, Nardini C. 2013. From desk to bed: computational simulations provide indication for rheumatoid arthritis clinical trials. BMC Syst. Biol. 7, 10 (doi:10.1186/1752-0509-7-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loeser RF, Long DL, Olex AL, Fetrow JS. 2013. A systems biology approach identifies heparin-binding EGF-like growth factor as a potential mediator in OA. Osteoarthritis Cartilage 21, S234–S235 (doi:10.1016/j.joca.2013.02.482) [Google Scholar]
- 23.Prasad TSK, et al. 2009. Human protein reference database: 2009 update. Nucleic Acids Res. 37, D767–D772 (doi:10.1093/nar/gkn892) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akiyama H, et al. 2004. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 18, 1072–1087 (doi:10.1101/gad.1171104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gobezie R, Kho A, Krastins B, Sarracino DA, Thornhill TS, Chase M, Millett PJ, Lee DM. 2007. High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res. Ther. 9, R36 (doi:10.1186/ar2172) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cillero-Pastor B, Ruiz-Romero C, Carames B, Lopez-Armada MJ, Blanco FJ. 2010. Proteomic analysis by two-dimensional electrophoresis to identify the normal human chondrocyte proteome stimulated by tumor necrosis factor alpha and interleukin-1 beta. Arthritis Rheum. 62, 802–814 (doi:10.1002/art.27265) [DOI] [PubMed] [Google Scholar]
- 27.Guo D, et al. 2008. Proteomic analysis of human articular cartilage: identification of differentially expressed proteins in knee osteoarthritis. Joint Bone Spine 75, 439–444 (doi:10.1016/j.jbspin.2007.12.003) [DOI] [PubMed] [Google Scholar]
- 28.Guo F, Lai Y, Tian Q, Lin EA, Kong L, Liu C. 2010. Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum. 62, 2023–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambrecht S, Verbruggen G, Verdonk PC, Elewaut D, Deforce D. 2008. Differential proteome analysis of normal and osteoarthritic chondrocytes reveals distortion of vimentin network in osteoarthritis. Osteoarthritis Cartilage 16, 163–173 (doi:10.1016/j.joca.2007.06.005) [DOI] [PubMed] [Google Scholar]
- 30.Jmeian Y, El Rassi Z. 2008. Micro-high-performance liquid chromatography platform for the depletion of high-abundance proteins and subsequent on-line concentration/capturing of medium- and low-abundance proteins from serum. Application to profiling of protein expression in healthy and osteoarthritis sera by 2-D gel electrophoresis. Electrophoresis 29, 2801–2811 (doi:10.1002/elps.200890074) [DOI] [PubMed] [Google Scholar]
- 31.Ling SM, et al. 2009. Serum protein signatures detect early radiographic osteoarthritis. Osteoarthritis Cartilage, 17, 43–48 (doi:10.1016/j.joca.2008.05.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pecora F, et al. 2007. A quantitative and qualitative method for direct 2-DE analysis of murine cartilage. Proteomics 7, 4003–4007 (doi:10.1002/pmic.200700276) [DOI] [PubMed] [Google Scholar]
- 33.Ma WJ, Guo X, Liu JT, Liu RY, Hu JW, Sun AG, Yu YX, Lammi MJ. 2011. Proteomic changes in articular cartilage of human endemic osteoarthritis in China. Proteomics 11, 2881–2890 (doi:10.1002/pmic.201000636) [DOI] [PubMed] [Google Scholar]
- 34.Ruiz-Romero C, Lopez-Armada MJ, Blanco FJ. 2005. Proteomic characterization of human normal articular chondrocytes: a novel tool for the study of osteoarthritis and other rheumatic diseases. Proteomics 5, 3048–3059 (doi:10.1002/pmic.200402106) [DOI] [PubMed] [Google Scholar]
- 35.Rosenthal AK, Gohr CM, Ninomiya J, Wakim BT. 2011. Proteomic analysis of articular cartilage vesicles from normal and osteoarthritic cartilage. Arthritis Rheum. 63, 401–411 (doi:10.1002/art.30120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruiz-Romero C, Lopez-Armada MJ, Carreira V, Fernandez MC, Blanco FJ. 2006. Proteomic differential expression analysis of osteoarthritis-related proteins in human articular chondrocytes. Mol. Cell. Proteomics 5, S70 [Google Scholar]
- 37.Ruiz-Romero C, Carreira V, Rego I, Remeseiro S, Lopez-Armada MJ, Blanco FJ. 2008. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics 8, 495–507 (doi:10.1002/pmic.200700249) [DOI] [PubMed] [Google Scholar]
- 38.Ruiz-Romero C, Calamia V, Mateos J, Carreira V, Martinez-Gomariz M, Fernandez M, Blanco FJ. 2009. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol. Cell. Proteomics 8, 172–189 (doi:10.1074/mcp.M800292-MCP200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu J, Liu W, Bemis A, Wang E, Qiu Y, Morris EA, Flannery CR, Yang Z. 2007. Comparative proteomic characterization of articular cartilage tissue from normal donors and patients with osteoarthritis. Arthritis Rheum. 56, 3675–3684 (doi:10.1002/art.22876) [DOI] [PubMed] [Google Scholar]
- 40.Vincourt JB, Lionneton F, Kratassiouk G, Guillemin F, Netter P, Mainard D, Magdalou J. 2006. Establishment of a reliable method for direct proteome characterization of human articular cartilage. Mol. Cell. Proteomics 5, 1984–1995 (doi:10.1074/mcp.T600007-MCP200) [DOI] [PubMed] [Google Scholar]
- 41.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E. 2008. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (doi:10.1088/1742-5468/2008/10/P10008) [Google Scholar]
- 42.Ghoshal G, Barabasi AL. 2011. Ranking stability and super-stable nodes in complex networks. Nat. Commun. 2, 2394 (doi:10.1038/ncomms1396) [DOI] [PubMed] [Google Scholar]
- 43.Newman MEJ. 2012. Communities, modules and large-scale structure in networks. Nat. Phys. 8, 25–31 (doi:10.1038/nphys2162) [Google Scholar]
- 44.Lancichinetti A, Fortunato S. 2009. Community detection algorithms: a comparative analysis. Phys. Rev. E 80, 056117. [DOI] [PubMed] [Google Scholar]
- 45.Guo DH, et al. 2004. A functional variant of SUMO4, a new I κ B α modifier, is associated with type 1 diabetes. Nat. Genet. 36, 837–841 (doi:10.1038/ng1391) [DOI] [PubMed] [Google Scholar]
- 46.Noso S, et al. 2005. Genetic heterogeneity in association of the SUMO4 M55 V variant with susceptibility to type 1 diabetes. Diabetes 54, 3582–3586 (doi:10.2337/diabetes.54.12.3582) [DOI] [PubMed] [Google Scholar]
- 47.Park Y, Park S, Kang JG, Yang SW, Kim D. 2005. Assessing the validity of the association between the SUMO4 M55 V variant and risk of type 1 diabetes. Nat. Genet. 37, 112 (doi:10.1038/ng0205-112a) [DOI] [PubMed] [Google Scholar]
- 48.Ikegami H, Fujisawa T, Kawabata Y, Noso S, Ogihara T. 2006. Genetics of type 1 diabetes: similarities and differences between Asian and Caucasian populations. Ann. NY Acad. Sci. 1079, 51–59 (doi:10.1196/annals.1375.008) [DOI] [PubMed] [Google Scholar]
- 49.Gibbons LJ, Thomson W, Zeggini E, Worthington J, Barton A, Eyre S, Donn R, Hinks A. 2005. The type 1 diabetes susceptibility gene SUMO4 at IDDM5 is not associated with susceptibility to rheumatoid arthritis or juvenile idiopathic arthritis. Rheumatology 44, 1390–1393 (doi:10.1093/rheumatology/kei041) [DOI] [PubMed] [Google Scholar]
- 50.Orozco G, et al. 2006. Study of the role of functional variants of SLC22A4, RUNX1 and SUMO4 in systemic lupus erythematosus. Ann. Rheum. Dis. 65, 791–795 (doi:10.1136/ard.2005.044891) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoo YJ, Gao G, Zhang K. 2007. Case-control association analysis of rheumatoid arthritis with candidate genes using related cases. BMC Proc. 1(Suppl. 1), S33 (doi:10.1186/1753-6561-1-s1-s33) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sharif M, Whitehouse A, Sharman P, Perry M, Adams M. 2004. Increased apoptosis in human osteoarthritic cartilage corresponds to reduced cell density and expression of caspase-3. Arthritis Rheum. 50, 507–515 (doi:10.1002/art.20020) [DOI] [PubMed] [Google Scholar]
- 53.Chen Q, Zhang B, Yi T, Xia C. 2012. Increased apoptosis in human knee osteoarthritis cartilage related to the expression of protein kinase B and protein kinase C-alpha in chondrocytes. Folia Histochem. Cytobiol. 50, 137–143 (doi:10.5603/FHC.2012.0020) [DOI] [PubMed] [Google Scholar]
- 54.Blain EJ. 2008. Involvement of the cytoskeletal elements in articular cartilage homeostasis and pathology. Int. J. Exp. Path 90, 1–15 (doi:10.1111/j.1365-2613.2008.00625.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anilkumar N, Parsons M, Monk R, Ng T, Adams JC. 2003. Interaction of fascin and protein kinase Calpha: a novel intersection in cell adhesion and motility. EMBO J. 22, 5390–5402 (doi:10.1093/emboj/cdg521) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu Q, Zhu M, Rosier RN, Zuscik MJ, O'Keefe RJ, Chen D. 2010. Beta-catenin, cartilage, and osteoarthritis. Ann. NY Acad. Sci. 1192, 344–350 (doi:10.1111/j.1749-6632.2009.05212.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo D, Han J, Adam BL, Colburn NH, Wang MH, Dong Z, Eizirik DL, She JX, Wang CY. 2005. Proteomic analysis of SUMO4 substrates in HEK293 cells under serum starvation-induced stress. Biochem. Biophys. Res. Commun. 337, 1308–1318 (doi:10.1016/j.bbrc.2005.09.191) [DOI] [PubMed] [Google Scholar]