

Abstract
The tomato (Solanum lycopersicum L.) is a model plant for genome research in Solanaceae, as well as for studying crop breeding. Genome-wide single nucleotide polymorphisms (SNPs) are a valuable resource in genetic research and breeding. However, to do discovery of genome-wide SNPs, most methods require expensive high-depth sequencing. Here, we describe a method for SNP calling using a modified version of SAMtools that improved its sensitivity. We analyzed 90 Gb of raw sequence data from next-generation sequencing of two resequencing and seven transcriptome data sets from several tomato accessions. Our study identified 4,812,432 non-redundant SNPs. Moreover, the workflow of SNP calling was improved by aligning the reference genome with its own raw data. Using this approach, 131,785 SNPs were discovered from transcriptome data of seven accessions. In addition, 4,680,647 SNPs were identified from the genome of S. pimpinellifolium, which are 60 times more than 71,637 of the PI212816 transcriptome. SNP distribution was compared between the whole genome and transcriptome of S. pimpinellifolium. Moreover, we surveyed the location of SNPs within genic and intergenic regions. Our results indicated that the sufficient genome-wide SNP markers and very sensitive SNP calling method allow for application of marker assisted breeding and genome-wide association studies.
Keywords: next generation sequencing (NGS), single nucleotide polymorphism (SNP), tomato
INTRODUCTION
Rapid progress in genome sequencing platforms, such as next-generation sequencing (NGS), provides much opportunity for developing DNA-based molecular markers (Davey et al., 2011; Shendure and Ji, 2008). Various molecular markers, including simple sequence repeats (SSR), random amplified polymorphic DNA (RAPD), and amplified fragment length polymorphisms (AFLP), have been developed for analysis of genetic diversity (Davey et al., 2011). Moreover, single nucleotide polymorphisms (SNPs) have been identified as powerful selection markers for use in genome-wide studies conducted after genome sequencing is completed (Altshuler et al., 2000).
These markers can be used routinely in crop breeding programs for such activities as genetic diversity analysis, cultivar identification, characterization of genetic resources, and association with agronomic traits (Edwards and Batley, 2010; Lu et al., 2012). In particular, SNPs represent the most frequent type of genetic polymorphism, and may provide high density of markers near a locus of interest (Edwards and Batley, 2010). They are finely resolved, highly stable and reliable, and compatible with ultra-high-throughput automation and detection. Often developed by re-sequencing a genome, a genome-wide set of SNPs is a valuable resource in genetic research and breeding (Davey et al., 2011).
Using NGS technologies, genome-wide SNPs have been discovered in many organisms, including several crop species, such as maize (Barbazuk et al., 2007), rice (McNally et al., 2009), sugarcane (Bundock et al., 2009), soybean (Hyten et al., 2010), durum wheat (Trebbi et al., 2011), and potato (Hamilton et al., 2011). Recently, many transcriptome analyses using NGS platforms have been reported for various crops such as chickpea (Agarwal et al., 2012) and tomato (Hamilton et al., 2012). The sequencing material for genome-wide SNPs discovery is typically selecting resequencing and transcriptome data (Trick et al., 2009).
The tomato genome has been sequenced and assembled, thereby enabling the identification of genome-wide SNPs (The Tomato Genome Consortium, 2012). SNPs are discovered by aligning raw data to a reference genome. However, genetic variation in reference genomes (e.g., heterozygosity) renders this analysis difficult or making a mistake. Although hundreds of validated SNPs have been reported in the tomato, this data is still not sufficient for identifying major genetic variations (Davey et al., 2011). Currently, a large amount of tomato NGS data is available for understanding the genetic variations in the tomato genome.
The objective of this study was to identify genetic variations in the reference genome and improve the SNP calling pipeline for discovery of genome-wide SNPs in the tomato using the modified SAMtools method (Li et al., 2009). We then demonstrated here that high and accurate numbers of genome-wide SNPs can be discovered by new pileup method from high- or low-depth NGS data. We also compared the sequencing materials between resequencing and transcriptome data for genome wide SNP discovery to incorporate of breeding strategies, such as marker-assisted and genome-wide association studies.
MATERIALS AND METHODS
Data collection and pre-processing
Raw sequencing data sets from nine tomato accessions were collected from the Short Read Archive at the National Center for Biotechnology Information (NCBI-SRA, http://www.ncbi.nlm.nih.gov/Traces/sra/). These sequences were produced by NGS platforms, such as 454, Illumina GA (Genome Analyzer)/HiSeq, and consisted of two whole genome shotgun sequencing (WGS) and seven transcriptome sequencing data sets (Table 1). Except for M82 sequenced Roche (Germany) 454 GS FLX, six transcriptomes were analyzed using the Illumina (USA) RNASeq paired-end protocol on a GAII (Table 1).
Table 1.
Summary of sequencing data and statistics obtained from mapping against the tomato reference genome
| Platform | Accession name | Accession no. (SRX#) | Total raw bases | Total raw reads | Reads after trimming | Reads mapped |
|---|---|---|---|---|---|---|
| Genome | ||||||
| HiSeq 2000 | S. lycopersicum cv. Heinz 1706 | 118405 | 40,049,336,282 | 396,528,082 | 378,384,282 | 366,065,700 |
| GAII | S. pimpinellifolium | 032869 | 39,527,019,832 | 391,356,632 | 320,611,344 | 285,949,799 |
| Transcriptome | ||||||
| GAIIx | PI212816 SE1 S. pimpinellifolium | 111861 | 921,292,920 | 15,354,882 | 12,807,056 | 12,223,430 |
| GAIIx | PI212816 SE2 S. pimpinellifolium | 111862 | 1,554,019,740 | 18,500,235 | 15,596,070 | 14,707,350 |
| GAIIx | PI114490 S. lycopersicum var. cerasiforme | 111858 | 1,809,075,540 | 30,151,259 | 26,055,403 | 25,613,628 |
| GAIIx | T5 S. lycopersicum | 111853 | 1,708,971,720 | 28,482,862 | 25,350,357 | 24,903,243 |
| GAIIx | OH9242 S. lycopersicum | 111849 | 1,353,093,900 | 22,551,565 | 20,993,463 | 20,369,579 |
| GAIIx | NC84173 S. lycopersicum | 111845 | 1,383,236,700 | 23,053,945 | 21,504,517 | 20,862,511 |
| GAIIx | FL7600 S. lycopersicum | 111557 | 1,702,107,120 | 28,368,452 | 25,681,817 | 25,130,637 |
| GS FLX | S. lycopersicum cv. M82 (unpollinated style) | 036616 | 39,349,666 | 150,688 | 47,513 | 19,093 |
| GS FLX | S. lycopersicum cv. M82 (pollen) | 036614 | 41,558,631 | 209,378 | 206,577 | 23,963 |
| GS FLX | S. lycopersicum cv. M82 (pericarp of fruit at 7 days post breaker stage) | 036612 | 23,888,768 | 46,661 | 45,909 | 21,949 |
After these sequences were converted into FASTQ format using the SRA Tool Kit (v. 2.1.16 centOS Linux 64-bit), the data were split into two paired-end files using Python script. The forward and reverse paired-end reads of two resequencing data sets were linked to each other and the indexed adapter sequences were trimmed using the Solexa QA package v. 1.13 (Cox et al., 2010). Because it is common for the quality of bases from either end of Illumina reads to decline, we trimmed either end when the Phred quality score dropped below Q = 20. In addition, we also removed reads shorter than 25 bp in length, as well as all 5′ and 3′ stretches of ambiguous ‘N’ nucleotides. These trimmed reads were used for downstream analysis.
The reference genome sequence of S. lycopersicum cv. Heinz (ITAG version 2.3) was downloaded from the SGN Tomato Genome Page (http://solgenomics.net/organism/Solanum_ly-copersicum/genome).
SNP discovery
We aligned two WGS data sets to the reference genome sequence using the Burrows-Wheeler Aligner (BWA) (v. 0.6.1-r104) program (Li and Dubin, 2009). The BWA default values for mapping were used, except that seed length (-l) was set to 28 and maximum differences in the seed (-k) equaled 1. To align short reads of the transcriptome to the reference tomato genome, we used TopHat (v. 1.3.3) (Trapnell et al., 2009) software, which considers gene splicing junctions and gene track information. TopHat was implemented with the option of mismatches (-n) set to 1 (tighter than the default) and the maximum and minimum intron lengths were set as 23,000 bp and 40 bp, respectively. Following alignment to the reference genome, data from S. pimpinellifolium PI212816 (accession no. SRX111861 and SRX111862) and M82 accessions (accession no. SRX036612, SRX036614 and SRX036616), which was composed of two and three data sets, respectively, were merged into one file.
After aligning with BWA or TopHat, only the reliable mapped reads were considered for SNP calling. The SNP positions within the aligned reads compared to the reference genome were identified using the pileup function in SAMtools utilities (v. 0.1.16) (Li et al., 2009). Using the various filter commands, SNPs were predicted for various positions with a minimum mapping quality (-Q) of 30. The minimum and maximum read depths were set to 3 and 100, respectively. These parameters ensure high-quality, reliable mapping of the reads, which is important for variant calling.
To confirm the accuracy and reliability of SNP genotypes, we developed scripts to process SNP validation. Programs were generated to analyze the depth, variation, and consensus quality of each SNP. Finally, a Perl script was written to select significant sites within the predicted SNP positions. The script can be downloaded at sourceforge (http://sourceforge.net/projects/seeders/files/open_script/snp_validation_script.zip).
Classification of intergenic, exonic and intronic SNPs
To determine whether the SNP location within the transcript structure is intronic, exonic, or intergenic, we tracked information from the reference genome sequence and annotated the exon or intron at which the SNP was located if it was not intergenic.
Gene Ontology (GO) was analyzed using a generic GO slim database composed of 366,327 proteins downloaded from the Gene Ontology website (http://archive.geneontology.org/lite/2013-01-26/), which lists high-level GO terms that provide a broad overview of the ontology content. The GO annotations of the genes were then mapped to the GO slim ontology database using the map2slim script (http://search.cpan.org/~cmungall/goperl/scripts/map2slim), and these results were used in the final classification of these genes.
RESULTS AND DISCUSSION
Data collection and pre-processing
In order to assess the quality of genome-wide SNP predictions from tomato, two whole genome shotgun (WGS) sequencing data sets, including a reference genome and seven RNA-seq data sets from tomato accessions, were collected from the NCBISRA. We pre-processed 954 mega raw reads over a total length of 90 Gb in length (Table 1). Approximately 85% of these raw reads across all samples were retained after filtering out, except unpollinated style M82, leaving 795 quality-filtered mega reads to be aligned to the reference genome.
Reference genome validation using raw data alignment
In order to assess its sequence variation, we examined the reference genome by aligning raw data of the tomato reference genome, S. lycopersicum cv. ‘Heinz 1706’, and predicted SNPs using the BWA (Li and Dubin, 2009) and SAMtools programs (Li et al., 2009) (Supplementary Table 1). Using default parameter values, a total of 87,929 SNPs were detected. Of these, 10,699 (12.2%) and 77,230 (87.8%) SNPs were classified as homo- and hetero-types, respectively. Not surprisingly, most of the predicted SNPs were hetero-type, suggesting that the consensus sequence of the reference genome could be generated from heterozygous loci.
However, homo-type SNPs were also discovered. To test the accuracy of this SNP analysis, we manually curated samples from the alignment using the T-view and pileup functions of SAMtools (Supplementary Figs. 1A and 1B). As shown in Supplementary Table 1, 6,052 of the 10,699 SNPs were identified as true homo-type SNP loci. The remaining 4,647 SNPs were falsely predicted because the SNP positions of the reference sequence was ‘N’. Therefore, a new pipeline for SNP calling was developed to optimize the read depth, mismatch, and mapping quality parameters. These data suggest that 10,699 homo-type SNPs loci of the reference genome could be corrected the nucleotide and 77,230 hetero-type SNP loci should be marked as heterozygote loci to take care while SNP calling against the reference genome.
Improved method for SNP calling
SAMtools is widely used because of its various modules for file conversion, mapping statistics, and variant calling (Li et al., 2009). Through manual curation, we found that many true SNP loci were filtered out because the current version of SAMtools is optimized with a sufficient amount of high-quality raw data. Therefore, in order to improve the SNP calling workflow for low-depth sequence coverage, we mapped raw data representing from 2× to 40× genome coverage of the reference genome, S. lycopersicum cv. ‘Heinz 1706’ to reference genome and then identified SNPs (Table 2). As shown in Table 2, calculating SNPs using SAMtools with a new pileup method resulted in the identification of at least 4,749 more homo-type SNPs than using previous pileup programs from the tomato reference genome (Li et al., 2009). Moreover, the number of hetero-type SNPs identified with the new pileup program was greater than that called by the original pileup program regardless of raw data coverage except 2× genome coverage data. These results demonstrate an improvement in the sensitivity of SNP calling by a new pileup in SAMtools.
Table 2.
Summary of the genetic diversity of the tomato reference genome according to sequence coverage
| Coverage | Pileup | Homo-type SNPs | Hetero-type SNPs | Total SNPs | ||
|---|---|---|---|---|---|---|
| 2× | Pileup | 4,492 | (5.1%) | 84,441 | (94.9%) | 88,933 |
| New_Pileup | 12,874 | (17.5%) | 60,645 | (82.5%) | 73,519 | |
| 10× | Pileup | 5,550 | (4.1%) | 129,732 | (95.9%) | 135,282 |
| New_Pileup | 11,727 | (4.7%) | 237,800 | (95.3%) | 249,527 | |
| 20× | Pileup | 5,753 | (5.1%) | 108,110 | (94.9%) | 113,863 |
| New_Pileup | 11,100 | (6.0%) | 172,871 | (94.0%) | 183,971 | |
| 40× | Pileup | 6,052 | (7.3%) | 77,110 | (92.7%) | 83,162 |
| New_Pileup | 10,801 | (6.3%) | 159,585 | (93.7%) | 170,386 | |
Next, we examined the specificity of the results produced by the new pileup program. Specificity was calculated as the number of true positives divided by the sum of true positives plus false positives. We defined a true positive as any SNP present in more than one sample while a false positive was any SNP present in only one sample (Fig. 1, Supplementary Table 2, and Fig. 2). Both pileup methods yielded a similar pattern in the specificity of SNP calls, namely the number of false positives decreased as raw data coverage increased. Examination of homo-type SNPs identified by the new pileup program revealed that 12,874 and 10,801 SNPs were called in the 2× and 40× genome coverage of the raw data, respectively (Supplementary Fig. 2). Of the 12,874 homo-type SNPs, 4,076 (31.7%) were in concordance with the results obtained from the 40× analysis. Furthermore, 6,258 (48.6%) were classified as hetero-type SNPs while 2,540 (19.7%) were not called by the 40× coverage analysis. A previous pileup program showed similar results in that 1,148 (25.5%) of 4,492 homo-type SNPs were identified in the 2× and 40× coverage, 1,023 (22.8%) homo-types SNPs were also classified as hetero-type SNPs, and 2,321 (51.7%) SNPs were detected in the 2× coverage only. Taken together, these data indicate that the new pipeline for SNP calling is sensitive and reliable for discovering homo- and heterozygote loci from high- and low-depth genome sequencing data.
Fig. 1.
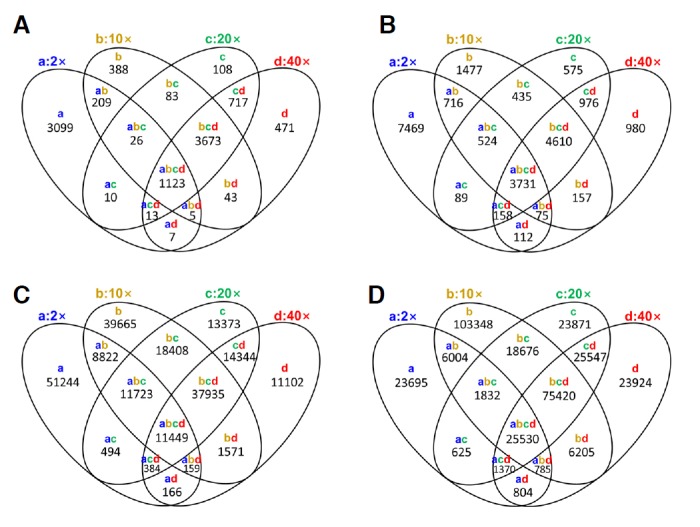
Venn diagram of SNPs according the raw data sequence coverage. (A) Homo-type SNPs in pileup, (B) homo-type SNPs in new pileup, (C) hetero-type SNPs in pileup, (D) hetero-type SNPs in new pileup. Colored lowercase letters a, b, c, and d indicate raw data sets representing 2×, 10×, 20× and 40× genome coverage, respectively. Numbers under the colored lowercase letters represent the number of SNPs.
Genome-wide SNP discovery from re-sequencing genome data
Using the improved BWA-SAMtools workflow (Li and Dubin, 2009; Li et al., 2009) identified novel genome-wide SNPs. In the WGS data from S. pimpinellifolium, 4,680,647 putative SNPs were detected, of which 4,210,454 (89.9%) and 470,193 (10.1%) homo- and hetero-type SNPs were classified (Table 3). Our analysis revealed that the number of SNPs present differs across the various chromosomes (Fig. 2A). Chromosome 1 (Chr1) had the greatest number of total SNPs (548,857), whereas Chr11 possessed the least number (244,544). The highest number of homo-type SNPs (495,231, 93.4%) was found on Chr7, while the greatest number of hetero-type SNPs (69,199, 12.6%) was predicted on Chr1.
Table 3.
Statistics of SNPs called from one resequencing and seven transcriptome data sets
| Accession name | Total # of SNP | SNP classified type
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Homo-type | Hetero-type | ||||||||
|
|
|
||||||||
| Total # of SNP | aIntergenic region |
bGenic region
|
Total # of SNP | aIntergenic region |
bGenic region
|
||||
| cExon | Intron | cExon | Intron | ||||||
| Genome | |||||||||
| S. pimpinellifolium | 4,680,647 | 4,210,454 (89.9%) | 3,853,232 (91.5%) | 108,637 (2.6%) | 248,585 (5.9%) | 470,193 (10.1%) | 432,796 (92.0%) | 17,491 (3.8%) | 19,906 (4.2%) |
| Transcriptome | |||||||||
| PI212816 | 71,637 | 66,410 (92.7%) | 14,568 (21.9%) | 49,987 (73.8%) | 2,855 (4.3%) | 5,227 (7.3%) | 1,129 (28.2%) | 4,008 (76.7%) | 90 (1.7%) |
| PI114490 | 23,902 | 17,868 (74.8%) | 4,211 (23.6%) | 12,877 (72.1%) | 780 (4.4%) | 6,034 (25.2%) | 1,344 (22.3%) | 4,557 (75.5%) | 133 (2.2%) |
| T5 | 9,544 | 4,780 (50.1%) | 1,210 (25.3%) | 3,339 (69.9%) | 231 (4.8%) | 4,764 (49.9%) | 1,090 (22.9%) | 3,593 (75.4%) | 81 (1.7%) |
| OH9242 | 8,313 | 5,712 (68.7%) | 1,222 (21.4%) | 4,254 (74.5%) | 236 (4.1%) | 2,601 (31.3%) | 552 (21.2%) | 1,989 (76.5%) | 60 (2.3%) |
| NC84173 | 7,744 | 5,203 (67.2%) | 1,218 (23.4%) | 3,766 (72.4%) | 219 (4.2%) | 2,541 (32.8%) | 508 (20.0%) | 1,977 (77.8%) | 56 (2.2%) |
| FL7600 | 10,466 | 6,501 (62.1%) | 1,665 (25.6%) | 4,537 (69.8%) | 299 (4.6%) | 3,965 (37.9%) | 844 (21.3%) | 3,048 (76.9%) | 73 (1.8%) |
| M82 | 179 | 80 (44.7%) | 10 (12.5%) | 68 (85.0%) | 2 (2.5%) | 99 (54.3%) | 16 (16.2%) | 82 (82.8%) | 1 (1.0%) |
Intergenic region is defined as DNA sequences located between genes within the genome.
Genic region consists of exons and introns.
Exon includes the 3′-UTR, 5′-UTR, and coding regions.
Fig. 2.
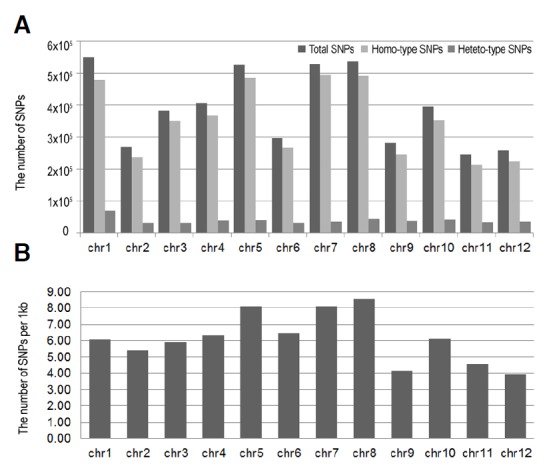
The SNPs distribution and density in S. pimpinellifolium. (A) The distribution of total SNPs in 12 chromosomes of S. pimpinellifolium: homo- and hetero-type SNPs of 12 chr. (B) The density of SNPs in 12 chr. of S. pimpinellifolium. The density was calculated as the average number of SNPs within a 1 kb region of each chromosome.
Next, we examined the SNP density for each chromosome size in S. pimpinellifolium by dividing the total number of SNPs in each chromosome by the chromosome length (Fig. 2B). An average SNP density of 6.1 SNPs/kb in the genome was observed. However, this analysis did not provide unequivocal evidence of a correlation between SNP density and chromosomal size (Fig. 2B). Our data also show that polymorphic variation for Chr5, 7, and 8 was significantly higher than Chr9, 11, and 12.
The SNP distribution within the genome structure of S. pimpinellifolium was also investigated. This analysis revealed that 8.5% and 91.5% of total genome-wide SNPs were found within genic and intergenic regions, respectively (Supplementary Fig. 3A). These results were quite similar to that of homo- (Supplementary Fig. 3B) and hetero-type SNPs (Supplementary Fig. 3C). We also found that a higher percentage of SNPs was observed in intergenic regions than in intragenic regions. In particular, more SNPs were identified in introns than in exons (Table 3 and Supplementary Fig. 3).
Genome-wide SNP discovery from transcriptome data
We next applied TopHat software to align transcriptome data to the reference genome sequence (Trapnell et al., 2009). Our analysis demonstrated that 95.4%, 94.3%, 98.3%, 98.2%, 97%, 97%, and 97.9% of the short reads from PI212816 (SE1, SE2), PI14490, T5, OH9242, NC84173, and FL7600, respectively, were mapped onto the reference genome. Moreover, 40.2%, 11.6%, and 47.8% of the reads from unpollinated, pollen, and the fruit pericarp at 7 days in M82 were mapped, respectively (Table 1).
As summarized in Table 3, 131,785 SNPs were identified from seven-accession tomato transcriptome data sets. As expected, S. pimpinellifolium PI212816 showed four to ten times more SNPs (71,637 SNPs) than other data sets, suggesting high diversity. Likewise, analysis of S. lycopersicum PI14490 revealed 23,902 SNPs, which was two to three times higher than other datasets. The distribution of PI14490 SNPs revealed the existence of SNP hot spots, implying the occurrence of introgression of a wild species genome fragment and possibly explaining the observed increase in SNP number (data not shown). In contrast, identification of 179 SNPs for the M82 accession was significantly lower than that for other accessions because this was the smallest data set (approximately 105Mb, 0.05–0.1% of the other data sets) and possessed lower genome coverage (0.06%). Overall, the ratio of homo- to hetero-type SNPs was quite diverse between the different accessions. PI212816 exhibited a high percentage of homo-type SNPs (92.7%), while T5 (49.9%) and M82 (54.3%) displayed a higher percentage of hetero-type SNPs.
In addition, to identify or predict the possible function of SNPs we performed gene ontology (GO) slim analysis (The Gene Ontology Consortium, 2013). The GO terms associated with biological processes such as re-production, stress and stimulus responses, signaling, and developmental processes were identified (Supplementary Fig. 4).
Comparing SNPs between transcriptome and resequencing data
Next, we compared the number and distribution of SNPs from transcriptome and resequencing data of S. pimpinellifolium (The Tomato Genome Consortium, 2012). SNPs in the exon regions of resequencing data were also compared against SNPs from transcriptome data (Table 3). From S. pimpinellifolium resequencing data, 4,680,647 SNPs were identified, of which 126,128 SNPs were detected in exon regions. From the transcriptome data of S. pimpinellifolium PI212816, 53,995 (75.3%) of 71,637 SNPs were detected within the exons of the reference genome and 15,697 SNPs were detected in intergenic regions. These results suggest that some expressed genes were not annotated or an unknown fragment of the genome could be expressed in transcriptome data set. Comparison of the number of SNPs from the S. pimpinellifolium transcriptome identified 72,133 SNPs that were also present within the exon region of resequencing data. To identify a sufficient number of SNPs among individuals in the same species or closely related lines, the resequencing method can be performed. However, if a reference sequence is unavailable or many samples (individuals) will be sequenced or SNP discovery is concerned with gene function, the transcriptome method can be selected (Shirasawa et al., 2010). RNA-Seq on an Illumina platform can generate redundant transcriptome sequences with high read depth, thereby guaranteeing the highest quality large-scale SNP identification.
SNP distribution along the chromosomes was also compared to gene distribution and SNPs from the transcriptome and re-sequencing data sets (Fig. 3). SNPs identified from the transcriptome coincided with the distribution of genes frequently discovered at chromosome ends (The Tomato Genome Consortium, 2012). However, SNPs from resequencing data showed a different pattern as they were either almost evenly distributed along the chromosome or clustered in gene-poor regions. These results demonstrate that intergenic regions possess more SNPs than genic regions. Therefore, to identify SNPs in gene-poor regions, the resequencing method is preferred.
Fig. 3.
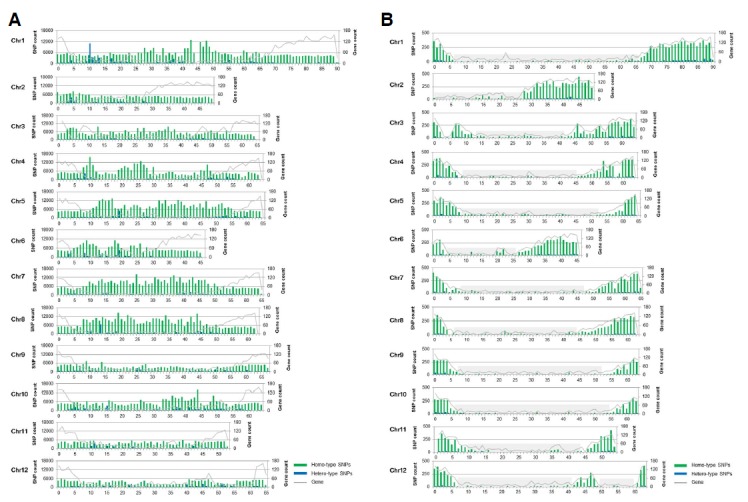
The distribution of SNPs detected with (A) resequencing and (B) transcriptome data along 12 chromosomes from the S. pimpinellifolium. Homo- and hetero-type SNPs exhibit varied distribution across different chromosomes. The left y-axis represents the number of SNPs while the right y-axis indicates gene count. The horizontal x-axis represents the length (Mb) of each chromosome. Gray shade boxes in (B) are regions identified low gene number.
In summary, we identified genome-wide SNPs and developed a novel method for sequence-based SNP validation. Using the improving sensitivity of SAMtools pileup (Li et al., 2009), we found more than 24,655 homo-type SNPs and 231,508 hetero-type SNPs in current version of tomato reference genome. We also identified 4,812,432 non-redundant SNPs with 50 Gb of raw sequence of NGS from a resequencing and seven transcriptome data sets of tomato accessions. Moreover, the SNP validation rates obtained from statistical analysis of SNP of the tomato reference genome using own raw data. These sufficient and qualified SNP markers will be used for application of crop breeding process.
Supplementary information
Acknowledgments
This work was supported by a grant from the Next-Generation BioGreen 21 Program (No.PJ009063), Rural Development Administration, Republic of Korea.
Note:
Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Agarwal G., Jhanwar S., Priya P., Singh V.K., Saxena M.S., Parida S.K., Garg R., Tyagi A.K., Jain M. Comparative analysis of kabuli chickpea transcriptome with desi and wild chickpea provides a rich resource for development of functional markers. PLoS One. 2012;7:e52443. doi: 10.1371/journal.pone.0052443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler D., Pollara V.J., Cowles C.R., Van Etten W.J., Baldwin J., Linton L., Lander E.S. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature. 2000;407:513–516. doi: 10.1038/35035083. [DOI] [PubMed] [Google Scholar]
- Barbazuk W.B., Emrich S.J., Chen H.D., Li L., Schnable P.S. SNP discovery via 454 transcriptome sequencing. Plant J. 2007;51:910–918. doi: 10.1111/j.1365-313X.2007.03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bundock P.C., Eliott F.G., Ablett G., Benson A.D., Casu R.E., Aitken K.S., Henry R.J. Targeted single nucleotide polymorphism (SNP) discovery in a highly polyploid plant species using 454 sequencing. Plant Biotechnol. J. 2009;7:347–354. doi: 10.1111/j.1467-7652.2009.00401.x. [DOI] [PubMed] [Google Scholar]
- Cox M.P., Peterson D.A., Biggs P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey J.W., Hohenlohe P.A., Etter P.D., Boone J.Q., Catchen J.M., Blaxter M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011;12:499–510. doi: 10.1038/nrg3012. [DOI] [PubMed] [Google Scholar]
- Edwards D., Batley J. Plant genome sequencing: applications for crop improvement. Plant Biotechnol. J. 2010;8:2–9. doi: 10.1111/j.1467-7652.2009.00459.x. [DOI] [PubMed] [Google Scholar]
- Hamilton J.P., Hansey C.N., Whitty B.R., Stoffel K., Massa A.N., Van Deynze A., De Jong W.S., Douches D.S., Buell C.R. Single nucleotide polymorphism discovery in elite North American potato germplasm. BMC Genomics. 2011;12:302. doi: 10.1186/1471-2164-12-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton J.P., Sim S.C., Stoffel K., Van Deynze A., Buell C.R., Francis D.M. Single nucleotide polymorphism discovery in cultivated tomato via sequencing by synthesis. Plant Genome. 2012;5:17–29. [Google Scholar]
- Hyten D.L., Cannon S.B., Song Q., Weeks N., Fickus E.W., Shoemaker R.C., Specht J.E., Farmer A.D., May G.D., Cregan P.B. High-throughput SNP discovery through deep re-sequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics. 2010;11:38. doi: 10.1186/1471-2164-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Dubin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 genome project data processing subgroup The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F.H., Kwon S.W., Yoon M.Y., Kim K.T., Cho M.C., Yoon M.K., Park Y.J. SNP marker integration and QTL analysis of 12 agronomic and morphological traits in F8 RILs of pepper (Capsicum annuum L.) Mol. Cells. 2012;34:25–34. doi: 10.1007/s10059-012-0018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally K.L., Childs K.L., Bohnert R., Davidson R.M., Zhao K., Ulat V.J., Zeller G., Clark R.M., Hoen D.R., Bureau T.E., et al. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. USA. 2009;106:12273–12278. doi: 10.1073/pnas.0900992106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J., Ji H. Next-generation DNA sequencing. Nat. Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Shirasawa K., Isobe S., Hirakawa H., Asamizu E., Fukuoka H., Just D., Rothan C., Sasamoto S., Fujishiro T., Kishida Y., et al. SNP discovery and linkage map construction in cultivated tomato. DNA Res. 2010;17:381–391. doi: 10.1093/dnares/dsq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium Gene ontology annotations and resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Tomato Genome Consortium The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485:635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trebbi D., Maccaferri M., de Heer P., Sørensen A., Giuliani S., Salvi S., Sanguineti M.C., Massi A., van der Vossen E.A.G., Tuberosa R. High-throughput SNP discovery and genotyping in durum wheat (Triticum durum Desf.) Theor. Appl. Genet. 2011;123:555–569. doi: 10.1007/s00122-011-1607-7. [DOI] [PubMed] [Google Scholar]
- Trick M., Long Y., Meng J., Bancroft I. Single nucleotide polymorphism (SNP) discovery in the polyploid Brassica napus using Solexa transcriptome sequencing. Plant Biotechnol. J. 2009;7:334–346. doi: 10.1111/j.1467-7652.2008.00396.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.