

Abstract
Altered glutamate signaling contributes to a myriad of neural disorders, including schizophrenia. While synaptic levels are intensely studied, nonvesicular release mechanisms, including cystine-glutamate exchange, maintain high steady-state glutamate levels in the extrasynaptic space. The existence of extrasynaptic receptors, including metabotropic group II glutamate receptors (mGluR), pose nonvesicular release mechanisms as unrecognized targets capable of contributing to pathological glutamate signaling. We tested the hypothesis that activation of cystine-glutamate antiporters using the cysteine prodrug N-acetylcysteine would blunt psychotomimetic effects in the rodent phencyclidine (PCP) model of schizophrenia. First, we demonstrate that PCP elevates extracellular glutamate in the prefrontal cortex; an effect that is blocked by N-acetylcysteine pretreatment. To determine the relevance of the above finding, we assessed social interaction and found that N-acetylcysteine reverses social withdrawal produced by repeated PCP. In a separate paradigm, acute PCP resulted in working memory deficits assessed using a discrete trial T-maze task, and this effect was also reversed by N-acetylcysteine pretreatment. The capacity of N-acetylcysteine to restore working memory was blocked by infusion of the cystine-glutamate antiporter inhibitor (S)-4-carboxyphenylglycine into the prefrontal cortex or systemic administration of the group II mGluR antagonist LY341495 indicating that the effects of N-acetylcysteine requires cystine-glutamate exchange and group II mGluR activation. Lastly, protein levels from post mortem tissue obtained from schizophrenic patients revealed significant changes in the level of xCT, the active subunit for cystine-glutamate exchange, in the dorsolateral prefrontal cortex. These data advance cystine-glutamate antiporters as novel targets capable of reversing the psychotomimetic effects of PCP.
Keywords: extrasynaptic, system Xc-, prefrontal cortex, nonvesicular, working memory, xCT
Introduction
Abnormal signaling in the prefrontal cortex has been proposed to contribute to schizophrenia (Weinberger, 1987; Bunney and Bunney, 2000; Chavez-Noriega et al., 2002), however, the precise role of glutamate is equivocal since evidence indicates that either an increase or a decrease in glutamate signaling may contribute to the symptoms of the disease. A hypoglutamatergic state is supported by functional imaging studies revealing diminished activation of the frontal cortex in schizophrenics (Barch et al., 2001; MacDonald et al., 2005). Further, noncompetitive NMDA receptor antagonists, including phencyclidine (PCP), exacerbate symptoms in schizophrenic patients and produce a broad range of schizophrenia-like symptoms in humans and rats (Luby et al., 1959; Pearlson, 1981; Javitt and Zukin, 1991; Krystal et al., 1994; Malhotra et al., 1997); thus, hypoglutamatergic function through NMDA receptors is sufficient to produce a schizophrenia-like state. In contrast, a hyperglutamatergic hypothesis stems from evidence that noncompetitive NMDA receptor antagonists increase extracellular glutamate levels in the prefrontal cortex which generates an increase in the firing rate of cortical neurons (Moghaddam and Adams, 1998; Lorrain et al., 2003; Jackson et al., 2004), and that these changes are necessary for the psychotomimetic effects of PCP (Moghaddam and Adams, 1998; Homayoun et al., 2005).
The uncertainty regarding glutamate dysfunction and schizophrenia may arise, in part, by the existence of multiple, distinct pools of glutamate that are present in the synaptic and extrasynaptic spaces. Continuous nonvesicular glutamate release has been detected following NMDA receptor activation, but only after sodium-dependent glutamate transporters were blocked (Jabaudon et al., 1999). This finding indicates that nonvesicular glutamate is released into the extrasynaptic compartment, but that sodium-dependent transporters block entry into the synapse. In support microdialysis experiments have repeatedly indicated the existence of extrasynaptic glutamate maintained in the microM range by nonvesicular release mechanisms (Herrera-Marschitz et al., 1996; Timmerman and Westerink, 1997; Baker et al., 2002). The distribution of glutamate receptors outside the synapse poses extrasynaptic glutamate as a novel mechanism capable of modulating glutamate signaling in the normal and pathological state.
Cystine-glutamate antiporters represent a mechanism capable of nonvesicular glutamate release into the extrasynaptic compartment following the exchange of extracellular cystine for intracellular glutamate (Baker et al., 2002; Moran et al., 2005{Melendez, 2005 #15). Studies utilizing microdialysis in the nucleus accumbens or electrophysiological recordings from acute prefrontal cortical or nucleus accumbens tissue slices indicate that nonvesicular glutamate release from cystine-glutamate exchange stimulates extrasynaptic group II metabotropic glutamate receptors (mGluR) (Baker et al., 2002; Xi et al., 2002a; Moran et al., 2005). Because group II mGluRs function as autoreceptors (Baskys and Malenka, 1991; Cochilla and Alford, 1998; Hu et al., 1999b; Schoepp, 2001; Valenti et al., 2002; Xi et al., 2002b), extrasynaptic glutamate maintained by cystine-glutamate exchange negatively modulates synaptic release of glutamate (Moran et al., 2005). Since most studies focus on synaptic content, the failure to account for the presence of high, steady-state levels of glutamate in the extrasynaptic space represents a critical gap in attempts to model the contribution of excitatory neurotransmission to brain functioning in both the normal and diseased states.
Extant data indicate that cystine-glutamate exchange may be altered in schizophrenic patients. An increase in the expression of group II mGluR has been linked to schizophrenia (Gupta et al., 2005), and this could arise in response to chronic understimulation of the receptor. Further, schizophrenics exhibit reduced glutathione levels in the prefrontal cortex (Do et al., 2000). This may reflect blunted cystine-glutamate exchange because cystine uptake into the cell represents the rate-limiting step in glutathione synthesis (Bannai, 1984; Meister, 1991; Sies, 1999). Alternatively, this could lead to blunted cystine-glutamate exchange because extracellular glutathione metabolism serves as a reservoir for cystine that is critical in maintaining cystine-glutamate exchange (Deneke and Fanburg, 1989; Sato et al., 1999; Sies, 1999; Kim et al., 2001; Shih et al., 2006). Collectively, these data would be expected following a reduction in cystine-glutathione activity.
To determine whether cystine-glutamate antiporters represent a novel target in the treatment of schizophrenia, we examined the impact of the cyst(e)ine prodrug N-acetylcysteine on the psychotomimetic effects of PCP in rodents. N-acetylcysteine-induced changes in the psychotomimetic effects of PCP are hypothesized to involve cystine-glutamate exchange and increased signaling through group II mGluRs, thus, the effects of N-acetylcysteine were also determined in the presence of the cystine-glutamate antiporter inhibitor (S)-4-carboxyphenylglycine (CPG) and the group II mGluR antagonist LY 341495. Because PCP may produce psychotomimetic effects through mechanisms distinct from schizophrenia, we sought to better link the present studies to schizophrenia by measuring the levels of xCT, the active subunit for cystine-glutamate exchange, in post mortem tissue obtained from schizophrenic patients. Collectively, these experiments may establish cystine-glutamate exchange as a novel mechanism capable of contributing to the psychotomimetic effects of PCP and possibly schizophrenia.
Materials and Methods
Animals and Surgeries
Male Sprague Dawley rats (Harlan, Indianapolis, IN) weighing 300–350 g were individually housed in a temperature-controlled colony room with a 12-h light/dark cycle with food and water available ad libitum. The housing conditions and care of the rats was in accordance with the Animal Welfare Act, and all procedures were approved by the Marquette University IACU Committee. Rats included in the microdialysis studies were anesthetized using pentobarbital (50 mg/kg, IP) with atropine sulfate (1 mg/kg, IP) pretreatment to limit tracheobronchial secretions. Bilateral guide cannula (20 gauge, 14 mm; Plastics One, Roanoke VA) were implanted using coordinates (+ 3.1 mm anterior and + 1.0 mm mediolateral to Bregma, and −0.75 mm ventral from the surface of the skull at a 6° angle from vertical) derived from Paxinos and Watson (1986). Rats were then given at least five days to recover from surgery prior to testing.
Drug Treatments
(S)-4-Carboxyphenylglycine (CPG; Tocris-Cooksin, Ellisville, MO) pretreatment, when used, was infused into the prefrontal cortex via reverse dialysis for 180 min prior to behavioral testing. N-acetylcysteine (Sigma, St. Louis, MO) pretreatments occurred 90 or 100 min prior to PCP administration in behavioral and microdialysis experiments, respectively. LY341495 (Tocris-Cookson, Ellisville, MO) pretreatments occurred 70 min prior to PCP administration. Phencyclidine (PCP; NIDA Drug Supply Program, Research Triangle, NC) was administered 60 min prior to commencement of behavioral testing. Drug treatments in the microdialysis and t-maze studies involved acute administration; treatments in the social interaction experiments involved administration over 14 consecutive days.
In vivo Microdialysis
Microdialysis experiments were conducted as previously described (Baker et al., 2002). Briefly, removable probes were inserted through the guide cannulae into the prefrontal cortex. The next day, dialysis buffer (5 mM glucose, 140 mM NaCl, 1.4 mM CaCl2, 1.2 mM MgCl2, and 0.15% phosphate buffer saline, pH 7.4) was pumped through the probes at 2 μl/min for at least 3 hr; afterwards 20 min samples were collected before and after drug treatments as described above. Glutamate concentrations in the samples were determined using HPLC coupled to fluorescence detection as reported previously (Baker et al., 2002). Reverse dialysis was conducted as described above except the buffer also contained CPG (1 μM) and the flow rate was 1 μl/min.
T-Maze Paradigm
Access to food was restricted to 15 g of rat chow/day during t-maze training and testing. Rats were trained until accuracy in a choice-alternation t-maze task exceeded 80% over three days. Daily sessions involved 10 trials per day with each trial consisting of a restricted and an unrestricted run. In the restricted run, a randomly determined arm was baited with cereal and the opposite arm was blocked. Ten seconds later, the unrestricted run was conducted whereby the rat had access to both arms of the maze but cereal was placed only in the arm blocked during the preceding restricted run. A correct choice occurred when the rat entered the baited arm during the unrestricted run. Training continued until performance exceeded 80% for at least three days.
Social Interaction
Social interaction was measured as described by Sams-Dodd (1999). Briefly, interaction was measured in rats that had received fourteen daily administrations of N-acetylcysteine (0–90 mg/kg, IP) followed by PCP (0–3 mg/kg, SC). On day 14, unfamiliar rat pairs receiving identical drug treatments were placed into an open-field maze (150 × 100 × 40 cm) and the amount of time spent within 20 cm was measured. The predictive validity of this procedure as a negative model of schizophrenia has previously been established (Sams-Dodd, 1999).
Brain Tissue preparation
Postmortem brain tissue from elderly, well diagnosed patients with schizophrenia and a comparison group was obtained from the Mount Sinai Medical Center Brain Bank (Table 1). Subjects were matched for age, postmortem interval (PMI) and pH, and did not show any signs of neurodegenerative disorders or other discernable neuropathologies, including Alzheimer’s disease (Purohit et al., 1998). Patients with a medication history had only been treated using typical antipsychotic medications. Tissue blocks containing dorsolateral prefrontal cortex (DLPFC), anterior cingulate cortex (ACC) and hippocampus were dissected from coronal sections, snap frozen and kept at −80°C until further processing. Approximately 1 cm3 of tissue from each region was grinded on dry ice and subsequently homogenized (10% W/V) in ice-cold 50 mM Tris-HCl (pH 7.0) for 30 sec with a polytron homogenizer and stored at −80°C. Protein concentration of tissue homogenates was determined by the Bradford method (Bradford et al., 1987).
Table 1. Subject characteristics.
Cohort characteristics for the subjects analyzed in this study.
| PMI (H) | Age | SEX | pH | Rx<6wk | Cause of death | |
|---|---|---|---|---|---|---|
| 1 | 4.7 | 86 | F | 6.5 | NA | Myocardial infarction |
| 2 | 4.2 | 90 | F | 6.0 | NA | Pneumonia |
| 3 | 3.0 | 74 | F | 6.0 | NA | Cardio respiratory failure |
| 4 | 5.1 | 88 | F | 6.4 | NA | COPD/Pneumonia |
| 5 | 1.4 | 98 | F | 6.6 | NA | Myocardial infarction, aortic aneurysm |
| 6 | 5.7 | 82 | F | 6.1 | NA | Cardiopulmonary arrest |
| 7 | 4.8 | 80 | F | 6.2 | NA | Sepsis |
| 8 | 3.4 | 73 | F | 6.3 | NA | Myocardial infarction |
| 9 | 18.5 | 84 | F | 6.2 | NA | Myocardial infarction |
| 10 | 2.3 | 89 | F | 6.7 | NA | Bronchopneumonia, Asthma, COPD |
| 11 | 6.2 | 83 | F | 6.8 | NA | Cardiopulmonary arrest |
| 12 | 6.5 | 75 | F | 6.0 | no | Cardiopulmonary arrest (received haldol 7 weeks prior to death) |
| 13 | 7.0 | 62 | F | 6.6 | NA | Myocardial infarction |
| 14 | 4.3 | 69 | M | 6.3 | NA | Cancer of lung |
| 15 | 4.7 | 101 | M | 6.8 | NA | Congestive Heart Failure, Cardio-Respiratory Failure, Bladder Cancer |
| 16 | 4.1 | 95 | M | 6.5 | NA | Chronic renal failure, N.I.D.D., hypertension |
| 17 | 3.8 | 65 | M | 6.8 | NA | Renal failure, Diabetes-Cirrhosis of liver |
| 18 | 7.6 | 66 | M | 6.6 | NA | Cardiac arrest |
| 19 | 7.4 | 69 | M | 6.7 | NA | Septic shock |
| 20 | 16.6 | 74 | M | 6.7 | NA | Cardiopulmonary arrest |
| 21 | 2.9 | 76 | M | 6.3 | NA | Bronchopneumonia bilateral. Artherosclerotic heart disease |
| 22 | 28.8 | 60 | M | 6.6 | NA | Myocardial infarction |
| 23 | 4.2 | 64 | M | 6.5 | NA | Myocardial infarction |
| 24 | 4.2 | 93 | M | 6.4 | NA | Myocardial infarction |
| 25 | 5.3 | 85 | M | 6.5 | NA | Transitional cell cancer of bladder with metastasis |
| 26 | 20.4 | 59 | M | 6.7 | NA | Coronary arterial disease; hypertension, deep venous thrombosis |
| 27 | 20.0 | 92 | M | 6.4 | NA | Arrhythmia |
| Subjects with schizophrenia
| ||||||
|---|---|---|---|---|---|---|
| PMI (H) | Age | SEX | pH | Rx<6wk | Cause of death | |
| 1 | 13.7 | 69 | F | 6.2 | yes | Cardio respiratory failure |
| 2 | 20.4 | 83 | F | 7.1 | no | Cardiopulmonary arrest, pancreatic cancer |
| 3 | 9.9 | 79 | F | 6.8 | no | Cardiac arrest |
| 4 | 7.0 | 74 | F | 6.3 | yes | Cardiopulmonary arrest |
| 5 | 12.5 | 81 | F | 5.9 | no | Myocardial infarction |
| 6 | 9.7 | 77 | F | 6.0 | yes | Cardiopulmonary arrest |
| 7 | 15.1 | 81 | F | 6.7 | no | Cardiopulmonary arrest |
| 8 | 6.7 | 58 | M | 6.2 | yes | Cardio pulmonary arrest |
| 9 | 29.5 | 52 | M | 5.9 | no | Cardio respiratory failure |
| 10 | 6.2 | 84 | M | 6.5 | no | Cardio respiratory failure |
| 11 | 13.3 | 58 | M | 6.9 | yes | cardiac papillary fibroelastoma |
| 12 | 30.3 | 57 | M | 6.1 | Expired during open heart surgery | |
| 13 | 6.2 | 63 | M | 6.3 | yes | Cardio pulmonary arrest |
| 14 | 11.2 | 87 | M | 6.5 | no | Cardio respiratory failure |
| 15 | 7.0 | 86 | M | 6.3 | yes | Cardio pulmonary arrest |
| 16 | 5.3 | 85 | M | 6.3 | yes | COPD with recurrent pneumonia |
| 17 | 7.9 | 73 | M | 6.5 | yes | Cardio respiratory failure |
| 18 | 14.1 | 86 | M | 6.7 | no | A.S.H.D. |
| 19 | 17.3 | 68 | M | 6.6 | yes | Congestive Heart Failure, Coronary Arterial disease |
| 20 | 40.2 | 69 | M | 6.7 | yes | Acute Renal Failure |
| 21 | 16.6 | 76 | M | 6.7 | no | CHF, Coronary Arterial Disease, A.S.H.D. |
| 22 | 21.4 | 57 | M | 6.4 | yes | Small cell cancer of Lung |
| 23 | 13.5 | 56 | M | 6.5 | no | Cardiopulmonary arrest |
| Summary
| |||||
|---|---|---|---|---|---|
| Age | N | PMI (H) | pH | Rx<6wk | |
| Comparison group | 79.0 | 27 (13F/14M) | 7.6 | 6.4 | 0/27 |
| Schizophrenia | 72.1 | 23 (7F/16M) | 14.6 | 6.4 | 12/22 |
| All Subjects | 75.8 | 50 (20F/30M) | 10.8 | 6.4 | 12/49 |
PMI = postmortem interval; N = number of subjects in the study; F = female; M = male; Rx<6wk = on antipsychotic medication within 6 weeks of death (only typical neuroleptics); COPD = Chronic obstructive pulmonary disease; N.I.D.D. = non-insulin dependent diabetes; A.S.H.D. = arteriosclerotic heart disease; CHF = Congestive Heart Failure.
Western blotting: Human Brain Tissue
Tissue homogenates from each subject (25 μg per lane) were mixed with 6x sample buffer (4.5% sodium dodecyl sulfate, 170 mM Tris-HCl (pH 6.8), 36% glycerol, 15% β-mercaptoethanol, 0.018% bromophenol blue) and heated at 95°C for 4 minutes. Samples were loaded in duplicate onto 10% SDS-PAGE (Bio-Rad Laboratories, Inc) and separated at 100 volts in electrophoresis buffer (25 mM Tris-base, 192 mM glycine, 0.1% SDS). After overnight transblotting (Trans-Blot Cell, Bio-rad Laboratories, Inc), protein-containing nitrocellulose membranes were blocked with 5% non-fat dry milk solution for one hour at room temperature and washed with TBS buffer containing 0.1 % Tween 20 (TBS-T). The blots were subsequently incubated with rabbit-anti-xCT antibody (TransGenic Inc., Japan; see figure 7) overnight at 4° C. Blots were washed with TBS-T (3X) for 5 min and incubated with HRP-conjugated secondary antibody (1hr). After washing (3X) in TBS-T buffer, the signal was detected with enhanced chemiluminescent substrate (Amersham Biosciences). Membranes were then stripped for 30 minutes at 55°C (62.5mM Tris-Cl (pH 6.7), 2% SDS, 100 mM β-mercaptoethanol), washed thoroughly in dH2O and blocked in phosphate buffered saline (PBS) containing 3% dry milk. Blots were again incubated overnight with mouse anti-β-tubulin antibody (Upstate Biotechnology), washed briefly, and incubated with horseradish peroxidase-coupled secondary antibody (1hr; Upstate Biotechnology) and developed using enhanced chemiluminescence.
Figure 7.

Knockdown or overexpression of xCT altered xCT RNA or protein levels as determined using semi-quantitative multi-plex RT-PCR (a), western blotting (b), and immunofluorescence (c). (a) The expression of hxCT RNA (724 bp) was equivalent between mock and control siRNA transfected cells. Cells transfected with hxCT siRNA (empty vector) exhibited a >90% reduction of hxCT transcript relative to mock controls. Cells co-transfected with mouse xCT (mxCT) cDNA plus hxCT siRNA expressed an increase in mxCT (182 bp) and a reduction in hxCT. The expression of the internal control, 18S rRNA, was equivalent or higher across all cells relative to mock controls. (b) The expression of xCT protein (55kD) was equivalent between mock and control siRNA transfected cells. Cells transfected with siRNA (+ empty vector) exhibited an 80% reduction of hxCT relative to mock controls. Transfection of cDNA mxCT restored xCT protein. β-actin levels were unchanged across all transfection conditions. (c) xCT labeling was evident in the mock and control siRNA transfected cells. Depletion of xCT labeling was observed in hxCT siRNA (+ empty vector) transfected cells. Transfection with mxCT cDNA along with hxCT siRNA restored xCT labeling. Collectively, these results demonstrated antibody specificity against the xCT protein for both human and mouse species.
Knockdown and overexpression of xCT in HEK cells
Approximately 50000 cells were split into a six-well plate. These studies utilized a siRNA duplex of human xCT [5′AAATGCCCAGATATGCATCGT-3′ (SLC7A11_1237)] which targets nucleotides 1237–1257 of the SLC7A11 mRNA sequence of NM_014331 and has previously been used to knockdown human xCT (hxCT) (Huang et al., 2005). HEK 293 cells were transfected with approximately 20 nM of siRNA using Lipofectamine 2000 (Invitrogen, Carlsbad, CA); the transfection efficiency exceeded 80% in all the experiments. The effect of RNAi was evaluated by multiplex RT-PCR, western blot, and immunofluorescence (see figure 7). In some cells, mouse xCT (mxCT) was overexpressed by cotransfection of 2 μg pCDNA3-xCT plasmid with hxCT siRNA; control cells received an equivalent amount of empty vector co-transfected with siRNA. Mock cells were incubated (48h) in Lipofectamine 2000 only. Additional cells were also incubated with a negative control siRNA (Ambion, Austin, Texas).
Semi-quantitative multiplex RT-PCR
After transfection (48 h), total RNA was isolated by Trizol (Invitrogen) reagent and approximately 100 ng of total RNA was used for RT. The first strand of cDNA was synthesized by Superscript Reverse Transcriptase (Invitrogen). The RT-product was amplified by PCR using appropriate gene-specific primers and a primer for 18S rRNA (Ambion Inc, TX) which served as an internal control in the same reaction (see figure 7). The conditions used to amplify the PCR products were 94° C for 40 seconds, 58° C for 45 seconds, and 72° C for 45 seconds for 30 cycles, and then 72° C for 7 min extension.
Western blotting: HEK 293 Cells
Transfected cells were washed with ice-cold PBS and lysed (10 mM Na2 PO4 pH 7.4, 150 mM NaCl, 5 mM EDTA, 5 mM EGTA, 1% Triton X-100, cock-tail protease inhibitor (Roche Biochemicals) and 1 mM PMSF). Protein concentration was determined using the BCA assay; protein (30 μg) was loaded onto 10% SDS-PAGE gels. Resolved proteins were transferred onto PVDF membrane and blocked with 5% non-fat dry milk solution (1hr). Blots were subsequently incubated with primary antibody against mouse xCT (figure 7) overnight at 4° C and then with HRP-conjugated secondary antibody (1hr). The signal was detected by enhanced chemiluminesence (Pierce Biotechnology Inc, IL). Blots were stripped (SignaGen, MD) and re-probed with β-actin antibody (Santacruz biotechnology Inc. CA).
Immunofluorescence analysis
Transfected cells were subjected to immunofluorescence analysis. Cells were grown on poly L-lysine coated cover slips and fixed with 4% paraformaldehyde in PBS for 10 min. Cells were washed with PBS (3X), permeabilized with 0.1% Triton X-100 for 10 min, washed again with PBS (3X) and blocked with 5% normal donkey serum. Cells were then labeled with xCT primary antibody (see figure 7) with a dilution of 1:10 overnight at 4°C. After washing with PBS (3X) cells were incubated with Cy-3 conjugated secondary antibody (Jackson Immunoresearch, PA) for one hour at room temperature and washed with PBS for (2–3X). Finally, cover slips were mounted in ProLong Gold antifade medium (Invitrogen) and cells were examined using a confocal microscope (Zeiss).
Histology
Tissue slices obtained from rats included in the microdialysis studies were stained with cresyl violet to verify probe placements. Three rats determined to have misplaced guide cannula were excluded from all analyses.
Statistics
Microdialysis and behavioral data were analyzed using analysis of variance (ANOVA) with drug treatment as a between subject factor and sample number as a repeated factor (microdialysis). Significant main effects and interactions were further evaluated using Tukey HSD. Measures of post mortem xCT were initially analyzed using multiple regression to exclude effects caused by age, PMI and pH. Analysis of covariance was utilized when significant associations were obtained between xCT expression and the above independent variables; univariate ANOVA was used in the absence of a significant association. For all tests α = 0.05.
Results
N-acetylcysteine blocks PCP-evoked glutamate release in the prefrontal cortex
The psychotomimetic effects of PCP are dependent upon elevated glutamate in the prefrontal cortex (Moghaddam and Adams, 1998). As a result, we examined the impact of N-acetylcysteine pretreatment on the neurochemical effects of PCP. Similar to earlier findings, figure 1 illustrates that systemic administration of PCP produces a dose-dependent increase in extracellular glutamate levels in the prefrontal cortex. Rats pretreated with N-acetylcysteine (90 mg/kg, IP) failed to exhibit this increase indicating that N-acetylcysteine blocked PCP-induced glutamate. Basal levels of glutamate did not differ between rats receiving 3 mg/kg PCP with or without N-acetylcysteine (mean ± SEM: 4.2 ± 1, 3.2 ± 0.8 pmole/μL, respectively) or 1 mg/kg PCP (mean ± SEM: 3.7 ± 0.8 pmole/μL). The region of the prefrontal cortex (figure 2) sampled in this study was selected because it corresponds to the human dorsolateral prefrontal cortex (Uylings et al., 2003) which has been implicated in schizophrenia.
Figure 1.

N-acetylcysteine (NAC) prevents phencyclidine-evoked glutamate release in the rodent prefrontal cortex. Extracellular glutamate in the prefrontal cortex is depicted as change from baseline across 20 min samples. (a) A low dose of phencyclidine (PCP; 1 mg/kg, SC, N=8) failed to alter extracellular glutamate levels in the prefrontal cortex. (b) Rats injected with saline followed by PCP (3 mg/kg, SC; n=13) exhibited an increase in extracellular glutamate that was not observed in rats pretreated with N-acetylcysteine (90 mg/kg, IP; n = 13). ANOVA yielded an interaction between time and NAC treatment (F15,360=1.74, P=.04) such that there was a main effect of time in rats pretreated with saline (0 mg/kg NAC; F15,180=1.97, P=.02) but not NAC (90 mg/kg; F15,180=0.87, P=0.596). (c) Extracellular glutamate levels in the prefrontal cortex expressed as mean (± SEM) area under the curve (AUC) following the PCP injection; ANOVA yielded a main effect of treatment (F1,24 = 4.86, P = 0.037); * indicates an increase in AUC in rats pretreated with NAC relative to saline pretreated rats, Tukey HSD, p<.05.
Figure 2.
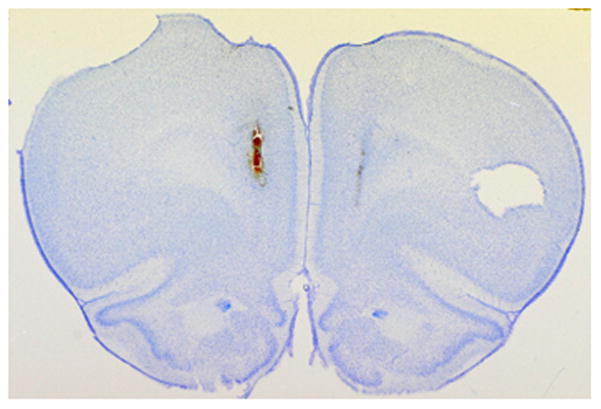
A representative coronal section illustrating the placement of microdialysis probes within the prefrontal cortex. The tract created by the insertion of a microdialysis probe is evident by the tissue damage on the medial aspect of each hemisphere. Note, the left hemisphere of each coronal section is marked with a tissue punch, which is evident as a cavity in the right side of the above, inverted section.
N-acetylcysteine attenuates PCP-evoked performance deficits in a t-maze task
Working memory deficits linked to schizophrenia have been modeled by measuring PCP-induced deficits in rats (Moghaddam and Adams, 1998; Krystal et al., 2003; Harvey and McClure, 2006). This paradigm has predictive validity in that cognitive deficits associated with schizophrenia and working memory deficits produced by noncompetitive NMDA receptor antagonists are at least partially responsive to clozapine (Faltus et al., 1973; Hauber, 1993; Breier et al., 1994). Figure 3a illustrates that PCP dose-dependently reduced performance in a t-maze task. Acute administration of the lowest dose (1 mg/kg, SC) of PCP failed to reduce performance while the highest dose (3 mg/kg, SC) produced the maximal attainable deficit since 50% accuracy represents chance performance. Interestingly, the low dose also failed to increase glutamate levels in the prefrontal cortex. N-acetylcysteine (90 mg/kg, IP) administered alone did not alter performance in a t-maze task (figure 3b). However, rats receiving N-acetylcysteine (90 mg/kg, IP) prior to PCP administration exhibited significantly higher accuracy than rats treated with PCP alone indicating that N-acetylcysteine attenuated PCP-induced t-maze deficits (figure 3b).
Figure 3.

N-acetylcysteine (NAC) blocks PCP-evoked t-maze deficits. (a) Mean (± SEM) accuracy in a discrete-trial T-maze task following an injection of PCP (0–3 mg/kg, SC; n = 4–13/group as indicated in the bars of the graph). Pretreatment (60 min) with PCP (1–3 mg/kg, SC) dose-dependently produced working memory deficits evident as a decrease in the percent of correct choices during ten daily trials; ANOVA yielded a main effect of treatment (F3,33 = 41.99, P < 0.001). * indicates a difference from controls, Tukey HSD, p<.05. (b) Mean (± SEM) accuracy in a discrete-trial T-maze task in rats pretreated (90 min) with NAC (90 mg/kg, IP; n = 7–29/group) prior to PCP (0–3 mg/kg, SC). NAC administration reverses PCP-induced working memory deficits; ANOVA yielded a main effect of treatment (F5,92 = 25.5, P < 0.001). * indicates a difference relative to rats receiving saline alone (sal + 0 PCP), Tukey HSD, p<.05. # indicates a difference relative to rats receiving saline and respective dose of PCP, Tukey HSD, p<.05.
N-acetylcysteine is used to target cystine-glutamate antiporters because it is a prodrug for cyst(e)ine (Williamson and Meister, 1981; Meister, 1985; Pileblad and Magnusson, 1992). However, cysteine can also be transported by the sodium-dependent glutamate transporter EAAC1 (Aoyama et al., 2006). To distinguish between these two mechanisms, we examined the capacity of the cystine-glutamate antiporter inhibitor CPG to block the effects of N-acetylcysteine (figure 4a). Infusion of CPG into the prefrontal cortex had no effect when administered alone or with PCP. However, rats receiving CPG along with N-acetylcysteine and PCP exhibited a significant reduction in performance relative to a drug-free (sham) session. In contrast, rats treated with N-acetylcysteine and PCP only did not exhibit a deficit in performance. Collectively, these data are consistent with the hypothesis that N-acetylcysteine targets cystine-glutamate antiporters in the prefrontal cortex to restore working memory.
Figure 4.

The capacity of N-acetylcysteine (NAC) to reverse PCP-evoked t-maze deficits is blocked by inhibitors of cystine-glutamate exchange and group II mGluRs. (a) Mean (± SEM) accuracy in a discrete-trial T-maze task assessed during a drug-free session (sham) and a test day in which the cystine-glutamate antiporter inhibitor (S)-4-carboxyphenylglycine (CPG; 1 μM) was infused into the dorsolateral prefrontal cortex for 180 min before testing. Rats also received a systemic injection of N-acetylcysteine (0 or 90 mg/kg, IP; 160 min prior to testing) and phencyclidine (PCP; 0 or 2 mg/kg, SC; 60 min prior to testing) on the test day. Rats treated with PCP alone exhibited a deficit in working memory evident as a reduction in t-maze performance on the test day relative to the sham day (PCP, 2 mg/kg, SC; n = 4); this deficit was reversed by co-administration of N-acetylcysteine (90 mg/kg, IP; n = 5). Rats receiving CPG + N-acetylcysteine + PCP (n = 6) exhibited deficits in performance comparable to rats receiving only PCP or PCP + CPG (n = 6). ANOVA yielded a day x treatment interaction (F5,25 = 5.235, P = .002). * indicates a difference relative to performance on respective sham day (T-test, P<.05). (b) Mean (± SEM) accuracy in a discrete-trial T-maze task in rats pretreated with NAC (90 mg/kg, IP; 90 min) and the group II mGluR antagonist LY341495 (1 mg/kg, IP; 70 min) prior to PCP (2 mg/kg, SC; n = 7–16/group). PCP produced a reduction in t-maze performance evident as a decrease in the percent of correct choices during ten daily trials, and this deficit was reversed by NAC pretreatment. Rats receiving NAC + LY341495 prior to PCP exhibited deficits in performance comparable to rats receiving PCP alone; ANOVA yielded a main effect of treatment (F4,47 = 14.42, P < 0.001). * indicates a difference relative to vehicle controls, Tukey HSD, p<.05; # indicates a difference from rats treated with PCP + NAC, Tukey HSD, p<.05.
Previous work indicates that cystine-glutamate antiporters regulate synaptic glutamate through activation of group II mGluRs (Baker et al., 2002; Moran et al., 2005). As a result, we examined whether the capacity of N-acetylcysteine to restore working memory requires group II mGluR signaling. Figure 4b illustrates that pretreatment with the group II mGluR antagonist LY341495 did not alter PCP-induced deficits in working memory. Rats pretreated with both N-acetylcysteine (90 mg/kg, IP) and LY341495 (1 mg/kg, IP) prior to PCP (2 mg/kg, SC) administration exhibited deficits in performance comparable to rats receiving PCP alone. This finding indicates that N-acetylcysteine blocks the capacity of acute PCP treatment to produce working memory deficits through a group II mGluR dependent mechanism.
N-acetylcysteine attenuates PCP-evoked social withdrawal
The impact of N-acetylcysteine on social withdrawal produced by repeated PCP treatment was assessed because this task has been used to model the negative symptoms of schizophrenia (Sams-Dodd, 1999). Similar to t-maze performance, this paradigm has predictive validity in that negative symptoms linked to schizophrenia or repeated PCP-induced behavioral deficits that are used to model these symptoms are at least partially responsive to clozapine (Breier et al., 1994; Corbett et al., 1995; Sams-Dodd, 1999). Social withdrawal was evident as a reduction in the amount of time that two unfamiliar rats spent in close proximity when treated with PCP (3 mg/kg, SC) relative to saline (figure 5). Rats pretreated with N-acetylcysteine prior to PCP administration spent a comparable amount of time within close proximity as saline controls, indicating that N-acetylcysteine administration blocked PCP-evoked social withdrawal. Similar to t-maze performance, administration of LY341495 blocked the effects of N-acetylcysteine indicating that group II mGluR activation is necessary for social interaction restored by N-acetylcysteine (figure 5).
Figure 5.

N-acetylcysteine (NAC) blocks PCP-evoked social withdrawal in a group II mGluR-dependent manner. Social interaction was assessed as the mean (± SEM) time unfamiliar rat pairs spent within 20 cm. PCP (3 mg/kg, SC; n = 6–14) produced a decrease in interaction time and this effect that was reversed by NAC (90 mg/kg, IP) pretreatment (90 min). LY341495 (1 mg/kg, IP) reversed the protective effects of NAC indicating a role for mGluR II receptors in the effects of NAC; ANOVA yielded a main effect of treatment (F3,34 = 5.217, P = 0.005). * indicates a difference relative to vehicle controls, Tukey HSD, p<.05; # indicates a difference relative to rats treated with NAC + PCP, Tukey HSD, p<.05.
Protein levels of xCT are altered in schizophrenic post mortem tissue
Post mortem tissue levels of xCT, the active subunit for cystine-glutamate antiporters, were measured in the human dorsolateral prefrontal cortex (DLPFC), anterior cingulate cortex (ACC) and hippocampus (HPC). Correlation analysis including age, pH, and PMI was performed for xCT expression in all regions. A positive correlation between xCT expression was identified for pH in DLPFC (r=0.30, P=0.036). For all other regions, correlation analysis was not significant for any of the variables tested. Figure 6 illustrates that tissue obtained from schizophrenic patients had higher xCT protein levels in the DLPFC relative to controls, without significant changes in ACC and HPC. Note, there was no significant effect of gender or medication history on xCT expression in DLPFC. In support of the latter, xCT protein levels in the DLPFC did not differ between patients treated within 6 weeks of death and unmedicated individuals (see figure 6c). This indicates that typical antipsychotics do not exert therapeutic effects by altering xCT levels. Figure 7 illustrates the specificity of the anti-xCT antibody used in the above study. Specifically HEK cells treated with xCT siRNA displayed a reduction in xCT RNA and protein. An increase in xCT RNA and protein was detected in HEK cells transfected with xCT cDNA (figure 7). Collectively, these studies provide the first measures of xCT in schizophrenic patients, and further support extant data that is consistent with a reduction of cystine-glutamate exchange in schizophrenia.
Figure 6.

Protein expression of xCT is significantly higher in the dorsal lateral prefrontal cortex (DLPFC) of schizophrenic patients. (a) Levels of the catalytic subunit for cystine-glutamate antiporters, xCT protein, are expressed as a change (mean ± SEM) from controls (n = 27) in the ratio of xCT/β-tubulin measured from human DLPFC (n = 23; ANCOVA: F1,45 = 5.6, P = 0.022), anterior cingulate cortex (ACC; n = 23; ANCOVA: F1,45 = 0.03, P = 0.870), and hippocampus (n = 23; ANCOVA: F1,45 = 0.69, P = 0.41). * indicates a difference from control tissue, p<.05. (b) Representative immunoblots illustrate bands with an apparent molecular mass of 55 kDa. (c) The mean (± SEM) levels of xCT in patients that received antipsychotic therapy within the last six weeks (n = 12) relative to those that had not (n = 10).
Discussion
As the primary excitatory neurotransmitter in the brain, glutamate is involved in most aspects of neurotransmission in the normal and diseased states. The complexity of glutamate signaling has begun to emerge over the past decade with discoveries detailing the existence of vesicular glutamate release from astrocytes and the distinction between functional glutamate receptors in extrasynaptic and synaptic regions. Unfortunately, a dearth of knowledge exists regarding the involvement of extrasynaptic glutamate to neurotransmission in the normal or pathological states, despite the importance of glutamate signaling to brain function. The primary finding of the present report is that cystine-glutamate antiporters, a mechanism capable of nonvesicular release (Baker et al., 2002), can be targeted to reverse the psychotomimetic effects of acute or repeated PCP. The importance of this, coupled with an earlier observation that altered cystine-glutamate antiporter activity contributes to compulsive cocaine seeking (Baker et al., 2003; Lu et al., 2004), is that nonvesicular glutamate release may contribute to neural disorders involving glutamate or represent novel targets in the development of pharmacotherapies. This conclusion is supported by our observation that post mortem tissue obtained from schizophrenic patients exhibited altered levels of xCT, the active subunit for cystine-glutamate antiporters, in the dorsolateral prefrontal cortex.
N-acetylcysteine reversal of psychotomimetic effects of PCP
The psychotomimetic effects of PCP in rodents require increased levels of synaptic glutamate in the rodent prefrontal cortex, stimulation of AMPA receptors, and subsequent increased burst firing of cortical neurons (Moghaddam et al., 1997; Moghaddam and Adams, 1998; Homayoun et al., 2005). Not surprisingly, increased levels of glutamate and burst firing of cortical neurons are blocked by stimulating group II mGluRs (Moghaddam and Adams, 1998; Homayoun et al., 2005) since these receptors function as autoreceptors on presynaptic glutamatergic terminals (Kilbride et al., 1998; Schoepp et al., 1999; Marek et al., 2000). Similar to agonists for group II mGluR, the present data demonstrate that administration of the cysteine prodrug N-acetylcysteine blocks PCP-evoked glutamate release in the prefrontal cortex in a manner that requires cystine-glutamate exchange and increased group II mGluR signaling. This finding is consistent with several studies demonstrating that cystine-glutamate antiporters regulate endogenous stimulation of these receptors (Baker et al., 2002; Baker et al., 2003; Moran et al., 2005). These studies reinforce the hypothesis that multiple pools of glutamate exist and that extrasynaptic pools provide negative feedback regulating synaptic release.
It may seem paradoxical that targeting cystine-glutamate exchange, which increases nonvesicular glutamate, is effective in reducing the psychotomimetic effects of PCP since these behaviors require increased synaptic glutamate (Moghaddam and Adams, 1998; Lorrain et al., 2003; Jackson et al., 2004), however, this may reflect the existence of multiple, functionally-distinct pools of glutamate. Noncompetitive NMDA receptor antagonists increase glutamate in the synaptic cleft thereby generating behaviors dependent upon AMPA receptor stimulation (Moghaddam et al., 1997; Takahata and Moghaddam, 2003). Conversely, cystine-glutamate exchange releases glutamate into the extrasynaptic compartment thereby stimulating proximal receptors such as group II mGluRs (Baker et al., 2002; Baker et al., 2003; Moran et al., 2005). By stimulating extrasynaptic group II mGluRs without exerting postsynaptic effects (Moran et al., 2005), extrasynaptic glutamate appears to negatively influence the synaptic pools (Baker et al., 2002; Moran et al., 2005). The compartmentalization of glutamate is likely the result of sodium-dependent glutamate transporters. These transporters are active in clearing glutamate released from cystine-glutamate exchange (Baker et al., 2002) and, prevents glutamate maintained by nonvesicular release to access synaptic receptors (Jabaudon et al., 1999). To the extent that NR2B receptors are located outside the synapse, this hypothesis is supported by the recent observation that glutamate release from astrocytes appears to stimulate extrasynaptic, but not synaptic NMDA receptors (D’Ascenzo et al., 2007). The existence of multiple pools may partially account for the ambiguity regarding the contribution of glutamate to schizophrenia in which both increased and decreased glutamate signaling have been hypothesized (Moghaddam and Adams, 1998; Barch et al., 2001; MacDonald et al., 2005). Specifically, decreased cystine-glutamate antiporter activity would lead to reduced extrasynaptic glutamate resulting in increased synaptic levels of glutamate due to diminished group II mGluR stimulation. Regardless, these data further establish cystine-glutamate exchange as a novel mechanism capable of regulating glutamate signaling.
The psychotomimetic effects of PCP were assessed in the present studies by measuring performance deficits in a t-maze task dependent upon working memory and social interaction. These behaviors were selected, in part, because they model cognitive deficits and negative symptoms of schizophrenia, respectively (Moghaddam and Adams, 1998; Sams-Dodd, 1999) and these symptoms are not adequately treated with existing medications (Lieberman et al., 2005). Further, the behavioral deficits in these paradigms are produced by acute (t-maze) and repeated (social interaction) administration of PCP, both of which have merits as screens for putative antipsychotics. The predictive validity of these behavioral deficits in identifying antipsychotic efficacy is evident from studies showing that existing medications that are at least partially effective in treating cognitive deficits and negative symptoms of schizophrenia in humans also interfere with these behaviors in rodents (Faltus et al., 1973; Hauber, 1993; Breier et al., 1994; Corbett et al., 1995; Sams-Dodd, 1999). Consistent with earlier findings, these data indicate that PCP produces deficits in t-maze performance and social withdrawal (Moghaddam and Adams, 1998; Sams-Dodd, 1999). Interestingly, a dose of PCP that failed to elevate extracellular glutamate in the prefrontal cortex also failed to produce working memory deficits further indicating a relationship between increased extracellular glutamate in the prefrontal cortex and the psychotomimetic effects of PCP. Moreover, N-acetylcysteine, which blocked PCP-induced glutamate overflow, also prevented both PCP-induced working memory deficits and social withdrawal. The capacity of N-acetylcysteine to reverse the psychotomimetic effects of PCP is blocked by inhibitors of cystine-glutamate exchange and group II mGluRs. These data are consistent with the hypothesis that the psychotomimetic effects of PCP arise, in part, due to elevated extracellular glutamate levels in the prefrontal cortex, and that increased cystine-glutamate exchange represents a strategy to reverse behavioral deficits used to model aspects of schizophrenia. As a result, cystine-glutamate antiporters may represent a novel target in the treatment of schizophrenia.
Cystine-Glutamate Exchange & Schizophrenia
Post mortem levels of xCT, the active subunit for cystine-glutamate exchange, are increased in the dorsolateral prefrontal cortex of human schizophrenic patients. The labeling of xCT in the prefrontal cortex is consistent with earlier studies demonstrating cystine-glutamate exchange in the rat medial prefrontal cortex (Moran et al., 2005), a region thought to be analogous to human dorsolateral prefrontal cortex (Uylings et al., 2003). Although the modest change in protein levels detected in the present study does not provide direct evidence that altered cystine-glutamate exchange occurs in schizophrenia, it is consistent with several lines of evidence that reinforce this hypothesis. Schizophrenics exhibit reduced glutathione levels in the prefrontal cortex (Do et al., 2000). This may reflect blunted cystine-glutamate exchange because cystine uptake into the cell represents the rate-limiting step in glutathione synthesis (Bannai, 1984; Meister, 1991; Sies, 1999). Alternatively, this could cause blunted cystine-glutamate exchange because extracellular glutathione metabolism serves as a reservoir for cystine that is critical in maintaining cystine-glutamate exchange (Deneke and Fanburg, 1989; Sato et al., 1999; Sies, 1999; Kim et al., 2001; Shih et al., 2006). Regardless, either would diminish glutamate release from cystine-glutamate exchange. In support, earlier work has also detected increased levels of mGluR II receptors in post mortem tissue from schizophrenic patients (Gupta et al., 2005), which would be expected following long-term understimulation of the receptor. Collectively, the changes in xCT, glutathione, and group II mGluRs provide indirect evidence of reduced cystine-glutamate antiporter activity in schizophrenia and should serve to stimulate direct measures of cystine-glutamate function in schizophrenia in future experiments.
Although indirect evidence of altered function, the above data are compelling because blunted cystine-glutamate exchange could account for a myriad of pathological events thought to underlie schizophrenia including increased AMPA receptor stimulation, decreased NMDA receptor stimulation, depletion of glutathione, increased dopamine release, and abnormal neurodevelopment. As noted above, a reduction in glutamate release from cystine-glutamate exchange would be expected to decrease stimulation of group II mGluRs (Baker et al., 2002; Moran et al., 2005). Diminished activity of group II mGluRs would result in augmented synaptic release of glutamate and dopamine leading to increased stimulation of postsynaptic receptors, including AMPA receptors (Hu et al., 1999a; Cartmell and Schoepp, 2000; Baker et al., 2002; Chaki et al., 2006). Despite the increase in synaptic glutamate, NMDA receptor hypofunction may arise through at least two mechanisms. First, a reduction in extracellular glutathione would decrease NMDA receptor sensitivity by decreasing the activation of extracellular redox sites (Kohr et al., 1994). Second, metabolism of extracellular glutathione by the membrane-bound enzyme gamma-glutamyl transpeptidase represents a source of extracellular glycine (Abbott and Meister, 1986), which is known to function as a coagonist for the NMDA receptor. In support, glutathione depletion has been shown to produce NMDA receptor hypofunction (Steullet et al., 2006) as well as several behavioral abnormalities used to model schizophrenia (Cabungcal et al., 2006).
Diminished cystine-glutamate exchange may also result in impaired neurodevelopment. In support, nonvesicular glutamate has been shown to be a key regulator of postsynaptic development in Drosophila by regulating the expression of postsynaptic glutamate receptors during synaptogenesis. Specifically, decreased nonvesicular glutamate release resulted in increased expression of postsynaptic glutamate receptors (Featherstone et al., 2002); an effect recently linked to cystine-glutamate exchange (Augustin et al., 2007). Interestingly, postsynaptic glutamate receptor expression influences synapse morphology and presynaptic neurotransmitter release in Drosophila (Petersen et al., 1997; Sigrist et al., 2000). Nonvesicular glutamate release has also been shown to regulate cortical neuron migration during neurodevelopment (Manent et al., 2005). Glutamate has been previously shown to regulate neuroblast migration by functioning to promote migration, and to act as an acceleratory or stop signal. Synaptic glutamate is unlikely to contribute to this effect since normal cortical development occurs in munc13–1/2 double knock-out mice that are incapable of vesicular release of neurotransmitters (Verhage et al., 2000). Rather, recent studies indicate that nonvesicular release is the source of glutamate involved in neuronal development (Manent et al., 2005). Collectively, these data indicate that a disruption of nonvesicular release could disrupt cortical migration during development, impact synapse formation, and lead to altered neurotransmitter release, all of which appear to occur in schizophrenia.
Given the importance of glutamate and the existence of extrasynaptic receptors and release mechanisms, models of brain functioning in the normal and diseased states need to account for the existence of high, steady state levels of glutamate in the extrasynaptic space. The present data reveal that cystine-glutamate exchange represents a novel target capable of reversing pathological changes in complex, cognitive-dependent behavioral tasks used to model aspects of schizophrenia. It is important to note, however, that the pathophysiology underlying the psychotomimetic effects of PCP may be distinct from those underlying the symptoms of schizophrenia. While our data indicating altered xCT expression in postmortem tissue obtained from schizophrenic patients is suggestive of a link between altered cystine-glutamate exchange and schizophrenia, certainly additional data are needed. Moreover, our data, in conjunction with earlier work linking diminished cystine-glutamate exchange to compulsive drug-seeking (Baker et al., 2003), indicates that it is reasonable to postulate involvement of nonvesicular glutamate in other disorders involving abnormal glutamate signaling.
Acknowledgments
MH71672 (DAB), MH53327 (JMW), MH-45212 (VH), MH64673 (VH), and a Young Investigator Award from the National Alliance for Research on Schizophrenia & Depression (DAB). The authors thank Hideyo Sato for donating mxCT cDNA.
Footnotes
Disclosure/Conflict of Interest
Dr. James Meador-Woodruff is the editor in chief of Neuropsychopharmacology and the ACNP website and receives a stipend from ACNP for these services. The remaining authors do not have a conflict of interest or any relationship with outside organizations to disclose.
References
- Abbott WA, Meister A. Intrahepatic transport and utilization of biliary glutathione and its metabolites. Proc Natl Acad Sci U S A. 1986;83:1246–1250. doi: 10.1073/pnas.83.5.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Augustin H, Grosjean Y, Chen K, Sheng Q, Featherstone DE. Nonvesicular release of glutamate by glial xCT transporters suppresses glutamate receptor clustering in vivo. J Neurosci. 2007;27:111–123. doi: 10.1523/JNEUROSCI.4770-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- Bannai S. Induction of cystine and glutamate transport activity in human fibroblasts by diethyl maleate and other electrophilic agents. J Biol Chem. 1984;259:2435–2440. [PubMed] [Google Scholar]
- Barch DM, Carter CS, Braver TS, Sabb FW, MacDonald A, 3rd, Noll DC, Cohen JD. Selective deficits in prefrontal cortex function in medication-naive patients with schizophrenia. Arch Gen Psychiatry. 2001;58:280–288. doi: 10.1001/archpsyc.58.3.280. [DOI] [PubMed] [Google Scholar]
- Baskys A, Malenka RC. Agonists at metabotropic glutamate receptors presynaptically inhibit EPSCs in neonatal rat hippocampus. J Physiol. 1991;444:687–701. doi: 10.1113/jphysiol.1991.sp018901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford HF, Young AM, Crowder JM. Continuous glutamate leakage from brain cells is balanced by compensatory high-affinity reuptake transport. Neurosci Lett. 1987;81:296–302. doi: 10.1016/0304-3940(87)90399-5. [DOI] [PubMed] [Google Scholar]
- Breier A, Buchanan RW, Kirkpatrick B, Davis OR, Irish D, Summerfelt A, Carpenter WT., Jr Effects of clozapine on positive and negative symptoms in outpatients with schizophrenia. Am J Psychiatry. 1994;151:20–26. doi: 10.1176/ajp.151.1.20. [DOI] [PubMed] [Google Scholar]
- Bunney WE, Bunney BG. Evidence for a compromised dorsolateral prefrontal cortical parallel circuit in schizophrenia. Brain Res Brain Res Rev. 2000;31:138–146. doi: 10.1016/s0165-0173(99)00031-4. [DOI] [PubMed] [Google Scholar]
- Cabungcal JH, Nicolas D, Kraftsik R, Cuenod M, Do KQ, Hornung JP. Glutathione deficit during development induces anomalies in the rat anterior cingulate GABAergic neurons: Relevance to schizophrenia. Neurobiol Dis. 2006 doi: 10.1016/j.nbd.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Chaki S, Yoshikawa R, Okuyama S. Group II metabotropic glutamate receptor-mediated regulation of dopamine release from slices of rat nucleus accumbens. Neurosci Lett. 2006;404:182–186. doi: 10.1016/j.neulet.2006.05.043. [DOI] [PubMed] [Google Scholar]
- Chavez-Noriega LE, Schaffhauser H, Campbell UC. Metabotropic glutamate receptors: potential drug targets for the treatment of schizophrenia. Curr Drug Target CNS Neurol Disord. 2002;1:261–281. doi: 10.2174/1568007023339337. [DOI] [PubMed] [Google Scholar]
- Cochilla AJ, Alford S. Metabotropic glutamate receptor-mediated control of neurotransmitter release. Neuron. 1998;20:1007–1016. doi: 10.1016/s0896-6273(00)80481-x. [DOI] [PubMed] [Google Scholar]
- Corbett R, Camacho F, Woods AT, Kerman LL, Fishkin RJ, Brooks K, Dunn RW. Antipsychotic agents antagonize non-competitive N-methyl-D-aspartate antagonist-induced behaviors. Psychopharmacology (Berl) 1995;120:67–74. doi: 10.1007/BF02246146. [DOI] [PubMed] [Google Scholar]
- D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, Moss SJ, Haydon PG. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci U S A. 2007;104:1995–2000. doi: 10.1073/pnas.0609408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneke SM, Fanburg BL. Regulation of cellular glutathione. Am J Physiol. 1989;257:L163–173. doi: 10.1152/ajplung.1989.257.4.L163. [DOI] [PubMed] [Google Scholar]
- Do KQ, Trabesinger AH, Kirsten-Kruger M, Lauer CJ, Dydak U, Hell D, Holsboer F, Boesiger P, Cuenod M. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci. 2000;12:3721–3728. doi: 10.1046/j.1460-9568.2000.00229.x. [DOI] [PubMed] [Google Scholar]
- Faltus F, Hynek K, Dolezalova V, Kumnickova Z, Zemek P. Experience in the treatment of schizophrenia with clozapine. Act Nerv Super (Praha) 1973;15:95. [PubMed] [Google Scholar]
- Featherstone DE, Rushton E, Broadie K. Developmental regulation of glutamate receptor field size by nonvesicular glutamate release. Nat Neurosci. 2002;5:141–146. doi: 10.1038/nn789. [DOI] [PubMed] [Google Scholar]
- Gupta DS, McCullumsmith RE, Beneyto M, Haroutunian V, Davis KL, Meador-Woodruff JH. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse. 2005;57:123–131. doi: 10.1002/syn.20164. [DOI] [PubMed] [Google Scholar]
- Harvey PD, McClure MM. Pharmacological approaches to the management of cognitive dysfunction in schizophrenia. Drugs. 2006;66:1465–1473. doi: 10.2165/00003495-200666110-00004. [DOI] [PubMed] [Google Scholar]
- Hauber W. Clozapine improves dizocilpine-induced delayed alteration impairment in rats. J Neural Transm Gen Sect. 1993;94:223–233. doi: 10.1007/BF01277027. [DOI] [PubMed] [Google Scholar]
- Herrera-Marschitz M, You ZB, Goiny M, Meana JJ, Silveira R, Godukhin OV, Chen Y, Espinoza S, Pettersson E, Loidl CF, Lubec G, Andersson K, Nylander I, Terenius L, Ungerstedt U. On the origin of extracellular glutamate levels monitored in the basal ganglia of the rat by in vivo microdialysis. J Neurochem. 1996;66:1726–1735. doi: 10.1046/j.1471-4159.1996.66041726.x. [DOI] [PubMed] [Google Scholar]
- Homayoun H, Jackson ME, Moghaddam B. Activation of metabotropic glutamate 2/3 receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats. J Neurophysiol. 2005;93:1989–2001. doi: 10.1152/jn.00875.2004. [DOI] [PubMed] [Google Scholar]
- Hu G, Duffy P, Swanson C, Ghasemzadeh MB, Kalivas PW. The regulation of dopamine transmission by metabotropic glutamate receptors. J Pharmacol Exp Ther. 1999a;289:412–416. [PubMed] [Google Scholar]
- Hu G, Duffy P, Swanson C, Ghasemzadeh MB, Kalivas PW. The regulation of dopamine transmission by metabotropic glutamate receptors. J Pharmacol Exp Ther. 1999b;289:412–416. [PubMed] [Google Scholar]
- Huang Y, Dai Z, Barbacioru C, Sadee W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005;65:7446–7454. doi: 10.1158/0008-5472.CAN-04-4267. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci U S A. 1999;96:8733–8738. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:8467–8472. doi: 10.1073/pnas.0308455101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- Kilbride J, Huang LQ, Rowan MJ, Anwyl R. Presynaptic inhibitory action of the group II metabotropic glutamate receptor agonists, LY354740 and DCG-IV. Eur J Pharmacol. 1998;356:149–157. doi: 10.1016/s0014-2999(98)00526-3. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kanai Y, Chairoungdua A, Cha SH, Matsuo H, Kim DK, Inatomi J, Sawa H, Ida Y, Endou H. Human cystine/glutamate transporter: cDNA cloning and upregulation by oxidative stress in glioma cells. Biochim Biophys Acta. 2001;1512:335–344. doi: 10.1016/s0005-2736(01)00338-8. [DOI] [PubMed] [Google Scholar]
- Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron. 1994;12:1031–1040. doi: 10.1016/0896-6273(94)90311-5. [DOI] [PubMed] [Google Scholar]
- Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr, Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Lorrain DS, Baccei CS, Bristow LJ, Anderson JJ, Varney MA. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117:697–706. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- Lu L, Hope BT, Shaham Y. The cystine-glutamate transporter in the accumbens: a novel role in cocaine relapse. Trends Neurosci. 2004;27:74–76. doi: 10.1016/j.tins.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Luby ED, Cohen BD, Rosenbaum G, Gottlieb JS, Kelley R. Study of a new schizophrenomimetic drug; sernyl. AMA Arch Neurol Psychiatry. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- MacDonald AW, 3rd, Carter CS, Kerns JG, Ursu S, Barch DM, Holmes AJ, Stenger VA, Cohen JD. Specificity of prefrontal dysfunction and context processing deficits to schizophrenia in never-medicated patients with first-episode psychosis. Am J Psychiatry. 2005;162:475–484. doi: 10.1176/appi.ajp.162.3.475. [DOI] [PubMed] [Google Scholar]
- Malhotra AK, Adler CM, Kennison SD, Elman I, Pickar D, Breier A. Clozapine blunts N-methyl-D-aspartate antagonist-induced psychosis: a study with ketamine. Biol Psychiatry. 1997;42:664–668. doi: 10.1016/s0006-3223(96)00546-x. [DOI] [PubMed] [Google Scholar]
- Manent JB, Demarque M, Jorquera I, Pellegrino C, Ben-Ari Y, Aniksztejn L, Represa A. A noncanonical release of GABA and glutamate modulates neuronal migration. J Neurosci. 2005;25:4755–4765. doi: 10.1523/JNEUROSCI.0553-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, Wright RA, Schoepp DD, Monn JA, Aghajanian GK. Physiological antagonism between 5-hydroxytryptamine(2A) and group II metabotropic glutamate receptors in prefrontal cortex. J Pharmacol Exp Ther. 2000;292:76–87. [PubMed] [Google Scholar]
- Meister A. Methods for the selective modification of glutathione metabolism and study of glutathione transport. Methods Enzymol. 1985;113:571–585. doi: 10.1016/s0076-6879(85)13077-6. [DOI] [PubMed] [Google Scholar]
- Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol Ther. 1991;51:155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–1352. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic Press; 1986. [DOI] [PubMed] [Google Scholar]
- Pearlson GD. Psychiatric and medical syndromes associated with phencyclidine (PCP) abuse. Johns Hopkins Med J. 1981;148:25–33. [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Pileblad E, Magnusson T. Increase in rat brain glutathione following intracerebroventricular administration of gamma-glutamylcysteine. Biochem Pharmacol. 1992;44:895–903. doi: 10.1016/0006-2952(92)90121-x. [DOI] [PubMed] [Google Scholar]
- Purohit DP, Perl DP, Haroutunian V, Powchik P, Davidson M, Davis KL. Alzheimer disease and related neurodegenerative diseases in elderly patients with schizophrenia: a postmortem neuropathologic study of 100 cases. Arch Gen Psychiatry. 1998;55:205–211. doi: 10.1001/archpsyc.55.3.205. [DOI] [PubMed] [Google Scholar]
- Sams-Dodd F. Phencyclidine in the social interaction test: an animal model of schizophrenia with face and predictive validity. Rev Neurosci. 1999;10:59–90. doi: 10.1515/revneuro.1999.10.1.59. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med. 1999;27:916–921. doi: 10.1016/s0891-5849(99)00177-x. [DOI] [PubMed] [Google Scholar]
- Sigrist SJ, Thiel PR, Reiff DF, Lachance PE, Lasko P, Schuster CM. Postsynaptic translation affects the efficacy and morphology of neuromuscular junctions. Nature. 2000;405:1062–1065. doi: 10.1038/35016598. [DOI] [PubMed] [Google Scholar]
- Steullet P, Neijt HC, Cuenod M, Do KQ. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience. 2006;137:807–819. doi: 10.1016/j.neuroscience.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Takahata R, Moghaddam B. Activation of glutamate neurotransmission in the prefrontal cortex sustains the motoric and dopaminergic effects of phencyclidine. Neuropsychopharmacology. 2003;28:1117–1124. doi: 10.1038/sj.npp.1300127. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Westerink BH. Brain microdialysis of GABA and glutamate: what does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Uylings HB, Groenewegen HJ, Kolb B. Do rats have a prefrontal cortex? Behav Brain Res. 2003;146:3–17. doi: 10.1016/j.bbr.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Valenti O, Conn PJ, Marino MJ. Distinct physiological roles of the Gq-coupled metabotropic glutamate receptors Co-expressed in the same neuronal populations. J Cell Physiol. 2002;191:125–137. doi: 10.1002/jcp.10081. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Williamson JM, Meister A. Stimulation of hepatic glutathione formation by administration of L-2-oxothiazolidine-4-carboxylate, a 5-oxo-L-prolinase substrate. Proc Natl Acad Sci U S A. 1981;78:936–939. doi: 10.1073/pnas.78.2.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Baker DA, Shen H, Carson DS, Kalivas PW. Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens. J Pharmacol Exp Ther. 2002a;300:162–171. doi: 10.1124/jpet.300.1.162. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Ramamoorthy S, Baker DA, Shen H, Samuvel DJ, Kalivas PW. Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharmacol Exp Ther. 2002b;303:608–615. doi: 10.1124/jpet.102.039735. [DOI] [PubMed] [Google Scholar]