

More and more structures of proteins and nucleic acids, including enzymes that catalyze chemical reactions, are known with high accuracy. Much less is known about the dynamics of these macromolecules and their role in biological function. For example, motions within an enzyme molecule may be necessary to lower the transition state barrier. On page 1520 of this issue, Eisenmesser et al.(1) use an elegant nuclear magnetic resonance (NMR) method to investigate the motions linked to substrate turnover in the enzyme cyclophilin A. Their approach should facilitate the search for motions linked to catalytic events in other enzymes.
A complete analysis of the dynamics of a typical macromolecule requires knowledge of thousands of atomic trajectories as a function of time. This remains challenging, but NMR and other methods have revealed some basic features of thermally driven macromolecular dynamics, particularly in proteins.
Early protein NMR experiments detected ring-flipping motions of aromatic side chains, providing some of the first evidence that proteins are dynamic structures (2). Modern spectroscopic, time-resolved crystallographic, and computational studies have detected complex side chain and backbone thermal motions over time scales ranging from picoseconds to seconds (3–8). Chemical methods such as hydrogen exchange and disulfide trapping have probed thermal motions on longer time scales of microseconds to hours, revealing motional amplitudes as large as 1.5 nm on the millisecond time scale (9–11).
A protein in solution thus undergoes constant random thermal motions within a stable equilibrium structure. These motions involve displacements of individual atoms, bonds, functional groups, side chains, local regions of the backbone, secondary structure elements, and entire folded domains. Many proteins also undergo thermally driven transitions, called conformational changes, between two or more equilibrium structures.
Both types of motions can play important functional roles (12–16). Random thermal motions act as a molecular lubricant during conformational changes, allowing the protein to sample conformational space. Random thermal motions and the average conformation can both change substantially when a protein is modified by substrate or ligand binding, docking to another macromolecule, or covalent modification (such as phosphorylation). Such changes often have important functional consequences for the tuning of binding affinities and the switching of regulatory proteins. Yet, in a given macromolecule, only a subset of motions is important for biological function. The challenge is to identify these functionally relevant motions.
Dynamics play a role in certain aspects of enzyme function, but the links between dynamics and catalysis remain unclear. In the first step of an enzyme-catalyzed reaction, substrate binding typically induces a conformational change within the enzyme, thereby enclosing the substrate in a cavity protected from solvent or places the catalytic residues near the substrate (14–16).
An even more fascinating structural rearrangement may occur during the catalytic step of the reaction (17), when the complex moves from the ground state to the transition state. Such a transient rearrangement could simply serve to accommodate the structural changes in the substrate as the transition state is reached or could actively contribute to catalysis by preferentially stabilizing the transition state. Yet, because the transition state exists only fleetingly, enzyme dynamics during transition state formation and decay have never been directly detected during an enzyme-catalyzed reaction in solution.
Eisenmesser et al. have used NMR relaxation methods to probe transition state movements in cyclophilin A. They monitored the motional frequencies in the environments of 160 amide nitrogen nuclei in the protein backbone. The new twist is that the relaxation studies were performed on the working enzyme undergoing catalytic turnovers. Relaxation methods that monitor very fast motions on pico- to nanosecond time scales detected no motional changes when substrate was added. However, a transverse relaxation measurement that monitors micro- to millisecond time scales (3, 4, 18, 19) revealed new motional frequencies upon substrate addition at 10 of the 160 probe nuclei.
Nine of the 10 affected nuclei probably detect a routine protein conformational change triggered by substrate binding and dissociation. For these nuclei, titration with substrate yielded a maximum transverse relaxation rate at an intermediate substrate concentration, characteristic of relaxation resulting from repetitive substrate binding and dissociation events. Fitting the data yielded a rate constant for substrate dissociation of 11,000 to 15,000 s−1, which agrees well with the rate measured by an independent method (1). In the crystal structure of the enzyme-substrate complex (20), only some of the nine residues physically contact the substrate molecule, but all nine lie in the vicinity of the substrate binding site (blue residues in the figure).
Figure. Residues in motion.
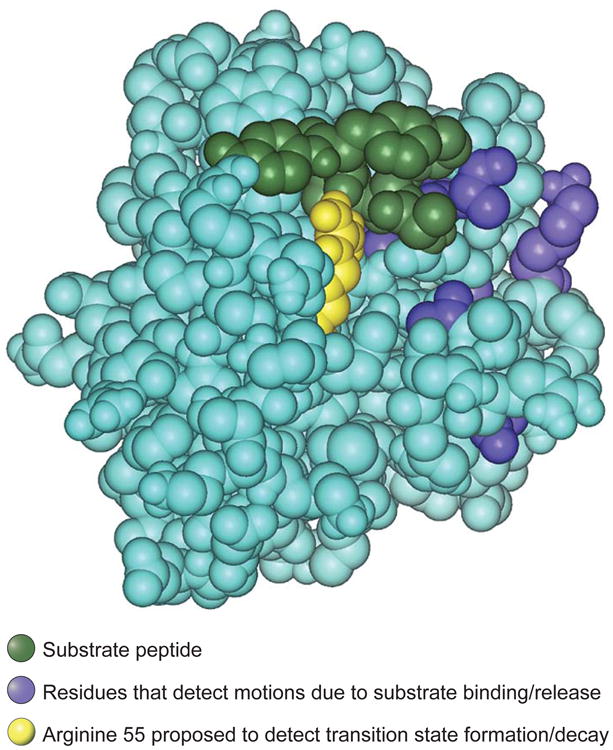
Space-filling atomic structure of the complex between the enzyme cyclophilin A and its substrate cis-Suc-Ala-Phe-Pro-4-NA (20). The substrate is green; residues whose backbone amides detect motional changes due to repetitive substrate binding and dissociation are blue. The catalytic residue arginine 55 is shown in yellow. This residue forms a hydrogen bond to the substrate and detects a motional frequency similar to the catalytic rate, suggesting that it could be involved in motions leading to the formation and decay of the transition state.
The most intriguing residue was arginine 55 (yellow residue in the figure), which is hydrogen bonded to the substrate and is essential for catalysis (20, 21). This residue exhibited two behaviors expected for a probe of transition state rearrangement. First, the transverse relaxation reached its maximum rate at a saturating substrate concentration, where the rate of transition state formation would be maximal. Second, and most striking, the excess transverse relaxation rate measured in the presence of saturating substrate roughly matched the frequency of transition state formation and decay, 9000 s−1, determined by an independent method (1).
The motion detected at arginine 55 thus occurs at about the same frequency as the chemical step of the reaction. The observed characteristics of arginine 55 suggest that its backbone amide could be sensing motional changes associated with transition state rearrangements in the protein or the substrate, or both.
The hypothesis that arginine 55 detects protein transition state rearrangements is plausible and testable. First, the frequency of the motion should be correlated with changes in the rate constant of the chemical step, not with the rate constant of substrate dissociation or any other parameter, as the enzyme is mutated or when natural variants of the enzyme are compared. Second, the transverse relaxation associated with transition state formation should vanish when a competitive inhibitor replaces the substrate.
The range of the motion detected by arginine 55 is more difficult to determine. As Eisenmesser et al. point out, motion linked to catalysis is not necessarily confined to arginine 55. Probe nuclei that do not experience a substantial chemical shift change during a structural transition will not give a signal even if they are located in a region of changing conformation. Moreover, Eisenmesser et al. have focused on backbone nuclei rather than side chains.
The range of the putative catalytic motions could extend to other residues within the active site or even to regions outside the active site. These possibilities can be examined by refining the relaxation measurements further and extending them to side chain nuclei.
The approach developed by Kern and co-workers will likely be applied to many other enzymes during catalysis. It should generate a wealth of new information about motions in working enzymes, particularly those correlated with transition state formation and decay. But once a structural rearrangement linked to transition state formation is identified and its range is characterized, the question remains whether the detected movement contributes to catalysis by lowering the transition state barrier.
In a dramatic substrate-to-product rearrangement such as proline cis-trans isomerization (as in cyclophilin A), the rearranging substrate may simply push certain side chains out of the way to reach the transition state. If these side chains are flexible and it costs negligible energy to brush them aside, then the accompanying movement will have little effect on catalysis. At the other extreme, rearrangements of the enzyme structure could be required to properly align the catalytic residues around the developing transition state, such that the protein dynamics that occur during transition state formation are essential to transition state stabilization and catalysis. The debate over these possibilities will likely occupy the field for years to come.
This is a commentary on article Eisenmesser EZ, Bosco DA, Akke M, Kern D.Enzyme dynamics during catalysis. Science. 2002;295(5559):1520-3.
References
- 1.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Science. 2002;295:1520. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 2.Wüthrich K, Wagner G. Trends Biochem Sci. 1978;9:152. [Google Scholar]
- 3.Palmer AG. Annu Rev Biophys Biomol Struct. 2001;30:129. doi: 10.1146/annurev.biophys.30.1.129. [DOI] [PubMed] [Google Scholar]
- 4.Wand AJ. Nature Struct Biol. 2001;8:926. doi: 10.1038/nsb1101-926. [DOI] [PubMed] [Google Scholar]
- 5.Hubbel WL, Cafiso DS, Altenbach C. Nature Struct Biol. 2000;7:735. doi: 10.1038/78956. [DOI] [PubMed] [Google Scholar]
- 6.Weiss S. Nature Struct Biol. 2000;7:724. doi: 10.1038/78941. [DOI] [PubMed] [Google Scholar]
- 7.Moffat K. Chem Rev. 2001;101:1569. doi: 10.1021/cr990039q. [DOI] [PubMed] [Google Scholar]
- 8.Daggett V. Curr Opin Struct Biol. 2000;10:160. doi: 10.1016/s0959-440x(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 9.Huyghues-Despointes BM, et al. Methods Mol Biol. 2001;168:69. doi: 10.1385/1-59259-193-0:069. [DOI] [PubMed] [Google Scholar]
- 10.Falke JJ, Koshland DE., Jr Science. 1987;237:1596. doi: 10.1126/science.2820061. [DOI] [PubMed] [Google Scholar]
- 11.Careaga CL, Falke JJ. J Mol Biol. 1992;226:1219. doi: 10.1016/0022-2836(92)91063-u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Pawley NH, Nicholson LK. J Mol Biol. 2001;313:873. doi: 10.1006/jmbi.2001.5083. [DOI] [PubMed] [Google Scholar]
- 13.Forman-Kay JD. Nature Struct Biol. 1999;6:1086. doi: 10.1038/70008. [DOI] [PubMed] [Google Scholar]
- 14.Joseph D, Petsko GA, Karplus M. Science. 1990;249:1425. doi: 10.1126/science.2402636. [DOI] [PubMed] [Google Scholar]
- 15.Faber HR, Matthews BW. Nature. 1990;348:263. doi: 10.1038/348263a0. [DOI] [PubMed] [Google Scholar]
- 16.Osborne MJ, Schnell J, Benkovic SJ, Dyson HJ, Wright PE. Biochemistry. 2001;40:9846. doi: 10.1021/bi010621k. [DOI] [PubMed] [Google Scholar]
- 17.Fersht A. Structure and Mechanism in Protein Science A Guide to Enzyme Catalysis and Protein Folding. Freeman; New York: 1999. [Google Scholar]
- 18.Kay LE. Nature Struct Biol. 1998;5:513. doi: 10.1038/755. [DOI] [PubMed] [Google Scholar]
- 19.Hoogstraten CG, Wank JR, Pardi A. Biochemistry. 2000;39:9951. doi: 10.1021/bi0007627. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y, Ke H. Biochemistry. 1996;35:7356. doi: 10.1021/bi9602775. [DOI] [PubMed] [Google Scholar]
- 21.Zydowsky LD, et al. Protein Sci. 1992;1:1092. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]