

Abstract
Background: Psychosis is a common and debilitating side effect of long-term dopaminergic treatment of Parkinson disease (PD). While clozapine is an effective treatment, the need for blood monitoring has limited its first-line use.
Objective: Since olanzapine shows similar receptor affinity to clozapine, we hypothesized that it might be an effective alternative to clozapine for treatment of drug-induced psychosis (DIP) in PD, and that lower doses than usual might make it tolerable.
Methods: In 1998-2003 we conducted a four-week, double-blind, placebo-controlled, parallel group, fixed-dose trial of olanzapine (0, 2.5mg, or 5mg) in 23 PD patients with DIP while allowing for clinically realistic dose adjustments of dopaminomimetic mid-study. The primary outcome measures were Brief Psychiatric Rating Scale (BPRS) ratings scored from videotaped interviews after study termination by an observer blinded to dose assignment and to interview timing, and CGI (Clinical Global Impression). The Unified Parkinson’s Disease Rating Scale motor subscale (UPDRS) was the primary measure of tolerability.
Results: Intention-to-treat analysis found no significant differences among treatment groups in study completion or serious adverse events. However, a disproportionate number of olanzapine vs. placebo subjects reported mild side effects (p<0.04), many citing motor worsening. Fourteen patients completed the study (seven on placebo, two on 2.5mg olanzapine, five on 5mg olanzapine). In study completers, analysis by repeated measures ANOVA revealed no significant difference between olanzapine and placebo groups in BPRS psychosis reduction (p=0.536), parkinsonism (p=0.608), or any other measured parameters (CGI, MMSE, Beck Depression Inventory, Hamilton Depression score, PDQ‑39, Schwab-England ADL assessment, and sleep scores).
Conclusion: This study adds to other evidence that olanzapine is ineffective in treating medication-induced psychosis in Parkinson disease.
Introduction
Drug-induced psychosis (DIP) is a significant and disabling complication of long-term treatment of Parkinson disease (PD), affecting a large minority of PD patients receiving chronic dopaminergic therapy 1. Visual hallucinations are the most commonly reported psychotic phenomena in this population, with auditory, tactile, somatic, and olfactory hallucinations being much less common. Delusions, when they occur, often antedate visual hallucinations and commonly are paranoid or persecutory in nature 2, 3. In addition to the increased caregiver burden caused by psychosis and its sequelae, hallucinations in the context of chronically treated PD tend to be progressive in nature, resulting in increased propensity for nursing home placement and subsequent higher mortality 4, 5. These sobering associations suggest aggressive management of DIP in this population. However, either dose reduction of antiparkinsonian medications or addition of traditional neuroleptics usually increases parkinsonian motor disabilities. Atypical antipsychotics, with their comparatively lower incidence of parkinsonism in schizophrenia, have potential advantages for treatment of hallucinations in this sensitive population 1.
Until recently, the only treatment proven with randomized, placebo-controlled studies to reduce DIP has been clozapine, an agent that does not worsen motor function 6– 8. Despite these favorable data, use of clozapine has been limited secondary to its rare but potentially serious risk of agranulocytosis and the consequent necessity for frequent blood draws 1. Thus alternative treatments have been eagerly sought.
Quetiapine has become the most commonly prescribed antipsychotic in DIP 9. Although double-blind, placebo-controlled trials of quetiapine in PD confirmed it is well tolerated in terms of motor side effects, it has not proven significantly more effective than placebo in treating psychosis 10– 15, and a head-to-head comparison found clozapine superior to quetiapine 16. Ziprasidone showed some benefit in open-label experience 17, including in a random-assignment open comparison to clozapine 18. However, ziprasidone can cause motor side effects in PD and is not generally considered standard therapy for DIP 1, 19. Other treatments, such as ondansetron, acetylcholinesterase inhibitors, and electroconvulsive therapy are supported by limited data in idiopathic Parkinson disease but are generally not viewed as first-line therapy 1, 19. Recently, a phase III clinical trial of a serotonin 5HT 2A inverse agonist, pimavanserin, showed benefit over placebo, but the drug will not be available in the U.S. at least until late 2014 20– 22.
Clozapine’s antipsychotic efficacy is often attributed to its D 4 receptor antagonism. It is also posited that its robust 5HT 2A receptor antagonism, especially in relation to its relatively weaker D 2 receptor blockade, actually increases dopamine transmission in prefrontal cortical and nigrostriatal projections 23. This may account for the cognitive improvement as well as paucity of extrapyramidal adverse events observed in clozapine-treated patients with dopaminomimetic-induced psychosis 23, 24. Olanzapine, therefore, with its ostensibly similar receptor binding profile to clozapine at D 2, D 4, and serotonergic receptors (especially 5HT 2A and 5HT 2C), and muscarinic sites, provides a theoretically encouraging alternative to clozapine in this fragile population 25.
An initial open study of olanzapine in Parkinson disease revealed antipsychotic benefit without motor deterioration when drug dosage was optimized in a slow titration (mean daily dose at end of study was 6.5mg) and dopaminomimetic dose adjustments were allowed 26. Aarsland and colleagues replicated these findings in a relatively more challenging population of Parkinson disease patients with and without dementia 27. Several other small, open-label studies of olanzapine, however, have demonstrated antipsychotic benefit but at the expense of intolerable worsening of gait and bradykinesia, frequently leading to premature termination of the drug 28– 30. Another small open-label trial and case report series suggested unacceptable Parkinsonian motor deterioration in the context of dubious antipsychotic efficacy 31, 32. Later, two double-blind placebo-controlled trials revealed equivocal antipsychotic benefit and problematic motor decline in PD patients with DIP treated with 2.5–15mg/day olanzapine (mean final doses 4.1–4.6mg/day) 33, 34. As a result, experts have recommended against the use of olanzapine in PD 1, 19.
None of these studies, however, were parallel-group fixed-dose trials, and some allowed for neuroleptic dose in the same range as approved for schizophrenia; experience with clozapine suggests that an effective antipsychotic dose in PD is often an order of magnitude less than that typical for schizophrenia treatment. In addition, the two double-blind placebo-controlled trials did not permit adjustments of subjects’ dopaminomimetics, which might have alleviated motoric side effects. Finally, some of the studies cited were terminated prematurely due to side effects. Given that the only marketed drug for which efficacy has been shown is clozapine, demonstrating efficacy for an alternative agent would be important, and a fixed low dose of olanzapine (2.5mg/day) may allow a reasonably low incidence of side effects if dopaminomimetic dose adjustments are allowed. We discuss here the findings of a double-blind, placebo-controlled study of fixed, low-dose olanzapine for treatment of DIP in the context of flexible dopaminomimetic dosing. The hypothesis was that olanzapine given in this fashion would reduce DIP in patients with idiopathic PD significantly more than would a placebo, without causing intolerable motor worsening.
Methods and materials
The completed CONSORT checklist 35, 36 and the original study protocol are available in the Data Files.
Ethics statement
All patients gave written informed consent to participate in the study, which was approved by the Washington University Human Studies Committee (approval # 97-0366). In most cases an appropriate surrogate decision maker also consented. FDA approval was through IND # 53,556. This trial concluded in 2003, so it is exempt from the current ICMJE requirement of prospectively registering clinical trials.
Patient selection
Twenty-four patients were recruited from the Washington University Movement Disorders Center from February 1998 to October 2003. Patients were examined by a movement disorders specialist and diagnosed with idiopathic PD based on presence of at least two of three cardinal manifestations of the disease (rigidity, bradykinesia, rest tremor), response to levodopa or a dopamine agonist, and absence of historical or examination features suggesting secondary parkinsonism. Subjects were treated with levodopa and were experiencing clinically significant hallucinations or delusions, as judged by their treating neurologist or psychiatrist and by the investigator (KJB). Subjects were required to be over 30 years old and have a caregiver who could provide a reliable report. At study entry, patients were required to be treated with the lowest clinically acceptable dose of dopaminomimetic. Patients treated only with a dopamine agonist were not entered in the study, as it was deemed more clinically appropriate to try a switch to levodopa before adding an antipsychotic. Exclusion criteria included a Folstein Mini-Mental State Examination (MMSE) score < 22 37, pregnancy, concurrent diagnosis of delirium (unless clearly explained by dopaminomimetics), catatonia or neuroleptic malignant syndrome (NMS)-like syndrome, other confounding central nervous system (CNS) illness or systemic illness with potential CNS effects, antipsychotic use within the last month predating study enrollment (within the past six months for depot neuroleptics), history of olanzapine sensitivity, or any expectation of significant medical or surgical intervention within six weeks after enrollment. Subjects were also excluded if severity of psychosis warranted hospitalization or if, in the investigator’s judgment, psychosis severity would have made randomization to placebo inappropriate.
Treatment protocol
Patients were randomized 1:1:1 to treatment with placebo or either of two doses of olanzapine. At study initiation, treatment groups consisted of a placebo arm, a 5mg arm, in which patients received this dosage nightly throughout the four weeks of investigation, and a 10mg arm, in which patients received 5mg for the first week and 10mg thereafter. Subjects received matched tablets or capsules provided by Lilly Research Laboratories (Indianapolis, IN), who provided the investigator with sealed, sequentially numbered envelopes containing the medication identity for each subject. The envelopes were not opened until after all data were collected and reviewed for accuracy, and after all decisions about statistical analysis were final, so that both investigators and patients were blind to intervention assignment. The randomization was done by Lilly. KJB enrolled subjects and patients were assigned to treatment packages sequentially by enrollment date.
After the first five patients were enrolled, an interim safety analysis was conducted by a reviewer otherwise not involved in the study, in light of reports published since the study initiation that higher olanzapine doses caused intolerable exacerbation of parkinsonism in PD. Though serious adverse events were no more common in the treatment groups than in the placebo group, it was decided at this time that the two active treatment arms would be changed to fixed doses of 2.5mg and 5mg olanzapine, maintained throughout the four weeks of study. New treatment packages were received and the blind was maintained until after data analysis, as above. No other changes to the protocol were made. See Table 1 for a summary of the final study design. The study was planned for 10 subjects in each of three dose arms. This would produce 90% power (at alpha = 0.05) to detect a change of the magnitude and variability seen in the Wolters et al. 26 report.
Table 1. Summary of final study design.
| Baseline | Weeks 1–2 | 2 week visit | Weeks 3–4 | 4 week visit |
|---|---|---|---|---|
| Clinical evaluation;
randomize |
Placebo
2.5mg 5mg |
Clinical evaluation; ↑
dopaminometic, if indicated |
Placebo
2.5mg 5mg |
Clinical evaluation; return to
routine clinical care |
This table summarizes the study design and timing of assessments and interventions for the last 19 subjects enrolled in the study. ↑ dopaminometic: dose increase allowed for antiparkinsonian medication, if parkinsonism had worsened since starting the study. See Methods and Figure 1 for further details.
Subjects received a baseline evaluation that involved a full psychiatric, neurologic, and medical history and examination, CGI (Clinical Global Impression) by MD 38, PDQ-39, a self-rated quality-of-life measure for PD 39, videotaped interview for later BPRS (Brief Psychiatric Rating Scale) rating blind to drug dose and blind to which visit was being rated 40, Schwab-England ADL assessment 41, UPDRS (Unified Parkinson’s Disease Rating Scale), section III (motor) 42, MMSE 37, HDRS (Hamilton Depression Rating Scale) 43, BDI (Beck Depression Inventory) 44, and patient/caretaker reported hours and quality of sleep. Repeated measures at the two-week interim visit and the final four-week evaluation included CGI (by MD, patient, and caretaker), videotaped interview for later blinded BPRS, Schwab-England ADL assessment, UPDRS, MMSE, PDQ-39, BDI, sleep questionnaire, and pill counts. All assessments were done at Washington University Medical Center.
Primary efficacy measures were CGI scores and BPRS ratings of psychosis. At each visit, the coordinator interviewed the patient during videotaping using a semi-structured interview designed to facilitate later scoring of psychopathology using the BPRS 40, 45, 46. After all subjects had completed participation, the videotaped segments were edited to remove references to date or study visit. Author KJB in consultation with a BPRS expert (John G Csernansky, MD) wrote rules for rating “motor retardation” and other BPRS items potentially influenced by parkinsonism (see Supplementary materials), and trained author MJN in BPRS ratings. Videotaped segments were reviewed in random order by MJN, who was unaware of drug assignment or treatment duration at the time of the visit. BPRS ratings used the anchored BPRS and each item was scored from 1–7 40. Secondary efficacy measures included the PDQ-39, ADL assessments (Schwab-England and UPDRS), BDI, and sleep log. Primary safety measures were UPDRS motor ratings, sleep logs, and MMSE in addition to clinical review of systems.
Statistical analysis
Prior to unblinding of drug codes, the decision was made to analyze data from weeks 0–2 and weeks 2–4 separately. This a priori decision was made since adjustment of dopaminomimetics was allowed at the interim (week 2) visit. Change from 0 to 2 weeks was chosen to be the primary test of efficacy. An intention-to-treat (ITT) analysis was performed on all enrolled subjects. However, since some subjects dropped out without completing outcome measures at a follow-up visit, the ITT analysis was limited to between-group comparisons of dropout rate, serious adverse events, and reported worsening of parkinsonism or other side effects judged to be at least mild in severity. Adverse events, side effects, and study withdrawal were compared between groups using the chi-squared test.
For those subjects with data at both time points of an epoch, primary and secondary efficacy measures were tested separately for the two epochs using repeated-measures ANOVA to compare the groups. The decision was made a priori to include any subject in these analyses if that patient had taken at least one week’s worth of drug during an epoch and returned for a follow-up visit. A secondary post hoc analysis of the data from trial completers was also performed across all three visits using repeated-measures ANOVA. Statistical computations used STATISTICA 7.1 (StatSoft, Inc., Tulsa, OK) or Excel (Microsoft, Redmond, WA).
Results
Baseline characteristics
A total of 24 patients were enrolled (see Figure 1). Though the original study design sought enrollment of 30 patients, the study was terminated early, secondary to the growing body of literature questioning the safety of olanzapine in the treatment of DIP as well as the increasing difficulty in enrolling antipsychotic-naive patients.
Figure 1. CONSORT flowchart.
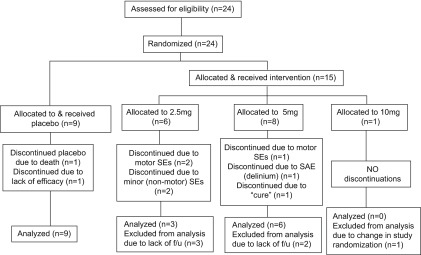
Only one subject was treated with 10mg (one other was randomized to the 10mg group, but was treated only for one week, so received only 5mg doses). His hallucinations were rated “very much improved” at the study end; he required no adjustment in dopaminomimetic dose mid-study and no side effects were observed. This 10mg subject was not included in statistical analyses. In the remaining 23 subjects, no significant imbalances were present at baseline between placebo and treatment groups on any demographic characteristic or any psychiatric or neurologic measure ( Table 2).
Table 2. Patient characteristics at baseline.
| Olanzapine | ||||
|---|---|---|---|---|
| Measure | Placebo (n=9) | 2.5mg (n=6) | 5mg (n=8) | p value |
| Age | 71.3 (6.5) | 70.7 (8.1) | 72.4 (4.8) | 0.882 |
| MMSE | 26 (2.6) | 27 (3.6) | 27 (2.7) | 0.976 |
| BPRS-T | 34.8 (5.9) | 34.3 (5.4) | 33.4 (3) | 0.874 |
| BPRS-P | 7.9 (2) | 9 (3) | 7.8 (2.1) | 0.633 |
| UPDRS, motor
score |
30 (11) | 27.5 (13.1) | 31 (11.6) | 0.855 |
| PDQ-39 | 53 (25.7) | 59 (15.9) | 59 (27.3) | 0.867 |
| BDI | 10.1 (6) | 9.8 (6) | 12.6 (9.2) | 0.738 |
| HAM-D | 8.7 (6.1) | 5.3 (1.6) | 11.6 (7.6) | 0.177 |
| CGI | 4.1 (0.9) | 3.2 (1) | 3.9 (0.8) | 0.161 |
| INS | 4.2 (4) | 4 (2.1) | 2.6 (2.6) | 0.566 |
| HYPINS | 1.5 (1) | 2.3 (1.9) | 2.6 (2.1) | 0.446 |
| SEADL | 76 (15) | 72 (24) | 75 (17) | 0.918 |
Values are given as mean (SD). MMSE, Folstein mini mental test examination; BPRS-T, Brief Psychiatric Rating Scale total score; BPRS-P, psychosis subscale; UPDRS, Unified Parkinson’s Disease Rating Scale; PDQ-39, Parkinson’s disease quality of life questionnaire; BDI, Beck depression inventory; HAM-D, Hamilton depression rating scale; CGI, Clinical global impression; INS, Insomnia score; HYP, Hypersomnia score; SEADL, Schwab-England ADL assessment.
Intention-to-treat analyses
The intention-to-treat analyses did not show significant differences between groups except for incidence of mild side effects (p<0.04) ( Table 3). While spontaneous report of motor side effects was not statistically significant between groups, a disproportionate number of olanzapine vs. placebo group subjects who withdrew did so secondary to reported motor side effects (0% of placebo withdrawers vs. 21% of olanzapine withdrawers). Nine subjects did not complete the study: two from the placebo group, four from the 2.5mg olanzapine group, and three from the 5mg olanzapine group. In the placebo group, one patient died of myocardial infarction and another withdrew from the study secondary to lack of efficacy. In the 5mg olanzapine group, two reported serious adverse events and a third discontinued her medication following the first dose, declaring herself “cured”. Of the 5mg subjects who withdrew for serious adverse events, one was hospitalized with delirium three weeks into the study; the other withdrew after day six due to hospitalization with hip fracture and pneumonia, and reported worsening PD symptoms prior to dropout. Of the four subjects who dropped out of the 2.5mg olanzapine group, two withdrew due to worsening parkinsonian symptoms, one secondary to unspecified side effects, and one secondary to “feeling confused”. Only two subjects in the 2.5mg group completed the study, both requiring increases of their levodopa dose at their interim visit. One each in the placebo and 5mg olanzapine arms also required levodopa adjustment at their two-week assessment. Retention and attrition of study subjects is summarized in Table 3 and Figure 1.
Table 3. Subject retention and side effects by group.
| Olanzapine | |||||
|---|---|---|---|---|---|
| Placebo | 2.5mg | 5mg | All | p value | |
| # enrolled | 9 | 6 | 8 | 23 | |
| # withdrew | 2 (22%) | 4 (66%) | 3 (38%) | 9 (39%) | 0.2232 |
| # withdrew for motor SEs | 0 (0%) | 2 (33%) | 1 (12%) | 3 (13%) | 0.1712 |
| # w/motor SE complaint | 1 (11%) | 2 (33%) | 1 (12%) | 4 (17%) | 0.4863 |
| # w/any mild SEs | 2 (22%) | 5 (83%) | 2 (25%) | 9 (39%) | *0.0356 |
| # w/serious adverse events | 1 (11%) | 0 (0%) | 2 (25%) | 3 (13%) | 0.3795 |
| # included in 1st epoch | 9 (100%) | 3 (50%) | 5 (63%) | 17 (74%) | 0.0640 |
| # included in 2nd epoch | 7 (78%) | 2 (33%) | 5 (63%) | 14 (61%) | 0.2232 |
| # w/dopaminomimetic ↑ | 1 (11%) | 2 (33%) | 1 (13%) | 4 (17%) | 0.4863 |
Side effects (SEs) were any complaint of drug spontaneously reported by the patient, independent of whether SE intensity was severe enough to prompt withdrawal from the study. Serious adverse events always prompted withdrawal. SE, side effects; ↑, increase; 1st epoch, week 0–2 analysis; 2nd epoch, week 2–4 analysis, *, p<0.05.
To assess adequacy of blinding, both the primary investigator and study subjects were asked on study completion (or drop-out) to guess the identity of administered medication (i.e., olanzapine vs. placebo). Both investigator and patient were much more likely than chance would predict to correctly guess the identity of administered medication (for investigator, χ 2=12.29, p=0.0021; for study subjects, χ 2=6.94, p=0.0312). However, the videotape rater had no information about side effects.
CONSORT checklist
Subject characteristics at study entry: S#, subject ID for this study; DRUG, olanzapine dose (mg) this patient took; ADJUST, whether the dose of antiparkinsonian medication was adjusted at the week 2 visit. For remaining columns, the trailing zero in the column heading refers to the score at the baseline (week 0) visit; see Methods section for references. BPRST, BPRS total score; BPRSP, BPRS psychosis items subscore; PDQ, PDQ39 score; BDI, Beck Depression Inventory; HamD, Hamilton Depression Rating Scale; CGIMDo/a, Clinical Global Impression score for overall clinical state as rated by investigator; INS, insomnia score from sleep rating scale; HYPIN, hypersomnia score from sleep rating scale; SEADLS, Schwab-England score; BlindKB, investigator guess at week 4 as to drug assignment; BlindPt., patient guess at week 4 as to drug assignment.
Outcomes other than blinded BPRS ratings: S#, subject ID for this study; DRUG, olanzapine dose (mg) this patient took; ADJUST, whether the dose of antiparkinsonian medication was adjusted at the week 2 visit; change, details of that change; use0-2, analyze this subject’s data in the ANOVA for weeks 0-2; use2-4, analyze this subject’s data in the ANOVA for weeks 2-4; WD, ended study participation early; WDSE, withdrew from study because of side effects; WDnowork, withdrew from study because of lack of benefit; WDcure, withdrew from study because subject pronounced self “cured” after one dose; SAEs, serious adverse events; mild SEs, mild side effects; other, other comments on efficacy or side effects; handed, right- or left-handed. For remaining columns, the trailing numeral in the column heading refers to the score at the visit from week 0, 2, or 4. See Methods section for additional information. VH, visual hallucinations present; AH, auditory hallucinations present; Del, delusions present; BPRST, BPRS total score; BPRSP, BPRS psychosis items subscore; 1UPDRS, UPDRS subscale 1; 2UPDRS, UPDRS subscale 2 (etc.); PDQ, PDQ39 score; BDI, Beck Depression Inventory; HamD, Hamilton Depression Rating Scale; CGI, Clinical Global Impression scale; CGIMD, CGI rated by investigator; CGI…overall, CGI score for overall clinical state; CGIPT, CGI rated by patient; CGI…hall, CGI for hallucinations; CGI…improve, CGI improvement from study initiation; INS, insomnia score from sleep rating scale; BlindJH, study RN guess at week 4 as to drug assignment; BlindKB, investigator guess at week 4 as to drug assignment; BlindKBdrug, same, collapsed to simply olanzapine vs placebo (no dose category); BlindPt., patient guess at week 4 as to drug assignment; BlindBR, guess of study neurologist at week 4 as to drug assignment; 0/5/10, whether this subject was enrolled under the initial study drug assignment (placebo vs 5 or 10mg); HYPIN, hypersomnia score from sleep rating scale; SEADLS, Schwab-England score.
Blinded BPRS ratings from videotape: video ID#, code by which blinded videotape reviewer scored each video segment; BPRS-T, BPRS total score; BPRS-P, BPRS psychosis items subscore.
Primary planned analyses
Analysis of the psychosis subscale of BPRS scores (the more sensitive of our primary efficacy measures) did not reveal a statistically significant difference between groups (drug doses) in severity of psychosis in either the week 0–2 epoch (p=0.433) or the week 2–4 epoch (p=0.393). Again, post hoc analysis in study completers revealed no statistical significance in psychosis reduction between olanzapine (combined groups) and placebo (p=0.536), as shown in Figure 2.
Figure 2. Brief Psychiatric Rating Scale (BPRS) scores across four week study revealed no significant difference between placebo and olanzapine groups among study completers.

Current effect: F(2, 24)=0.64064, p=0.53573. Effective hypothesis decomposition. Vertical bars denote 0.95 confidence intervals. Olanzapine-blue; placebo-red.
Data from the first and second epochs revealed no statistically significant difference in parkinsonian signs across treatment groups, as measured by the UPDRS III (week 0–2 epoch, placebo vs. 2.5mg olanzapine group p=0.172; week 2–4 epoch p=0.677). Post hoc analysis of UPDRS motor scores comparing olanzapine (combined groups) versus placebo across the duration of study found no significant difference in parkinsonism among study completers (p=0.608) ( Figure 3).
Figure 3. Unified Parkinson’s Disease Rating Scale (UPDRS) scores across four week study revealed no significant difference between placebo and olanzapine groups among study completers.

Current effect: F(2, 24)=0.50826, p=0.60787. Effective hypothesis decomposition. Vertical bars denote 0.95 confidence intervals. Olanzapine-blue; placebo-red.
Analyses were repeated in like fashion for all other psychiatric and neurological parameters (CGI impression, CGI improvement, BPRS total, BDI, MMSE, insomnia score, hypersomnolence score, PDQ-39, and Schwab-England ADL assessment), none of which revealed statistical significance between olanzapine groups and placebo.
Discussion
The study failed to reject the null hypothesis. This could be a Type II error, but larger studies of olanzapine also failed to demonstrate antipsychotic efficacy of this drug in the PD population 14, 33. In study completers, we did not observe the motoric exacerbation documented in several studies in the literature 28– 34, but perhaps this is a function of our allowance for dopaminomimetic increase mid-study as well as a selection bias in some analyses for those subjects who best tolerated the medication and therefore completed the study. After all, of the nine subjects who withdrew from the study, a third identified a worsening of their motor disability prior to dropout, all of whom were discovered on unblinding to have been randomized to olanzapine. Therefore the good retrospective accuracy of investigator and patient guesses of study drug identity is not surprising.
The subjects enrolled are relatively typical of PD patients with psychotic symptoms with a few exceptions. Subjects with urgent need for treatment were not enrolled for ethical reasons. Although mild dementia was allowed, this sample had relatively high cognitive functioning, with a mean MMSE score > 26 ( Table 2). Finally, at this center, some of the patients are referred for subspecialty movement disorders consultation, though a large fraction of the patients are not referred and are typical of PD patients treated in the community. With these caveats, the results appear to be generally applicable to patients with PD and psychosis.
One methodological innovation in this study was the use of videotape to record semi-standardized interviews for later analysis by a rater blind not only to drug assignment but also to time (i.e., week 0, week 2, or week 4). The rationale was to minimize rater expectation of improvement over time that might reduce our power to detect significantly greater improvement in the active treatment groups. It also reduced the likelihood of rater unblinding.
This trial supports other evidence suggesting that olanzapine is ineffective for relieving dopaminomimetic-induced psychotic symptoms in Parkinson disease and that it may cause intolerable worsening of motor disability 1, 19. This trial also underscores the importance of rigorous study design for the assessment of drug effectiveness in special populations, as we and others have not replicated the early, positive open-label experience reported for olanzapine in this population. If clozapine’s prominence in the clinical management of DIP in PD is to be usurped, antipsychotic agents will have to meet the burden of proof of double-blind, randomized, placebo-controlled trials.
Acknowledgements
Joel S. Perlmutter, M.D., gave valuable advice on study design and was instrumental in patient referral. Special thanks to Colleen Taylor, Jonathan Koller, Maria Chushak, Tamara Hershey, Ph.D., and John G. Csernansky, M.D., for their assistance with this project.
Funding Statement
This study was funded by Lilly Research Laboratories (Investigator-Initiated Trial F1D-MC-I012).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
v1; ref status: indexed
Supplementary materials
Guidelines for rating selected BPRS items in a treatment study of psychosis in Parkinson disease.
1. Emotional withdrawal = interpersonal relatedness during interview.
2. Tension:
a. Ignore: rest tremors, postural tremors, chorea, athetosis, dystonia.
b. Include: tardive dyskinesia and akathisia.
3. Depressive mood rating does not consider “pure apathy” ( i.e., apathy w/o other depressive signs or symptoms), but apathy can contribute to the total judgment of depressive mood if other signs or symptoms are present.
4. Hallucinatory behavior:
a. 2 = illusions and “shadow in the corner of the eye”.
b. 3 = e.g., colors on the wall.
c. ≥ 4 = definitively abnormal sensory perceptions.
5. Motor retardation: Speed of movement, not amplitude (also, depressive retardation is not substantially helped by external cues; if slowed movement is substantially helped by external cues, then it may be more parsimoniously attributed to PD).
6. Unusual thought content: Ratings ≥ 5 require action on delusion.
7. Blunted affect: Rate according to scale, considering emotional variance, regardless of amplitude; remember that flat/blunted affect is not equivalent to depressed affect.
8. Disorientation: Off by one day of week = 3.
Motor hyperactivity: Limit rating to pressured speech and voluntary movement; festination does not count.
Kevin J Black MD consulted with John G Csernansky MD to write these additional rules for scoring BPRS items potentially influenced by motor signs in Parkinson disease patients.
References
- 1.Friedman JH: Parkinson disease psychosis: Update. Behav Neurol. 2013;27(4):469–77 10.3233/BEN-129016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papapetropoulos S, Mash DC: Psychotic symptoms in Parkinson's disease. From description to etiology. J Neurol. 2005;252(7):753–64 10.1007/s00415-005-0918-5 [DOI] [PubMed] [Google Scholar]
- 3.Chou KL, Messing S, Oakes D, et al. : Drug-induced psychosis in Parkinson disease: phenomenology and correlations among psychosis rating instruments. Clin Neuropharmacol. 2005;28(5):215–9 10.1097/01.wnf.0000180228.77802.32 [DOI] [PubMed] [Google Scholar]
- 4.Goetz CG, Stebbins GT: Mortality and hallucinations in nursing home patients with advanced Parkinson's disease. Neurology. 1995;45(4):669–671 10.1212/WNL.45.4.669 [DOI] [PubMed] [Google Scholar]
- 5.Goetz CG, Leurgans S, Pappert EJ, et al. : Prospective longitudinal assessment of hallucinations in Parkinson's disease. Neurology. 2001;57(11):2078–82 10.1212/WNL.57.11.2078 [DOI] [PubMed] [Google Scholar]
- 6.The Parkinson Study Group. Low-dose clozapine for the treatment of drug-induced psychosis in Parkinson's disease. N Engl J Med. 1999;340(10):757–763 10.1056/NEJM199903113401003 [DOI] [PubMed] [Google Scholar]
- 7.The French Clozapine Parkinson Study Group. Clozapine in drug-induced psychosis in Parkinson's disease. Lancet. 1999;353(9169):2041–2042 10.1016/S0140-6736(99)00860-0 [DOI] [PubMed] [Google Scholar]
- 8.Pollak P, Tison F, Rascol O, et al. : Clozapine in drug induced psychosis in Parkinson's disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry. 2004;75(5):689–695 10.1136/jnnp.2003.029868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weintraub D, Chen P, Ignacio RV, et al. : Patterns and trends in antipsychotic prescribing for Parkinson disease psychosis. Arch Neurol. 2011;68(7):899–904 10.1001/archneurol.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kurlan R, Cummings J, Raman R, et al. : Quetiapine for agitation or psychosis in patients with dementia and parkinsonism. Neurology. 2007;68(17):1356–63 10.1212/01.wnl.0000260060.60870.89 [DOI] [PubMed] [Google Scholar]
- 11.Fernandez HH, Okun MS, Rodriguez RL, et al. : Quetiapine improves visual hallucinations in Parkinson disease but not through normalization of sleep architecture: results from a double-blind clinical-polysomnography study. Int J Neurosci. 2009;119(12):2196–205 10.3109/00207450903222758 [DOI] [PubMed] [Google Scholar]
- 12.Shotbolt P, Samuel M, Fox C, et al. : A randomized controlled trial of quetiapine for psychosis in Parkinson's disease. Neuropsychiatr Dis Treat. 2009;5:327–32 10.2147/NDT.S5335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shotbolt P, Samuel M, David A: Quetiapine in the treatment of psychosis in Parkinson's disease. Ther Adv Neurol Disord. 2010;3(6):339–50 10.1177/1756285610389656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ondo WG, Tintner R, Voung KD, et al. : Double-blind, placebo-controlled, unforced titration parallel trial of quetiapine for dopaminergic-induced hallucinations in Parkinson's disease. Mov Disord. 2005;20(8):958–63 10.1002/mds.20474 [DOI] [PubMed] [Google Scholar]
- 15.Rabey JM, Prokhorov T, Miniovitz A, et al. : Effect of quetiapine in psychotic Parkinson's disease patients: A double-blind labeled study of 3 months' duration. Mov Disord. 2007;22(3):313–318 10.1002/mds.21116 [DOI] [PubMed] [Google Scholar]
- 16.Merims D, Balas M, Peretz C, et al. : Rater-blinded, prospective comparison: quetiapine versus clozapine for Parkinson's disease psychosis. Clin Neuropharmacol. 2006;29(6):331–7 10.1097/01.WNF.0000236769.31279.19 [DOI] [PubMed] [Google Scholar]
- 17.Gomez-Esteban JC, Zarranz JJ, Velasco F, et al. : Use of ziprasidone in parkinsonian patients with psychosis. Clin Neuropharmacol. 2005;28(3):111–4 10.1097/01.wnf.0000164297.91643.ff [DOI] [PubMed] [Google Scholar]
- 18.Pintor L, Valldeoriola F, Baillés E, et al. : Ziprasidone versus clozapine in the treatment of psychotic symptoms in Parkinson disease: A randomized open clinical trial. Clin Neuropharmacol. 2012;35(2):61–66 10.1097/WNF.0b013e31824d5115 [DOI] [PubMed] [Google Scholar]
- 19.Seppi K, Weintraub D, Coelho M, et al. : The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson's disease. Mov Disord. 2011;26(Suppl 3):S42–S80 10.1002/mds.23884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meltzer HY, Mills R, Revell S, et al. : Pimavanserin, a serotonin(2A) receptor inverse agonist, for the treatment of parkinson's disease psychosis. Neuropsychopharmacology. 2010;35(4):881–92 10.1038/npp.2009.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.ACADIA announces presentation of data from its pivotal Phase III Parkinson’s Disease Psychosis study with pimavanserin at the American Academy of Neurology annual meeting. Business Wire,2013. Reference Source [Google Scholar]
- 22.ACADIA announces expedited path to NDA filing for pimavanserin following meeting with FDA. Business Wire,2013. Reference Source [Google Scholar]
- 23.Meltzer HY: An overview of the mechanism of action of clozapine. J Clin Psychiatry. 1994;55(Suppl B):47–52 [PubMed] [Google Scholar]
- 24.Seeman P, Van Tol HH: Dopamine receptor pharmacology. Curr Opin Neurol Neurosurg. 1993;6(4):602–8 [PubMed] [Google Scholar]
- 25.Bymaster FP, Calligaro DO, Falcone JF, et al. : Radioreceptor binding profile of the atypical antipsychotic olanzapine. Neuropsychopharmacology. 1996;14(2):87–96 10.1016/0893-133X(94)00129-N [DOI] [PubMed] [Google Scholar]
- 26.Wolters EC, Jansen EN, Tuynman-Qua HG, et al. : Olanzapine in the treatment of dopaminomimetic psychosis in patients with Parkinson's disease. Neurology. 1996;47(4):1085–1087 10.1212/WNL.47.4.1085 [DOI] [PubMed] [Google Scholar]
- 27.Aarsland D, Larsen JP, Lim NG, et al. : Olanzapine for psychosis in patients with Parkinson's disease with and without dementia.The dopamine motor system. J Neuropsychiatry Clin Neurosci. 1999;11(3):392–394 [DOI] [PubMed] [Google Scholar]
- 28.Friedman J: Olanzapine in the treatment of dopaminomimetic psychosis in patients with Parkinson's disease. Neurology. 1998;50(4):1195–1196 10.1212/WNL.50.4.1196 [DOI] [PubMed] [Google Scholar]
- 29.Graham JM, Sussman JD, Ford KS, et al. : Olanzapine in the treatment of hallucinosis in idiopathic Parkinson's disease: a cautionary note. J Neurol Neurosurg Psychiatry. 1998;65(5):774–777 10.1136/jnnp.65.5.774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molho ES, Factor SA: Worsening of motor features of parkinsonism with olanzapine. Mov Disord. 1999;14(6):1014–1016 [DOI] [PubMed] [Google Scholar]
- 31.Jiménez-Jiménez FJ, Tallón-Barranco A, Ortí-Pareja M, et al. : Olanzapine can worsen parkinsonism. Neurology. 1998;50(4):1183–1184 10.1212/WNL.50.4.1183-a [DOI] [PubMed] [Google Scholar]
- 32.Marsh L, Lyketsos C, Reich SG: Olanzapine for the treatment of psychosis in patients with Parkinson's disease and dementia. Psychosomatics. 2001;42(6):477–481 10.1176/appi.psy.42.6.477 [DOI] [PubMed] [Google Scholar]
- 33.Breier A, Sutton VK, Feldman PD, et al. : Olanzapine in the treatment of dopaminomimetic-induced psychosis in patients with Parkinson's disease. Biol Psychiatry. 2002;52(5):438–45 [DOI] [PubMed] [Google Scholar]
- 34.Ondo WG, Levy JK, Vuong KD, et al. : Olanzapine treatment for dopaminergic-induced hallucinations. Mov Disord. 2002;17(5):1031–1035 10.1002/mds.10217 [DOI] [PubMed] [Google Scholar]
- 35.Schulz KF, Altman DG, Moher D, et al. : CONSORT 2010 Statement: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c332 10.1136/bmj.c332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moher D, Hopewell S, Schulz KF, et al. : CONSORT 2010 Explanation and elaboration: updated guidelines for reporting parallel group randomised trial. BMJ. 2010;340:c869 10.1136/bmj.c869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Folstein MF, Folstein SE, McHugh PR: “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189–198 10.1016/0022-3956(75)90026-6 [DOI] [PubMed] [Google Scholar]
- 38.Guy W, Bonato RR: CGI: clinical global impressions, in ECDEU Assessment Battery Manual, revised ed. NIMH: Chevy Chase, MD1970. Reference Source [Google Scholar]
- 39.Jenkinson C, Peto V, Fitzpatrick R, et al. : Self-reported functioning and well-being in patients with Parkinson's disease: comparison of the short-form health survey (SF-36) and the Parkinson's Disease Questionnaire (PDQ-39). Age Ageing. 1995;24(6):505–9 10.1093/ageing/24.6.505 [DOI] [PubMed] [Google Scholar]
- 40.Woerner MG, Mannuzza S, Kane JM: Anchoring the BPRS: an aid to improved reliability. Psychopharmacol Bull. 1988;24(1):112–7 [PubMed] [Google Scholar]
- 41.Gancher ST: Quantitative measures and rating scales, in Parkinson's Disease: Diagnosis and Clinical Management. S.A. Factor and W.J. Weiner, Editors. Demos Medical Publishing: New York2002. Reference Source [Google Scholar]
- 42.Fahn S, Elton RL: Members of the UPDRS Development Committee Unified Parkinson's Disease Rating Scale, in Recent developments in Parkinson's disease. S. Fahn, et al., Editors Macmillan: New York,1987;153–164 [Google Scholar]
- 43.Hamilton M: A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62 10.1136/jnnp.23.1.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beck AT, Ward CH, Mendelson M, et al. : An inventory for measuing depression. Arch Gen Psychiatry. 1961;4(6):561–571 10.1001/archpsyc.1961.01710120031004 [DOI] [PubMed] [Google Scholar]
- 45.Overall JE: Rating session. Video taped interviews and BPRS ratings. Psychopharmacol Bull. 1975;11(1):15 [PubMed] [Google Scholar]
- 46.Rhoades HM, Overall JE: The semistructured BPRS interview and rating guide. Psychopharmacol Bull. 1988;24(1):101–4 [PubMed] [Google Scholar]