

Abstract
Genetic factors significantly influence addiction-related phenotypes. This is supported by the successful bidirectional selective breeding of two replicate sets of mouse lines for amount of methamphetamine consumed. Some of the same genetic factors that influence methamphetamine consumption have been previously found also to influence sensitivity to the conditioned rewarding and aversive effects of methamphetamine. The goal of the current studies was to determine if some of the same genetic factors influence sensitivity to the conditioned rewarding and aversive effects of cocaine. Cocaine conditioned reward was examined in methamphetamine high drinking and low drinking line mice using a conditioned place preference procedure and cocaine conditioned aversion was measured using a conditioned taste aversion procedure. In addition, a general sensitivity measure, locomotor stimulant response to cocaine, was assessed in these lines; previous data indicated no difference between the selected lines in sensitivity to methamphetamine-induced stimulation. In contrast to robust differences for methamphetamine, the methamphetamine high and low drinking lines did not differ in sensitivity to either the rewarding or aversive effects of cocaine. They also exhibited comparable sensitivity to cocaine-induced locomotor stimulation. These data suggest that the genetic factors that influence sensitivity to the conditioned rewarding and aversive effects of methamphetamine in these lines of mice do not influence sensitivity to these effects of cocaine. Thus, different genetic factors may influence risk for methamphetamine versus cocaine use.
Keywords: methamphetamine, cocaine, mice, conditioned place preference, conditioned taste aversion, locomotor activation
1. Introduction
Methamphetamine (MA) and cocaine (COC) have been classified as having similar pharmacological and behavioral profiles. However, COC and MA exhibit differences in mechanisms of action and pharmacokinetic profiles that could lead to differences in abuse potential. Thus, genetic factors that influence risk for their use may also differ.
COC can be generally characterized as a monoamine transporter blocker that prevents the reuptake of released monoamines, whereas MA can be generally characterized as a monoamine releaser. The end result of treatment with either drug is higher synaptic levels of these neurotransmitters [1]. COC has a shorter half-life than MA [2], which likely influences frequency of use. The subjective and cardiovascular effects also differ in onset and duration, with those after MA being more profound [3]. In addition, some users have reported a better “high” from MA than COC [4], which may have an impact on addiction potential. COC has been found to substitute for MA in drug discrimination paradigms [5], suggesting these two drugs have some similar subjective effects. However, habitual abusers tend to use COC or MA fairly exclusively [6, 7], suggesting individual drug preferences. Although genetic influence on sensitivity and risk for abuse or dependence has been found for both MA [8, 9] and COC [10, 11], and mechanisms associated with their effects have been widely investigated, there is a lack of research addressing whether common genetic factors influence risk and responses to these two drugs. This has implications for prevention and treatment.
Our lab has created selected lines of mice for high and low MA drinking (MADR). Our data indicate that MA low drinking (MALDR) mice are insensitive to rewarding and reinforcing effects of MA, and are highly sensitive to aversive effects of MA, whereas the MA high drinking (MAHDR) mice show an opposite sensitivity profile [12, 13, 14, 15]. Thus, some common genetic factors influence MA consumption and sensitivity to the rewarding and aversive effects of MA. The MADR lines provide a genetic model for testing the hypothesis that common genetic factors influence sensitivity to MA and COC. Selective breeding for MADR may have altered the frequency of genes relevant to unique effects of MA; for example, MA and COC differentially regulate vesicular monoamine transporter-2 (VMAT-2), involved in storage of dopamine in synaptic vesicles [1]. Alternatively, selection could have impacted common mechanisms, perhaps monoamine effects.
In the current studies, the MADR lines were tested for COC responses, using the same procedures previously used to examine MA conditioned responses [12, 13, 14]. Sensitivity to the aversive effects of COC was measured using a conditioned taste aversion procedure (CTA) and sensitivity to the rewarding effects of COC was measured using a conditioned place preference (CPP) procedure. In addition, sensitivity to the stimulant effect of COC was examined for comparison to previous data that showed no difference between the MADR lines in the acute stimulant response to MA [14]. We hypothesized that if a line difference was found, the MAHDR line would be more sensitive to the rewarding and less sensitive to the aversive effects of COC compared to the MALDR mice, but they would not differ in sensitivity to the acute stimulant effects.
2. Materials and methods
2.1. Animals
Male and female mice from the second consecutive replicate of the short-term selectively bred MALDR and MAHDR lines were used. Short-term selected lines are bred using mass selection for only 4 to 5 generations [16]. This avoids excessive inbreeding at genetic loci not relevant to the selection phenotype (i.e., random drift). Consecutive replicates are created to test hypotheses derived from previous sets of the same type of selected line. We have demonstrated excellent replication of results for two sets of MADR lines bred two years apart [12, 13, 14]. The methods used to create the two sets of MADR lines are published [12, 13]. Briefly, these lines were created from the F2 cross of the C57BL/6J (B6) and DBA/2J (D2) inbred mouse strains. Mice from the F2 were chosen for breeding (i.e., selected) based on amount of consumption of a 40 mg MA per liter of tap water solution, when it was offered along with a separate drinking tube containing plain tap water. The highest MA consuming F2 mice served as breeders for the MAHDR lines and the lowest MA consuming F2 mice served as the breeders for the MALDR lines. Selection was terminated after 5 generations and after this time, mice were randomly chosen for breeding to produce additional mice for testing. Mice used in the current studies were offspring of the replicate 2 lines of the fifth (S5) selection generation. Mice were weaned at 20–22 days of age and subsequently group housed with same sex littermates, 2–5 mice per cage, in standard mouse shoebox cages (28.5 × 17.5 × 12 cm) lined with Bed-o’Cobs® bedding (The Andersons, Inc., Maumee, OH, USA). Mice were given ad libitum access to water and food (LabDiet® 5001, PMI Nutrition International LLC, St. Louis, MO, USA) that was purchased from Animal specialties Inc. (Hubbard, OR, USA). All mice were experiment- and drug-naïve prior to testing. All behavioral testing was conducted during the light phase of the 12:12 h light:dark cycle (lights on at 0600 h), between 0800 and 1600 h. Additional details regarding the mice that were used for each study are described with the results.
2.2. Drugs
Cocaine HCl (Sigma Aldrich; St. Louis, MO, USA) was prepared on the day of testing in 0.9% saline (Baxter Healthcare Corporation, Deerfield, IL) and administered by i.p. injection.
2.3. Conditioned place preference (CPP)
Sensitivity to the rewarding effects of COC was measured using a standard unbiased CPP procedure, as previously described [12, 13]. This CPP procedure was unbiased, since the assignment of the floor type paired with COC for each individual animal was not based on that individual’s initial floor preference. The current study was designed to match the CPP methods used to assess MA CPP in the MADR lines. Previous studies have found no initial bias for these conditioning cues (grid or hole floor) in either the D2 or B6 strains [17], the progenitors of the MADR lines, or in large panels of inbred BXD strains derived from the B6 and D2 strains [18]. The 30 × 15 ×15 cm CPP chambers (San Diego Instruments, San Diego, CA, USA) consisted of clear plastic walls and exchangeable floor panels. Three different floor types were used in this study: a solid black plastic acrylic floor; a “grid” floor constructed of 2.3 mm stainless steel rods mounted 6.4 mm apart; and a “hole” floor constructed of a stainless steel panel with 6.4 mm round holes aligned with 9.5 mm staggered centers. A removable black plastic divider was used to confine animals to the right or left side of the chamber on conditioning sessions. Conditioning boxes were housed in illuminated and ventilated sound attenuating chambers during testing. During test sessions, activity and location of the mouse was measured by photocell interruptions recorded by a fully automated system.
The COC-induced CPP procedure matched that used for our published work for MA [12, 13], with the exception that conditioning trial durations were 30 min, instead of 15 min, long. The longer trial duration was used because DBA/2J mice, one of the progenitor strains for the MADR lines, did not develop a COC-induced CPP using 15 min conditioning trials [19]. On day 1, to habituate the mice to handling and the CPP apparatus, all mice were given one, 5-min habituation session, during which the mouse was injected with saline and placed in the chamber with access to both sides (black plastic flooring on both sides). This solid black flooring was only used during the habituation session to allow the mice to acclimate to the CPP procedure and apparatus without exposing them to the floor types (grid and hole) used during subsequent conditioning sessions. On the next 12 alternating days, mice were conditioned with 10 mg/kg COC and saline, each paired with a distinct floor type (grid or hole); thus, there were 6 COC conditioning and 6 saline conditioning sessions. For each conditioning session, the mouse was injected with COC or saline and immediately placed on the appropriate floor type on one side of the apparatus for 30 min (floor type associated with COC, side of apparatus and whether COC or saline was given prior to the first conditioning session were counterbalanced). A 10 mg/kg dose of COC was chosen based on the results of previously published data using similar conditions [19]. Multiple doses of COC were not tested, as previous work has shown that COC is not effective at inducing dose-effect curves in the CPP procedure [20]. Twenty-four hours after the last conditioning session, mice were tested for floor preference in a drug-free state. All mice were injected with saline to match handling conditions during conditioning, and then placed into the CPP apparatus for 30 min with both floor types (grid and hole) present; the COC-conditioned floor was placed on the side of the apparatus matched to its location during conditioning, to control for possible non-floor cues that could have been conditioned to COC. On the following experimental session (day 22), drug state-dependent effects were assessed in a second preference test under the same conditions, except that mice were injected with 10 mg/kg COC immediately before being placed into the CPP apparatus. Consistent with previous methods for MA [12, 13], the dependent variable used to determine if COC induced a CPP was second/min on the grid floor, with the expectation that mice for which COC was paired with the grid floor (G+ group) would spend more time on that floor compared to mice for which COC had been paired with the hole floor (G− group) if they were sensitive to the rewarding effects of COC.
2.4. Conditioned taste aversion (CTA)
The COC-induced CTA procedure matched that used for our published work with MA [12, 14]. Mice were individually housed and familiarized to novel sipper tubes on 2 consecutive days. For the next four days, mice were acclimated to restricted water access (2 hr access/day) to motivate drinking during a specific period of time as necessary for the next phase of the study. Mice were weighed daily during water restriction, to monitor for excessive weight loss, which was not observed in any of the mice. On the next day, instead of water, mice were presented with a solution of 0.2M NaCl during a 1-h period to familiarize them with the taste of this novel solution. Mice were then given five conditioning sessions that occurred every other day, during which they were presented with the NaCl solution for 1 hour and then injected with saline or COC (15 or 30 mg/kg), immediately following the removal of the drinking tube. These doses of COC were chosen because they have been shown to induce moderate and robust CTA in mice [21]. To prevent dehydration, mice were also given a 30-min session with access to water, starting 4 hours after the initiation of the NaCl drinking session. On alternate days, mice were given 2-h access to water with no injections. Consumption of the NaCl solution across days was used as the measure of COC-induced CTA, with the expectation that consumption would decrease across days if the mice were sensitive to COC-induced CTA.
2.5. Acute locomotor activation
Sensitivity to the locomotor stimulant effects of COC was assessed using sixteen automated locomotor activity monitors made by AccuScan Instruments, Inc. (Columbus, OH, USA). Each monitor was equipped with eight photocell beams located 2 cm above the 40 × 40 × 30 cm clear acrylic chamber floor, with corresponding photo detectors located on opposite sides. A computer was used to record beam breaks, which were converted into horizontal distance traveled (in centimeters) using VERSADAT version 1.8 software (AccuScan Instruments, Inc.). Each monitor was enclosed in an Environmental Control Chamber (ECC) constructed from PVC/lexan (AccuScan Instruments, Inc.), was equipped with a fan that provided ventilation and background noise, and illuminated by a 3.3 Watt incandescent light bulb.
Mice were tested on three consecutive days as previously described [22, 23, 24]. On each day, mice were moved into the testing room 45 minutes prior to the start of the experiment to acclimate to the test room environment. Mice were then weighed, placed briefly in holding cages while injection syringes were filled, injected, and immediately placed into individual activity monitors, where behavior was recorded for 30 min. On days 1 and 2, mice received saline injections; on day 3, mice received an injection of saline or cocaine (5, 10, 20 or 30 mg/kg). Day 1 testing familiarized the animals with all handling and testing procedures; day 2 testing provided a measure of baseline activity collected under now familiar conditions. To eliminate the impact of possible differences in baseline activity level on the drug response measure, saline day 2 baseline activity data were subtracted from drug day 3 activity data for each individual animal. Use of this difference score as the dependent variable is consistent with our previous work and provides a measure of drug response above (or below) baseline [22, 23, 24, 25, 26].
2.6. Data analysis
All statistical analyses were performed using Statistica 9 software (StatSoft, Tulsa, OK, USA). Data were analyzed by factorial ANOVA, with repeated measures (RM-ANOVA) when appropriate. Significant interactions involving multiple factors were followed by ANOVA including fewer factors to determine the sources of interaction. Two-way interactions were interpreted using simple main effects analysis and Newman-Keuls post-hoc mean comparisons when appropriate. For all analyses, sex was first included as a factor and then follow-up analyses were performed with data from the sexes combined when sex did not interact with other factors. Effects were considered significant at an alpha level of 0.05 or less.
3. Results
3.1. Cocaine-induced CPP
COC-induced CPP was measured in 60–90 day old MADR mice (n = 10–14 per sex, line and conditioning floor type). For the drug-free preference test on day 19 (Figure 1A), there was a statistically significant difference between the G+ and G− groups for sec/min on the grid floor (F(1,83) = 97.8, p < 0.001), indicating that mice developed a CPP to 10 mg/kg COC. However, there were no significant main or interaction effects of sex or line. Thus, both lines developed a COC-induced place preference and there was no difference between the lines in magnitude of COC-induced CPP.
Figure 1. MAHDR and MALDR mice did not differ in the expression of a COC-induced CPP.
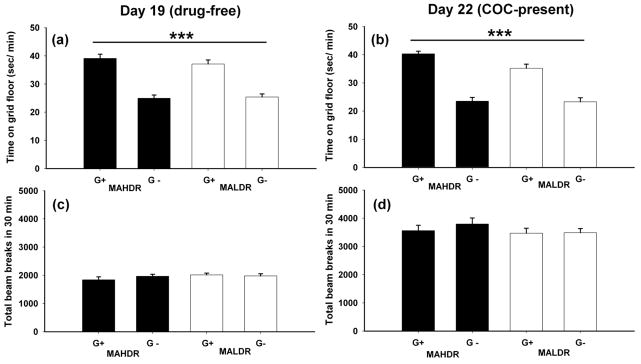
Shown are means ± SEM sec/min spent on the grid floor during a 30-min preference test after saline treatment (A) and after COC treatment (B), as well as means ± SEM level of locomotor activity during each of the preference tests (C, D). ***: p<0.001 for the main effect of floor type (G+ or G−), indicating that COC induced a significant CPP. Data are combined for the two sexes, because sex did not significantly influence the results.
During the COC present preference test on day 22 (Figure 1B), there was a statistically significant difference between the G+ and G− groups for sec/min on the grid floor (F(1,83) = 120.1, p < 0.001), indicating that mice also expressed a CPP when they were experiencing COC effects. There were no significant main or interaction effects of sex or line. Thus, the MADR lines did not differ in the expression of a COC-induced CPP when they were in the COC treated state.
Locomotor data were collected during both conditioning trials and the preference test sessions. As expected, activity was higher during the 10 mg/kg COC, compared to the saline conditioning sessions; however, the lines did not differ in locomotor response to COC or in activity level after saline on any day (Figure 2). In addition, locomotor response did not significantly change over the 6 COC trials, indicating that these mice did not develop locomotor sensitization to this dose of COC. During the drug-free and COC-present preference test sessions (day 19 and day 22), there were no significant differences in locomotor activity between the lines (Figure 1C and 1D). A RM-ANOVA was used to compare activity on days 19 and 22. There was a main effect of day (F(1,87) = 433.6, p < 0.001), indicating that mice were significantly more active on day 22 after COC treatment than on day 19 after saline; there were no line differences in activity level on these days. Activity counts were lower during the CPP conditioning trials which had the same duration as the preference test sessions. This could be due to the fact that the chamber size was smaller during the conditioning sessions as mice were confined to one side of the chamber in the one compartment CPP procedure that was used.
Figure 2. Level of locomotor activity in MADR mice during the saline and COC conditioning trials was comparable.

Locomotor activity was measured as photocell beam breaks in the CPP apparatus. Shown are means ± SEM total beam breaks for each of the lines and conditions (saline or COC). Data are combined for the two conditioning floor types and for the two sexes, because sex did not significantly influence the results.
3.2. Cocaine-induced CTA
COC-induced CTA was measured in 60–83 day old MADR mice (n = 10–11 per sex, line and COC dose). NaCl consumption across the 5 sessions was analyzed using a RM-ANOVA with sex, line and COC dose as factors. There was a significant day x COC dose interaction (F(8,436) = 32.4, p < 0.001), but no significant interactions involving line. COC dose-dependently reduced NaCl consumption across days (Figure 3) indicating a significant COC-induced CTA. There was also a significant day x sex x COC dose interaction (F(8,436) = 2.3, p < 0.05). To follow up this interaction, the effects within each sex were analyzed. There was a significant day x COC dose interaction in both males (F(8,220) = 8.4, p < 0.001) and females ((8,216) = 4.2, p < 0.001). Further analyses examining the effects of sex within each dose revealed that the source of the sex effect was at the higher dose of COC (30 mg/kg), with greater CTA in females compared to males; otherwise, the patterns were similar. In summary, COC dose-dependent CTA was found, with no significant differences between the MAHDR and MALDR lines.
Figure 3. MAHDR (A) and MALDR (B) mice did not differ in the development of a COC-induced CTA.

Shown are means ± SEM ml of 0.2 M NaCl consumed prior to COC conditioning (day 10) and on each of the COC conditioning days (days 12, 14, 16, and 18). Data are combined for the two sexes because sex did not significantly influence the results.
3.3. Cocaine-induced locomotor activation
This study was performed to complement locomotor activity data collected during CPP testing. It extended the evaluation to multiple COC doses and corrected for individual differences in baseline activity level. The acute locomotor response to COC was measured in 61–95 day old MADR mice (n=6–11 per sex, line and COC dose). There were no statistically significant effects of line, dose or sex for day 2 (habituated baseline) activity data. Similar results were also found for day 1, with the exception of a significant effect of sex (p=0.04); females were more active than males. To eliminate individual differences in baseline locomotor activity in the evaluation of COC effects, day 3 test scores were corrected by subtracting day 2 habituated baseline scores. For day 3-day 2 activity scores during the total 30-min test (Fig. 4), there was a significant main effect of dose (F(4,149) = 64.0, p < 0.001), but no significant effects of line or sex. COC dose-dependently stimulated activity, but the MAHDR and MALDR lines did not differ in sensitivity, similar to results obtained during the conditioning trials of the CPP study. Results were similar when earlier time periods were examined (data not shown). These results are similar to our published findings for MA in the MADR lines [13].
Figure 4. MAHDR and MALDR mice did not differ in sensitivity to COC-induced locomotor activation.

Shown are means ± SEM difference scores, expressed as cm traveled. Baseline data collected on day 2 after saline treatment were subtracted from saline or COC data collected on day 3 for each individual animal (Day 3 – Day 2). Activity sessions were 30-min in duration, beginning immediately after treatments. Data are combined for the two sexes, because sex did not significantly influence the results.
4. Discussion
MAHDR and MALDR mice had similar sensitivities to the rewarding and aversive effects of COC. This is in stark contrast to our previously published data that showed robust differences in sensitivity of the MADR lines to MA-induced CTA and CPP [12, 13, 14]. These results indicate that genetic differences that alter MA intake and sensitivity to the rewarding and aversive effects of MA do not similarly influence sensitivity to the rewarding and aversive effects of COC. A single dose of COC was studied because dose-dependent effects of drugs in the CPP procedure have been difficult to demonstrate, within the range of doses that produce CPP [20]. It remains possible that there is a dose of COC for which the MADR lines would show a difference. However, at doses from 0.5 to 4 mg/kg of MA, under both drug-free and MA-present conditions, the MALDR line exhibited no MA-induced CPP, whereas here they exhibited a preference for the COC-paired place. These results are markedly different from those for MA, as they indicate that the MALDR line is sensitive to the rewarding effects of COC, whereas they are not sensitive to the rewarding effects of MA. Further, the MAHDR line was completely insensitive to MA-induced CTA at doses up to 4 times higher than a dose that induced CTA in MALDR mice, but the lines exhibited a similar level of sensitivity to COC-induced CTA.
An alternative interpretation of drug effects in the CTA procedure is that avoidance of the conditioned stimulus reflects reward value of the paired unconditioned stimulus (i.e., drug) and anticipation of the rewarding, rather than the aversive, properties of the drug [27]. There are several reasons why we do not think this could explain the results seen in the MADR lines. MAHDR line mice develop MA-induced CPP, but are completely insensitive to the conditioned effects of MA in the CTA procedure [12, 14]. MALDR line mice do not develop MA-induced CPP and are highly sensitive to the conditioned aversive effects of MA in a conditioned place aversion (CPA) procedure [13, 14] and CTA procedure [12, 14]. In addition, differences in sensitivity to the conditioned effects of MA, classified as rewarding and aversive, correspond with differences in oral and intracerebroventricular operant self-administration of MA that indicate strong reinforcement from MA in the MAHDR line and little reinforcement in the MALDR line [15]. For these reasons, this alternative interpretation does not likely explain our CTA data in the MADR lines for MA. Furthermore, the data are clear in showing the absence of a line difference in the CTA procedure using COC, leading to the conclusion that genetic factors that influence the line difference in sensitivity to MA-induced CTA do not influence sensitivity to COC-induced CTA.
The MADR lines displayed similar acute locomotor responses to COC during COC conditioning and the CPP test after COC treatment, and to multiple doses of COC in a larger activity arena. These results for acute treatment are similar to those found for MA in the MADR lines [13]. In the current study, the 10 mg/kg dose of COC did not induce locomotor sensitization. Under similar conditions, a low dose of 0.5 mg/kg MA did not produce acute stimulation or result in sensitization in either line, whereas 2 mg/kg MA induced similar levels of sensitization, and 4 mg/kg MA induced sensitization in only the MAHDR line [13]. It is possible that line differences exist for the development of sensitization to other doses of COC. Overall, these data indicate that sensitivities to acute locomotor stimulant effects of these drugs are not genetically related to preference for MA or to sensitivity to the rewarding or aversive effects of MA.
Differences in the expression of the serotonin (SERT) and norepinephrine (NET), but not dopamine (DAT), transporter genes have been found in nucleus accumbens tissue from non-MA treated MADR line mice [12]. These results could explain the different results for MA and COC, with regard to line differences in sensitivity to rewarding effects. MA is more potent at NET, compared to SERT or DAT, whereas COC has similar effects at all three monoamine transporters [28]. The actions of MA on NE release have been suggested to play a larger role in causing the subjective effects of MA, compared to its DA-releasing effects [29], whereas the discriminative stimulus effects of COC were found to better generalize to the effects of a DA uptake inhibitor than to MA [30]. Further, the DAT has been shown to be important for the formation of a COC-induced CPP [31], but does not appear to play a significant role in the aversive effects of COC [21, 32]. Studies using transporter blockers and single gene mutant mice indicated that NET was most substantially involved in COC-induced CTA, with some contribution of SERT and minimal if any contribution of DAT [21, 33]. Similar studies examining the role of the monamine transporters in the aversive effects of MA have not been reported. These differences in interactions with the specific types of monoamine transporters may play a role in genetically-determined differences in sensitivity to the conditioned rewarding and aversive effects of MA and COC.
COC and MA also differ in their actions at VMAT-2. MA interferes with VMAT-2 activity [34, 35]. In contrast, studies have found COC either had no effect at VMAT-2 [36], or increased VMAT-2 activity [37, 38]. The actions of COC and MA on VMAT-2 are likely the result of drug-specific actions causing differential redistribution of vesicles containing VMAT-2 [39]. It is possible that MA effects at VMAT-2 contribute to the selected line difference in sensitivity to the effects of MA, compared to COC. Supporting this hypothesis, VMAT-2 heterozygous knockout mice were found to be less sensitive than wild-type mice to the conditioned rewarding affects of amphetamine, but not COC [40]. VMAT-2 heterozygous knockouts were also found to be more sensitive to neurotoxic effects of MA [41], which could contribute to heightened aversion. After MA administration, there was reduced expression in nucleus accumbens tissue of the gene coding for VMAT-2 in MALDR, but not MAHDR mice. This difference could contribute to the increased sensitivity of MALDR mice to the aversive effects of MA. Investigation of VMAT-2 function in response to MA in MADR mice is planned for a future study.
Selection for differential MA consumption could also have resulted in genetic differences between the MADR lines that influence other, non-monoaminergic effects of MA that are not shared in common with COC. One difference between the MADR lines that has been identified is in one or more of the mechanisms relevant to μ–opioid responses. We previously identified a negative genetic correlation between MA consumption and sensitivity to the stimulant effects of drugs that act as μ–opioid receptor agonists, but not analgesic effects of these drugs [42]. This work was pursued, in part, as a candidate gene approach based on quantitative trait locus (QTL) mapping that identified a QTL on proximal mouse chromosome 10 in both sets of the replicate MADR lines that contains the μ–opioid receptor gene (Oprm1). In addition, Oprm1 expression was greater in prefrontal cortex tissue from MALDR, compared to MAHDR mice (Phillips et al., unpublished). Another potentially relevant mechanism is suggested by the results of gene network analysis that identified a number of genes in the neuroimmune pathway for which expression was different at baseline or differentially affected in the MADR lines by MA treatment [12]. For example, there was lower baseline expression of NFκB, a transcription factor that is part of the immune system signaling pathway, in the MAHDR line. MA has been previously shown to induce NFκB activity [43]. The expression of Nfkb2 in nucleus accumbens tissue was not different in untreated MAHDR versus MALDR mice, but MA treatment resulted in downregulation of Nfkb2 in MALDR and upregulation in MAHDR mice [12]. Recent data suggest that MA may have effects that result in persistent immune system dysregulation that crosses species lines (e.g., [44]). Work being pursued in the MA Abuse Research Center (MARC) at Oregon Health & Science University and the VA in Portland, OR is examining a panel of immune factors, such as cytokines and chemokines, in the MADR lines and in humans with MA dependence vs controls to help to clarify the role of the neuroimmune system in MA dependence.
Advantages of short-term selected lines, such as the MADR lines, are that they can be produced relatively quickly, compared to long-term selected lines, and can be replicated in series. Replication in series allows for significant findings from one set of lines to be reexamined or extended in subsequent independently selected lines. We have shown excellent replicability for key addiction-related MA traits in two sets of MADR selected lines, even though data were collected two years apart [12, 13, 14]. One disadvantage of short-term selected lines is that they are not perpetual and thus, follow-up, particularly to test hypotheses about differences between the lines, requires one to wait until the next set of lines is produced. This is because these lines are produced by mass selection and significant inbreeding of trait irrelevant alleles will amass across generations, but remains at an acceptable level up to 4–5 generations. Production of the current replicate of MADR lines for the purpose of examining genetically correlated responses and mechanisms has been terminated. However, a third set of lines is currently under construction within the MARC and will be available to test some of the hypotheses arising from the current work.
The MADR lines are the only genetic model that has been created specifically for an MA intake trait. Their strengths as a model to study genetic influences relevant to MA addiction have been discussed in our previous papers [12, 15], and include their differences in (1) voluntary MA consumption [12, 13], (2) operant intracranial self administrations of MA [15], (3) operant oral self administration of MA [15], (4) sensitivity to conditioned rewarding effects of MA [12, 13], and (5) sensitivity to conditioned aversive effects of MA [12, 13, 14]. In addition, published [12] and ongoing (Belknap et al., submitted) gene expression and mapping studies are identifying genes and gene networks that influence MA consumption. This information will have relevance to the traits mentioned above because of common genetic influences (pleiotropic effects) across the traits. These data have the potential to inform translational research into genetic factors that influence human MA addiction. However, limitations of our animal model must also be considered, including that we have not yet assessed binge or escalating patterns of drug intake in the MADR lines that may better model addiction phenotypes seen in human MA users. One future direction is to test the MADR lines using methods that have been shown lead to escalating or binge consumption of other drugs, such as drinking in the dark [45], multiple bottle access [46], or intermittent access [47] procedures. Another limitation of the MADR lines is that they are derived from an F2 cross of two inbred strains and thus, the genetic information about the MA drinking and genetically correlated traits is limited to the existing genetic variation present in the two progenitor strains. However, this genetic background was chosen (1) because the two strains are genetically diverse [48], (2) several previous selection studies [16, 26, 49, 50, 51] and a large panel of recombinant inbred strains [e.g. 11, 52, 53, 54] have been derived from this F2 cross and used for genetic investigation of drug-related traits, allowing for comparisons across drugs and traits for the same genetic variants, and (3) having only two potential allele forms at each locus simplifies gene mapping and expression analyses designed to lead to trait relevant gene identification. Research focused on identification of the specific genetic differences that influence MA intake in the MAHDR and MALDR lines is underway. Identification will allow for more targeted research on specific mechanisms and allow for increased translation to human MA addiction.
In summary, the current studies show that MAHDR and MALDR mice have similar sensitivities to the conditioned rewarding and aversive effects of COC, unlike their markedly different sensitivities to the same effects of MA. These data indicate that genes that influence both MA consumption and sensitivity to the conditioned rewarding and aversive effects of MA do not influence sensitivity to the conditioned rewarding and aversive effects of COC. Thus, the underlying mechanisms that contribute to magnitude of conditioned drug effects appear to be drug-specific in the case of MA and COC. These findings generate the hypothesis that different genetic factors contribute to individual differences in sensitivity to some rewarding and aversive effects of MA vs COC, which may be relevant to risk for MA vs COC use and dependence.
Highlights.
Mice bred for high and low methamphetamine (MA) intake were used to study cocaine responses.
These lines differ in sensitivity to MA conditioned reward and aversion.
These lines did not differ in sensitivity to cocaine conditioned reward and aversion.
A similar locomotor response previously found for MA was also found for cocaine.
Unique genetic factors influence sensitivity to hedonic effects of MA and cocaine.
Acknowledgments
We thank Sue Burkhart-Kasch and Harue Baba for their assistance with selective breeding of the MADR-2 lines. This work was supported by a grant from the Department of Veterans Affairs, NIDA Center grant P50 DA018165 and NIAAA F31AA020732.
Footnotes
All authors report no conflicts of interest or biomedical financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riddle EL, Fleckenstein AE, Hanson GR. Role of monoamine transporters in mediating psychostimulant effects. AAPS J. 2005;7:E847–51. doi: 10.1208/aapsj070481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fowler JS, Volkow ND, Logan J, Alexoff D, Telang F, Wang GJ, et al. Fast uptake and long-lasting binding of methamphetamine in the human brain: comparison with cocaine. Neuroimage. 2008;43:756–63. doi: 10.1016/j.neuroimage.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newton TF, De La Garza R, 2nd, Kalechstein AD, Nestor L. Cocaine and methamphetamine produce different patterns of subjective and cardiovascular effects. Pharmacol Biochem Behav. 2005;82:90–7. doi: 10.1016/j.pbb.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 4.Haile CN, De La Garza R, 2nd, Newton TF. Methamphetamine Cured my Cocaine Addiction. J Addict Res Ther. 2010;1 doi: 10.4172/2155-6105.1000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tidey JW, Bergman J. Drug discrimination in methamphetamine-trained monkeys: agonist and antagonist effects of dopaminergic drugs. J Pharmacol Exp Ther. 1998;285:1163–74. [PubMed] [Google Scholar]
- 6.Simon SL, Richardson K, Dacey J, Glynn S, Domier CP, Rawson RA, Ling W. A comparison of patterns of methamphetamine and cocaine use. J Addict Dis. 2002;21:35–44. doi: 10.1300/j069v21n01_04. [DOI] [PubMed] [Google Scholar]
- 7.Rawson R, Huber A, Brethen P, Obert J, Gulati V, Shoptaw S, Ling W. Methamphetamine and cocaine users: differences in characteristics and treatment retention. J Psychoactive Drugs. 2000;32:233–8. doi: 10.1080/02791072.2000.10400234. [DOI] [PubMed] [Google Scholar]
- 8.Bousman CA, Glatt SJ, Everall IP, Tsuang MT. Genetic association studies of methamphetamine use disorders: A systematic review and synthesis. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:1025–49. doi: 10.1002/ajmg.b.30936. [DOI] [PubMed] [Google Scholar]
- 9.Phillips TJ, Kamens HM, Wheeler JM. Behavioral genetic contributions to the study of addiction-related amphetamine effects. Neurosci Biobehav Rev. 2008;32:707–59. doi: 10.1016/j.neubiorev.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kendler KS, Prescott CA. Cocaine use, abuse and dependence in a population-based sample of female twins. Br J Psychiatry. 1998;173:345–50. doi: 10.1192/bjp.173.4.345. [DOI] [PubMed] [Google Scholar]
- 11.Phillips TJ, Huson MG, McKinnon CS. Localization of genes mediating acute and sensitized locomotor responses to cocaine in BXD/Ty recombinant inbred mice. J Neurosci. 1998;18:3023–34. doi: 10.1523/JNEUROSCI.18-08-03023.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler JM, Reed C, Burkhart-Kasch S, Li N, Cunningham CL, Janowsky A, et al. Genetically correlated effects of selective breeding for high and low methamphetamine consumption. Genes Brain Behavior. 2009;8:758–71. doi: 10.1111/j.1601-183X.2009.00522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shabani S, McKinnon CS, Reed C, Cunningham CL, Phillips TJ. Sensitivity to rewarding or aversive effects of methamphetamine determines methamphetamine intake. Genes Brain Behav. 2011;10:625–36. doi: 10.1111/j.1601-183X.2011.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shabani S, McKinnon CS, Cunningham CL, Phillips TJ. Profound reduction in sensitivity to the aversive effects of methamphetamine in mice bred for high methamphetamine intake. Neuropharmacology. 2012;62:1134–41. doi: 10.1016/j.neuropharm.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shabani S, Dobbs LK, Ford MM, Mark GP, Finn DA, Phillips TJ. A genetic animal model of differential sensitivity to methamphetamine reinforcement. Neuropharmacology. 2012;62:2169–77. doi: 10.1016/j.neuropharm.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belknap JK, Richards SP, O’Toole LA, Helms ML, Phillips TJ. Short-term selective breeding as a tool for QTL mapping: ethanol preference drinking in mice. Behav Genet. 1997;27:55–66. doi: 10.1023/a:1025615409383. [DOI] [PubMed] [Google Scholar]
- 17.Cunningham CL, Niehus DR, Malott DH, Prather LK. Genetic differences in the rewarding and activating effects of morphine and ethanol. Psychopharmacology (Berl) 1992;107:385–93. doi: 10.1007/BF02245166. [DOI] [PubMed] [Google Scholar]
- 18.Cunningham CL. Localization of genes influencing ethanol-induced conditioned place preference and locomotor activity in BXD recombinant inbred mice. Psychopharmacology (Berl) 1995;120:28–41. doi: 10.1007/BF02246142. [DOI] [PubMed] [Google Scholar]
- 19.Cunningham CL, Dickinson SD, Grahame NJ, Okorn DM, McMullin CS. Genetic differences in cocaine-induced conditioned place preference in mice depend on conditioning trial duration. Psychopharmacology. 1999;146:73–80. doi: 10.1007/s002130051090. [DOI] [PubMed] [Google Scholar]
- 20.Bardo MT, Rowlett JK, Harris MJ. Conditioned place preference using opiate and stimulant drugs: a meta-analysis. Neurosci Biobehav Rev. 1995;19:39–51. doi: 10.1016/0149-7634(94)00021-r. [DOI] [PubMed] [Google Scholar]
- 21.Jones JD, Hall FS, Uhl GR, Riley AL. Dopamine, norepinephrine and serotonin transporter gene deletions differentially alter cocaine-induced taste aversion. Pharmacol Biochem Behav. 2010;94:580–7. doi: 10.1016/j.pbb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmer AA, McKinnon CS, Bergstrom HC, Phillips TJ. Locomotor activity responses to ethanol, other alcohols, and GABA-A acting compounds in forward- and reverse-selected FAST and SLOW mouse lines. Behav Neurosci. 2002;116:958–67. doi: 10.1037//0735-7044.116.6.958. [DOI] [PubMed] [Google Scholar]
- 23.Kamens HM, Phillips Tn. A role for neuronal nicotinic acetylcholine receptors in ethanol-induced stimulation, but not cocaine- or methamphetamine-induced stimulation. Psychopharmacology. 2008;196:377–87. doi: 10.1007/s00213-007-0969-7. [DOI] [PubMed] [Google Scholar]
- 24.Gubner NR, McKinnon CS, Reed C, Phillips TJ. Accentuating effects of nicotine on ethanol response in mice with high genetic predisposition to ethanol-induced locomotor stimulation. Drug Alcohol Depend. 2013;127:108–14. doi: 10.1016/j.drugalcdep.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips TJ, Huson M, Gwiazdon C, Burkhart-Kasch S, Shen EH. Effects of acute and repeated ethanol exposures on the locomotor activity of BXD recombinant inbred mice. Alcohol Clin Exp Res. 1995;19:269–78. doi: 10.1111/j.1530-0277.1995.tb01502.x. [DOI] [PubMed] [Google Scholar]
- 26.Kamens HM, Burkhart-Kasch S, McKinnon CS, Li N, Reed C, Phillips TJ. Sensitivity to psychostimulants in mice bred for high and low stimulation to methamphetamine. Genes Brain Behav. 2005;4:110–125. doi: 10.1111/j.1601-183X.2004.00101.x. [DOI] [PubMed] [Google Scholar]
- 27.Grigson PS, Twining RC. Cocaine-induced suppression of saccharin intake: a model of drug-induced devaluation of natural rewards. Behav Neurosci. 2002;116:321–33. [PubMed] [Google Scholar]
- 28.Han DD, Gu HH. Comparison of the monoamine transporters from human and mouse in their sensitivities to psychostimulant drugs. BMC Pharmacol. 2006;6:6. doi: 10.1186/1471-2210-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki T, Mori T, Tsuji M, Misawa M, Onodera K. Generalization of D-, L- and DL-chlorpheniramine and zolantidine to the discriminative stimulus effects of cocaine and methamphetamine. Behav Pharmacol. 1997;8:718–24. doi: 10.1097/00008877-199712000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Tilley MR, O’Neill B, Han DD, Gu HH. Cocaine does not produce reward in absence of dopamine transporter inhibition. Neuroreport. 2009;20:9–12. doi: 10.1097/WNR.0b013e32831b9ce4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Neill B, Tilley MR, Gu HH. Cocaine produces conditioned place aversion in mice with a cocaine-insensitive dopamine transporter. Genes Brain Behav. 2013;12:34–8. doi: 10.1111/j.1601-183X.2012.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones JD, Hall FS, Uhl GR, Rice K, Riley AL. Differential involvement of the norepinephrine, serotonin and dopamine reuptake transporter proteins in cocaine-induced taste aversion. Pharmacol Biochem Behav. 2009;93:75–81. doi: 10.1016/j.pbb.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eyerman DJ, Yamamoto BK. A rapid oxidation and persistent decrease in the vesicular monoamine transporter 2 after methamphetamine. J Neurochem. 2007;103:1219–27. doi: 10.1111/j.1471-4159.2007.04837.x. [DOI] [PubMed] [Google Scholar]
- 35.Sandoval V, Riddle EL, Hanson GR, Fleckenstein AE. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J Pharmacol Exp Ther. 2003;304:1181–7. doi: 10.1124/jpet.102.045005. [DOI] [PubMed] [Google Scholar]
- 36.Wilson JM, Kish SJ. The vesicular monoamine transporter, in contrast to the dopamine transporter, is not altered by chronic cocaine self-administration in the rat. J Neurosci. 1996;16:3507–10. doi: 10.1523/JNEUROSCI.16-10-03507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown JM, Hanson GR, Fleckenstein AE. Regulation of the vesicular monoamine transporter-2: a novel mechanism for cocaine and other psychostimulants. J Pharmacol Exp Ther. 2001;296:762–7. [PubMed] [Google Scholar]
- 38.Schwartz K, Nachman R, Yossifoff M, Sapir R, Weizman A, Rehavi M. Cocaine, but not amphetamine, short term treatment elevates the density of rat brain vesicular monoamine transporter 2. J Neural Transm. 2007;114:427–30. doi: 10.1007/s00702-006-0549-8. [DOI] [PubMed] [Google Scholar]
- 39.Riddle EL, Topham MK, Haycock JW, Hanson GR, Fleckenstein AE. Differential trafficking of the vesicular monoamine transporter-2 by methamphetamine and cocaine. Eur J Pharmacol. 2002;449:71–4. doi: 10.1016/s0014-2999(02)01985-4. [DOI] [PubMed] [Google Scholar]
- 40.Takahashi N, Miner LL, Sora I, Ujike H, Revay RS, Kostic V, et al. VMAT2 knockout mice: heterozygotes display reduced amphetamine-conditioned reward, enhanced amphetamine locomotion, and enhanced MPTP toxicity. Proc Natl Acad Sci U S A. 1997;94:9938–43. doi: 10.1073/pnas.94.18.9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fumagalli F, Gainetdinov RR, Wang YM, Valenzano KJ, Miller GW, Caron MG. Increased methamphetamine neurotoxicity in heterozygous vesicular monoamine transporter 2 knockout mice. J Neurosci. 1999;19:2424–31. doi: 10.1523/JNEUROSCI.19-07-02424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eastwood EC, Phillips TJ. Opioid sensitivity in mice selectively bred to consume or not consume methamphetamine. Addict Biol. 2013 doi: 10.1111/adb.12003. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Asanuma M, Cadet JL. Methamphetamine-induced increase in striatal NF-kappaB DNA-binding activity is attenuated in superoxide dismutase transgenic mice. Brain Res Mol Brain Res. 1998;60:305–9. doi: 10.1016/s0169-328x(98)00188-0. [DOI] [PubMed] [Google Scholar]
- 44.Loftis JM, Choi D, Hoffman W, Huckans MS. Methamphetamine causes persistent immune dysregulation: a cross-species, translational report. Neurotox Res. 2011;20:59–68. doi: 10.1007/s12640-010-9223-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 46.Tordoff MG, Bachmanov AA. Mouse taste preference tests: why only two bottles? Chem Senses. 2003;28:315–24. doi: 10.1093/chemse/28.4.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahmed SH, Koob GF. Transition from moderate to excessive drug intake: change in hedonic set point. Science. 1998;282:298–300. doi: 10.1126/science.282.5387.298. [DOI] [PubMed] [Google Scholar]
- 48.Petkov PM, Ding Y, Cassell MA, Zhang W, Wagner G, Sargent EE, Asquith S, Crew V, Johnson KA, Robinson P, Scott VE, Wiles MV. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome Res. 2004;14:1806–11. doi: 10.1101/gr.2825804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linsenbardt DN, Boehm SL., 2nd Determining the heritability of ethanol-induced locomotor sensitization in mice using short-term behavioral selection. Psychopharmacology. 2013 doi: 10.1007/s00213-013-3151-4. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Metten P, Belknap JK, Crabbe JC. Drug withdrawal convulsions and susceptibility to convulsants after short-term selective breeding for acute ethanol withdrawal. Behav Brain Res. 1998;95:113–22. doi: 10.1016/s0166-4328(97)00216-7. [DOI] [PubMed] [Google Scholar]
- 51.Scibelli AC, McKinnon CS, Reed C, Burkhart-Kasch S, Li N, Baba H, Wheeler JM, Phillips TJ. Selective breeding for magnitude of methamphetamine-induced sensitization alters methamphetamine consumption. Psychopharmacology. 2011;214:791–804. doi: 10.1007/s00213-010-2086-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hitzemann R, Hitzemann B, Rivera S, Gatley J, Thanos P, Shou LL, Williams RW. Dopamine D2 receptor binding, Drd2 expression and the number of dopamine neurons in the BXD recombinant inbred series: genetic relationships to alcohol and other drug associated phenotypes. Alcohol Clin Exp Res. 2003;27:1–11. doi: 10.1097/01.ALC.0000047862.40562.27. [DOI] [PubMed] [Google Scholar]
- 53.Kirstein SL, Davidson KL, Ehringer MA, Sikela JM, Erwin VG, Tabakoff B. Quantitative trait loci affecting initial sensitivity and acute functional tolerance to ethanol-induced ataxia and brain cAMP signaling in BXD recombinant inbred mice. J Pharmacol Exp Ther. 2002;302(3):1238–45. doi: 10.1124/jpet.302.3.1238. [DOI] [PubMed] [Google Scholar]
- 54.Phillips TJ, Crabbe JC, Metten P, Belknap JK. Localization of genes affecting alcohol drinking in mice. Alcohol Clin Exp Res. 1994;18:931–41. doi: 10.1111/j.1530-0277.1994.tb00062.x. [DOI] [PubMed] [Google Scholar]