

Abstract
Plasma cell leukemia (PCL) is a rare, yet aggressive plasma cell (PC) neoplasm, variant of multiple myeloma (MM), characterized by high levels of PCs circulating in the peripheral blood. PCL can either originate de novo (primary PCL) or as a secondary leukemic transformation of MM (secondary PCL). Presenting signs and symptoms are similar to those seen in MM such as renal insufficiency, hypercalcemia, lytic bone lesions, anemia, and thrombocytopenia, but can also include hepatomegaly and splenomegaly. The diagnostic evaluation of a patient with suspected PCL should include a review of the peripheral blood smear, bone marrow aspiration and biopsy, serum protein electrophoresis (SPEP) with immunofixation, and protein electrophoresis of an aliquot from a 24h urine collection (UPEP). The diagnosis is made when a monoclonal population of PCs is present in the peripheral blood with an absolute PC count exceeding 2000/μL and PC comprising 20% or more of the peripheral blood white cells. The prognosis of PCL is poor with a median survival of 7 to 11 months. Survival is even shorter (2 to 7 months) when PCL occurs in the context of refractory or relapsing MM. There have been no prospective randomized trials investigating the treatment of PCL. Recommendations are primarily based upon data from small retrospective series, case reports, and extrapolation of data from patients with MM. In general, patients are treated with induction therapy followed by hematopoietic cell transplantation (HCT) in those who are appropriate candidates for this approach. The best induction regimen for PCL is not known and there is great variability in clinical practice. Newer agents that are being incorporated into frontline and salvage therapy for MM have also demonstrated activity in PCL such as Immunomodulatory agents and the use of bortezomib with different combinations.
Keywords: Plasma cell leukemia
1. Definition and epidemiology
PCL is the most aggressive presentation of the PC neoplasms and is characterized by circulating PCs >2×109/L in peripheral blood and a peripheral blood plasmacytosis >20%.1 Primary PCL (pPCL) is defined as a malignant PC proliferation first diagnosed in the leukemic phase, while secondary PCL (sPCL) corresponds to the leukemic transformation of a previously diagnosed MM, probably as a consequence of clonal transformation (Table 1).2,3 Secondary PCL occurs as a progression of disease in 1 to 4% of all cases of MM, although it is now suspected to be a more common complication of patients exhibiting greater longevity.4 Altogether 60% of all PCL is pPCL.5 In the United States, the incidence is approximately 0.02 to 0.03 cases per 100,000 population accounting for less than 0.2% of all leukemia cases diagnosed from 1997 to 2002.6 As with MM, PCL is more common in African Americans than Caucasians.6 There appears to be a 3:2 male to female sex distribution in both primary and secondary PCL.7
Table 1.
Diagnostic criteria for plasma cell leukemia.
|
International Myeloma Working Group.3
2. Clinical and pathologic features
As compared to MM, pPCL tends to be observed in younger individuals. The median age of diagnosis of pPCL is 55, a decade younger than the average age of MM diagnosis.7 The median time to leukemic transformation for patients with MM who evolve to sPCL is 21 months.7 Patients with pPCL present without a MM prodrome, and are characterized by higher prevalence of renal insufficiency and elevated β2-microglobulin as opposed to newly diagnosed MM. Patients with sPCL are also frequently afflicted with renal insufficiency.7
In addition to the high number of circulating PCs extramedullary deposits at other sites are common in PCL. For instance up to 15% of patients will have hepatomegaly, splenomegaly or lymphadenopathy.4 An interesting feature of pPCL is that the majority of these individuals do not have clinical evidence of overt bone destruction. Osteolytic lesions are more common in sPCL as they are also more common in pre-existing MM (sPCL 53% vs. pPCL 18%).7 Because of extensive involvement of the bone marrow patients with PCL have a higher prevalence of anemia and thrombocytopenia.2,7,8 Likewise other laboratory parameters of disease aggressiveness such as an elevated LDH or β2-microglobulin are common in PCL cases. (Table 2).7–9
Table 2.
Clinical and biological differences between PCL and MM.
| Characteristic | MM | sPCL | pPCL | p-value | |
|---|---|---|---|---|---|
| Patient numbers | 439 | 39 | 41 | ||
| Age (years) | median | 66 | 65.7 | 54.5 | <0.001 |
| Extraosseous plasmacytoma | % cases | 6 | 22 | 0.21 | |
| Osteolytic lesions | % cases | 81 | 53 | 35 | 0.19 |
| Splenomegaly | % cases | 8 | 18 | 0.29 | |
| Hepatomegaly | % cases | 11 | 32 | 0.04 | |
| WBC (×109/L) | median | 5.8 | 15.7 | 21.5 | 0.04 |
| PB plasmacytosis(%) | median | 0 | 52 | 46 | 0.017 |
| PB plasmacytosis (×109/L) | median | 0 | 6.3 | 7.2 | 0.19 |
| Hemoglobin (g/dL) | median | 10.9 | 9.1 | 9.4 | 0.05 |
| Platelets (×109/L) | median | 242 | 53 | 98 | 0.05 |
| Serum creatinine (mg/dL) | median | 1.2 | 1.4 | 1.9 | 0.03 |
| Kappa (excl non-secretors) | % cases | 66 | 58 | 42 | 0.24 |
| Lambda (excl non-secretors) | % cases | 34 | 42 | 58 | 0.24 |
| PCLI | median | 0.4 | 3.0 | 1.4 | 0.47 |
| BM plasmacytosis | %BM | 46 | 63 | 78 | 0.11 |
Abbreviations: BM, bone marrow; PB, peripheral blood.
Modified from Tiedemann et al. Leukemia. 2008; 22, 1044–1052.7
The type of monoclonal protein produced by PCL is reflective of the underlying genetic abnormalities associated with this disease (see below). The most common proteins are of the IgG subtype (33%) followed by IgA (20%), IgD (3%) and IgE (1%). Notably 35% of patients do not produce a heavy chain (light chain only disease) and less than 10% will be non-secretors. 7
3. Blood and bone marrow
The morphological features of PCs can differ depending upon their maturity. Mature PCs are oval with abundant basophilic cytoplasm. The nucleus is round and eccentrically located and the chromatin arranged in pyramidal blocks against the nuclear membrane, giving the characteristic “cartwheel” appearance. Immature PCs have dispersed nuclear chromatin, prominent nucleoli and high nuclear to cytoplasmic ratio (Fig. 1a and b). The findings on bone marrow aspiration and biopsy are similar to those seen in MM without PCL and demonstrate an increased number of monoclonal PCs (Fig. 1c and d). The pattern of infiltration is mostly diffuse in all cases and this infiltration is able to disrupt normal hematopoiesis.
Fig. 1.

a and b. Peripheral blood smear showing the morphology of patient’s leukemic cells. Chan et al. case report.51 c and d. Bone marrow aspirate smears from two different patients with multiple myeloma, illustrating a preponderance of mostly mature-appearing plasma cells with eccentrically placed nuclei and prominent Golgi zones (arrow) (Wright Giemsa stain). From Brunning, RD, McKenna, RW. Tumors of the bone marrow. Atlas of tumor pathology (electronic fascicle), Third series, fascicle 9, 1994, Washington, DC. Armed Forces Institute of Pathology.
4. Immunophenotypic characteristics
CD38 and CD138 antigen expression are excellent PC markers and does not differ between MM and PCL, while CD2, CD3 and CD16 are consistently negative. The frequency of CD10+, CD13+ and CD15+ is similar in both groups. Nevertheless significant differences were observed for the expression of CD20, CD56, CD9, CD117 and HLA-DR antigens showing there is some overlap in antigen expression from the pattern for MM. The CD20 antigen displayed higher reactivity in PCL, whereas the other four, including the CD56 (NCAM) antigen, have a lower expression.8 Negative expression of CD56 has been associated with extramedullary MM.8–10 Recent observations in MM patients made by our own team showed by GEP (gene expression profile) analysis that there is practically null expression of CD56 in the MAF group (t(14;16)(q32:q23)), when compared with other group of translocations and with hyperdiploid patients (Fig. 2). These observations could be in favor of a MM cell more independent of bone marrow in the MAF group. CD28 is more frequently expressed on malignant PCs in sPCL, which is consistent with an observation made in MM, that is, that acquisition of the CD28 antigen on PCs appears to correlate with an increased proliferative rate and disease progression.11
Fig. 2.
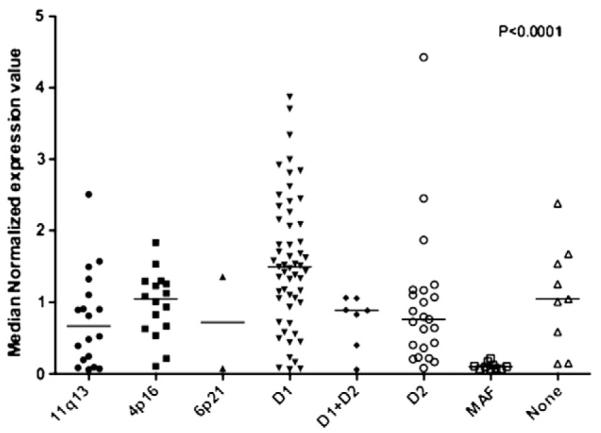
CD 56 expression in multiple myeloma. Images provided by Esteban Braggio.
5. Differential diagnosis
The diagnosis of PCL is usually straightforward and only involves differentiation from other conditions where circulating plasmacytoid or lymphocytic cells are observed. These include B-cell chronic lymphocytic leukemia, hairy cell leukemia and marginal zone lymphomas with circulating lymphocytes. Rarely PCL needs to be differentiated from reactive polyclonal plasmacytosis related to infectious or autoimmune disorders. The majority of cases will be easily distinguished from other forms of leukemia and lymphoma by morphology with confirmation by flow cytometry or immunohistochemistry. Patients with MM are arbitrarily distinguished from PCL by lower numbers of circulating PC, if present at all. The reason why such cut off value has been used for the absolute number and percent of circulating PC in PCL is completely arbitrary. Some authors have studied together strictly defined PCL with other MM cases of MM with many circulating PC, but yet not enough to fulfill diagnostic criteria. It is important for the treating physician to recognize that such patients have similar challenges as do patients with PCL, and several studies have shown the negative prognostic implications of having many circulating PCs in MM. Reactive polyclonal plasmacytosis can be excluded based upon absence of kappa or lambda light chain restriction.
6. Genetics of PCL
6.1. Ploidy status
The cytogenetic abnormalities observed in PCL are usually consisted on complex hypodiploid or pseudodiploid karyotypes. In two of the largest studies where informative karyotypes were obtained in 34 and 38 patients with PCL, 23 and 24 cases, respectively showed most cases were non-hyperdiploid (NH-MM) consisting mostly of complex hypodiploid or pseudodiploid karyotypes (67 and 63% respectively).7,12 Very few patients with hyperdiploid MM (H-MM) karyotypes in PCL have been reported, a finding that contrast with those published in MM in which hyperdiploidy is observed in approximately 60% of patients (Table 3).4,13
Table 3.
Ploidy categories in PCL.
| Ploidy | sPCL | pPCL |
|---|---|---|
| Hypodiploid (<45, >75) | 42 | 60 |
| Pseudo/diploid (45–47) | 42 | 40 |
| Hyperdiploid (48–75) | 17 | 0 |
| • Avet-Loiseau; 23 of 34 had abnormal karyotype and only 3 had hyperdiploidy | ||
| • Garcia-Sanz low rate H-MM | ||
7. Chromosome abnormalities identified by FISH
Table 4 summarizes commonly reported chromosome abnormalities by FISH (fluorescent in situ hybridization) in various published series of PCL.
Table 4.
Genetic aberrations in PCL.
| Tiedemann |
Avet-Loiseau |
|||||
|---|---|---|---|---|---|---|
| Abnormality | N | pPCL | sPCL | p | N | pPC |
| IgH Trx | 26 | 87 | 82 | 0.62 | 40 | 80% |
| t(11;14) | 21 | 65 | 49 | 0.60 | 40 | 33% |
| t(4;14) | 20 | 0 | 16 | 0.35 | 40 | 12% |
| t(14;16) | 20 | 0 | 16 | 0.35 | 40 | 16% |
| t(6;14) | 7 | 0 | 0 | – | ||
| t(11;14) | 30 | 71 | 23 | 0.03 | ||
| −13q | 19 | 85 | 67 | 0.56 | 40 | 68% |
7.1. IgH translocations
Chromosome 14q32 translocations are found in a great number of pPCL and sPCL patients (82–87%), as would be expected in NH-MM cases. In pPCL IgH translocations almost exclusively involve 11q13 (CCND1), supporting a central etiological role, while in sPCL multiple partner oncogenes are involved, including 11q13, 4p16 (FGFR3/MMSET) and 16q23 (MAF), recapitulating MM.7 Avet-Loiseau and colleagues described a higher prevalence of t(11;14) and t(14;16) in PCL as compared with a stage III MM population (33 and 13% versus 16 and 1%; p=0.025 and p=0.002, respectively), although in his study the incidence of t(4;14) was identical (12%).12
Genomic aberrations such as t(4;14), del 13q14, del 17p, del 1p21 and 1q21 gains are adverse risk factors in MM but their significance in PCL is unclear (as they usually indicate more aggressive behavior which is ubiquitous in PCL).14–21 Recently, Chang and colleagues investigated 41 PCL cases aiming to detect chromosome 1q amplifications and 1p deletions, and compared the genetic aberrations with those in 220 MM patients.22 They report del17p, del13q14, del1p21, 1q21 amplifications and t(4;14) were more frequent in PCL than MM. Deletions of 1p21 were associated with 1q21 amplification (p=0.03) and has a marginal association with del17p (p=0.06). They showed patients with 1p21 deletions had a shorter OS than those without such deletions (6.2 versus 33.5 months, p=0.006).22 Notably patients with t(4;14) had a shorter survival than those without t(4;14) (1.5 vs. 21.6 months, p=0.003). The presence of del 13q14, del17p, t(11;14) and 1q21 amplification did not influence survival in this cohort. In a multivariate analysis adjusting for all above genetic risk factors as well as CRP, calcium and β2-microglobulin levels, only t(4;14) was an independent predictor for a worse OS (p=0.008), 1p21 deletions did not retain the prognostic significance (p=0.14).22
7.2. Chromosome 13
In a series of 26 patients Garcia Sanz reported −13 in 84% of patients with PCL versus 26% for those with MM.8 In the Mayo study we found that loss of 13q by FISH was very common in pPCL (85%), more so than in MM (54%) (p=0.02); but with no difference in prevalence between pPCL and sPCL.7 As would be expected in NH-MM Avet-Loiseau reported a high frequency of monosomy 13 (85%).12
7.3. Deletions of 17p13 and TP53 inactivation
Previous studies reported a prevalence of deletion in MM 17p13.1 as a late event found only in 10% of patients,23 and TP53 coding mutations in only 3%.24 In the group of 80 patients reported by us there was a prevalence of deletion of 17p13.1, causing allelic loss of TP53, in 50% of pPCL and a remarkable 75% of sPCL. Moreover, TP53 deletion was complemented by functionally relevant TP53 coding mutations in 24% of PCL patients tested, contributing to a substantial prevalence of allelic TP53 inactivation of 56% in pPCL and 83% in sPCL. Eleven percent of pPCL and 33% of sPCL tumors showed biallelic inactivation of TP53 with simultaneous allelic deletion and mutation.7 Interestingly, monoallelic or biallelic inactivation of TP53 did not correlate significantly with survival in sPCL, unlike MM, where −17p13.1 predicts adverse survival.25,26 Lack of correlation between TP53 status and survival may reflect ubiquitous targeting of the TP53 pathway in sPCL.
Inactivation of p53 function may also occur through overexpression of negative regulatory elements such as MDM2, or by decreased activity of CDKN2A (p14ARF), a negative regulator of MDM2.27,28 Therefore we also screened PCL samples for mechanisms leading to MDM2 activation or suppression of p14ARF.7 No focal amplicons of MDM2 were detected indicating that p53 pathway inactivation in PCL is rare, if ever caused by MDM2 gene copy number change. However, the upstream tumor suppressor p14ARF was targeted by promoter methylation in 29% of sPCL cases, demonstrating a second mechanism by which p53 activity can be inhibited in PCL (Table 5).29,30
Table 5.
TP53 in plasma cell leukemia.
| p53 pathway | N | pPCL | sPCL | P value |
|---|---|---|---|---|
| Allelic p53 mutation | 25 | 25 | 23 | – |
| Allelic p53 deletion | 20 | 50 | 75 | 0.37 |
| Combined | 15 | 56 | 83 | 0.58 |
| biallelic p53 loss | 15 | 11 | 33 | 0.52 |
| MDM2 amplification | 18 | 0 | 0 | – |
| PTEN Deletion | 18 | 8 | 33 | 0.24 |
| RAS mutation | 24 | 27 | 15 | 0.63 |
| p16 methylation | 24 | 27 | 38 | 0.68 |
| p14 methylation | 27 | 0 | 29 | 0.10 |
Modified from Tiedemann et al 2008.7
7.4. MYC abnormalities and RAS mutation
Rearrangement of MYC has been identified by 3’FISH break apart in 33% of pPCL and sPCL tumors and is been complemented by MYC amplification or 5’MYC translocations in 8 and 17% of patients, respectively.7 MYC rearrangements are associated with a trend toward inferior OS in pPCL (median OS of 8.6 vs. 27.8 months without rearrangements, p=0.006).
On the other hand, mutations of K-RAS or N-RAS at codons 12, 13 or 61, previously characterized as functionally activating,31–34 are found in 27% pPCL and 15% sPCL.7 Activating mutations of RAS are associated with a trend toward poorer outcome in pPCL (p=0.069). However, the prevalence of K- or N-RAS mutation in sPCL is comparable to that reported in MM (21%),33 suggesting little, if any, selective pressure for RAS activation in secondary leukemic transformation from MM.
8. Survival
PCL is extremely aggressive associated with short survival; with treatment of only 7 to 11 months, with up to 28% of patients dying within the first month after diagnosis in different studies.2,7,12 Survival is even shorter when PCL occurs in the context of refractory or relapsing MM. In one of the largest series we reported and overall survival of 1.3 months for sPCL and 11.2 months for pPCL (Fig. 3).7 Accurate prognostication of PCL is hampered by the always low number of cases studied, and the realization that the disease is always associated with an adverse outcome. Most studies confirm that predictable variables of disease progression predict more aggressive course.8 Garcia-Sanz shows in a very small series that many variable predict outcome, all of them associated with greater tumor bulk and cell proliferation.8 In our study we could not find major prognostic implications for the various genetic factors tested.7 Avet-Loiseau reported longer survival, but still limited survival for patients with a t (11;14) (12 months, p=0.001),12 but other series have not reached similar conclusions.35,36
Fig. 3.
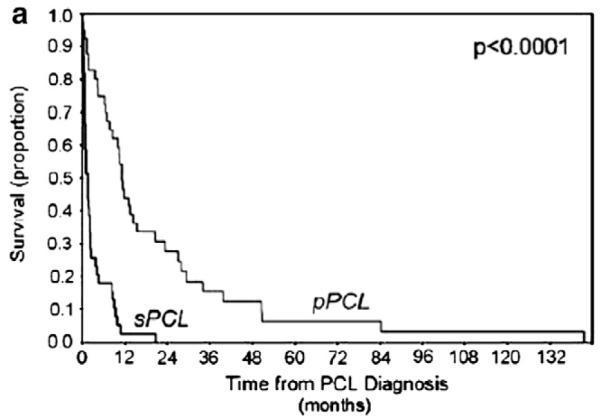
Overall survival (OS) in pPCL and sPCL, showing superior survival of pPCL vs sPCL from the time of leukaemia diagnosis. Abbreviations: sPCL, secondary plasma cell leukaemia; pPCL, primary plasma cell leukaemia. Tiedemann et al. Leukemia 2008, 22:1044.7
9. Treatment in PCL
Treatment of both pPCL and sPCL, as in MM, is aimed at prolonging survival and maximizing quality of life, as there are no recognized curative regimens.10 In general, patients are treated with aggressive induction therapy followed by HCT in those who are appropriate candidates for this approach. Chemotherapy alone is the principal option for those ineligible for HCT.
The best induction regimen for PCL is not known and there is great variability in clinical practice. Several combinations that include an alkylating agent have been used as sole treatments of PCL or as induction therapy prior to anticipated transplant. Combinations that include melphalan are generally avoided as induction therapy in potential transplant candidates to allow adequate stem cell collection.
Most patients need to be treated with the most effective agents currently available38–40 and historic data with older agents show poor rates of control. Patients with PCL have had a median overall survival of 4 months when treated with melphalan plus prednisone.7,8,37 Multi-agent infusional chemotherapy (e.g. vincristine, doxorubicin, plus dexamethasone) results in a superior, yet still poor median overall survival of approximately 15 to 18 months.7,8 Another study reported similar results by showing a median OS of 6.8 months for patients treated with VAD (vincristine, doxorubicin and dexamethasone) versus 2 months for those who received MP (p<0.05).5
9.1. Immunomodulatory (IMID) agents
The IMiDs agents have also been used for the treatment of PCL based on observation that in PCL the response to standard therapy is extremely poor. Thalidomide has been reported to be temporally effective in a small group of patients.41,42 Two case reports showed activity of thalidomide in PCL.43,44 Similar case reports have shown activity of lenalidomide in these patients.39 Despite of having a good initial efficacy, the duration of the responses were brief or with a short follow-up duration.39,40,45
9.2. Bortezomib
The efficacy of bortezomib as single agent or in combination with other chemotherapies has been reported both in pPCL as well as in sPCL.46,47 Complete remission (CR) and successful stem cell mobilization also have been reported in patients with refractory PCL.48 Musto reported on 12 patients with pPCL or sPCL, using different combinations and found that the majority of them entered into a high-quality remission phases, including 5 partial remissions (PR), 4 very good partial remissions (VGPR) and 2 complete remissions (CR). Interesting, some of them who have previously developed disease recurrence after HCT achieved a response. Overall, the median progression free survival (PFS) and OS were 8 and 12 months, respectively. Eight patients were alive at 6 to 21 months after therapy and as expected the best results were obtained in patients with pPCL.38 A case series of 3 patients with pPCL, all with poor risk cytogenetic features, were treated with bortezomib plus dexamethasone followed by maintenance bortezomib with excellent and sustained response at a single institution.49
There have been some reports using bortezomib with other associations. Bortezomib in combination with doxorubicin and dexamethasone has been used in combination used at front line or as salvage therapy was able to achieve VGPR and CR with sustained responses in some case reports.50,51 Al-Nawakil recently reported four case reports of patients with PCL treated with the PAD combination, three of whom achieved VGPR and one a CR.50 These data altogether represents evidence to suggest bortezomib and combinations schemes to be highly active as induction therapy in PCL.
9.3. Hematopoietic stem cell transplantation
Using more intensive approaches like autologous and allogeneic HCT have been also explored.7,52,53 An observational study showed that 30% of 21 patients with pPCL who received multiagent intravenous chemotherapy proceeded to SCT. Patients who received SCT had a longer median OS when compared with those who received multiagent intravenous chemotherapy alone (34 vs 11 months). Although part of this survival benefit is likely selection bias with younger patients who survived initial therapy proceeding to SCT, it seems reasonable to offer HCT to suitable individuals with PCL.7
Another study showed a median OS of 36 months (range, 1–106 months) after autologous HCT and 20 months (range, 1–84 months) after allogeneic HCT in 38 evaluable patients with pPCL.52 A recently retrospective review of 160 patients with pPCL was reported to show the CIBMTR experience.53 After a median follow-up of 38 months, 20 of the 52 patients (38%) were alive in the allogeneic HCT group and 68 of the 107 pts (64%) were alive in the autologous HCT group (Fig. 4). Progressive disease accounted for 38% of the deaths in the allogeneic HCT group and 86% of the deaths in the autologous HCT group.
Fig. 4.
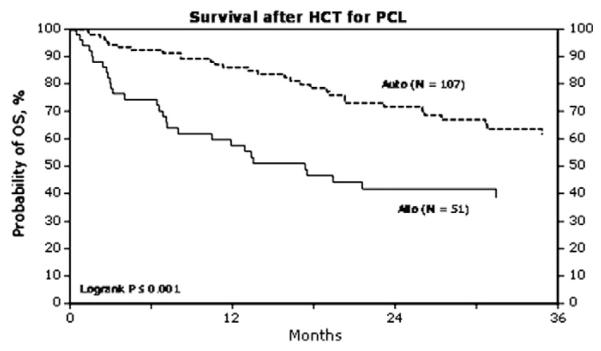
Mahindra A, et al. Abstract 532. Blood. ASH Annual Meeting Abstracts. 2009. Abstract 532; 114:22.53
10. Conclusions
PCL represents a unique subset of patients with an aggressive PC proliferative disorder, which is associated to a poor prognosis, with a shorter survival than for patients presenting with MM. Genomic and clinical differences between PCL and MM have been recognized. The presence of p53 deletion in a high level, 13q deletions, karyotypic complexity, hypodiploidy and 1q gains could define an advance stage on PC disease progression characterized by therapy resistance and a dismal prognosis. PCL requires intensive treatment that nowadays includes combinations with novel therapies such proteasome inhibitors, and IMiDs (that at least in preliminary reports appear to be promising), and bone marrow transplant with different approaches including allogeneic HCT.
Acknowledgements
Rafael Fonseca is a Clinical Investigator of the Damon Runyon Cancer Research Fund. This work is supported by grants SPORE CA90297052, P01 CA62242, R01 CA83724, ECOG CA 21115T, Predolin Foundation, Mayo Clinic Cancer Center and the Mayo Foundation.
Rafael Fonseca: Relevant to this work Dr. Fonseca has received a patent for the prognostication of MM based on genetic categorization of the disease. Non-relevant, he has received consulting fees from Medtronic, Otsuka, Celgene, Genzyme, BMS and AMGEN. He also has sponsored research from Cylene and Onyx.
Footnotes
Conflict of interest disclosure Flavio Albarracin: no conflicts of interest to declare.
References
- 1.Kyle RA, Maldonado JE, Bayrd ED. Plasma cell leukemia. Report on 17 cases. Arch Intern Med. 1974;133:813–8. doi: 10.1001/archinte.133.5.813. [DOI] [PubMed] [Google Scholar]
- 2.Noel P, Kyle RA. Plasma cell leukemia: an evaluation of response to therapy. Am J Med. 1987;83:1062–8. doi: 10.1016/0002-9343(87)90942-9. [DOI] [PubMed] [Google Scholar]
- 3.Criteria for the classification of monoclonal gammopathies. multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121:749–57. [PubMed] [Google Scholar]
- 4.Blade J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematol Oncol Clin North Am. 1999;13:1259–72. doi: 10.1016/s0889-8588(05)70125-8. [DOI] [PubMed] [Google Scholar]
- 5.Jimenez-Zepeda VH, Dominguez VJ. Plasma cell leukemia: a rare condition. Ann Hematol. 2006;85:263–7. doi: 10.1007/s00277-005-0054-4. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997-2002. Cancer Causes Control. 2008;19:379–90. doi: 10.1007/s10552-007-9097-2. [DOI] [PubMed] [Google Scholar]
- 7.Tiedemann RE, Gonzalez-Paz N, Kyle RA, et al. Genetic aberrations and survival in plasma cell leukemia. Leukemia. 2008;22:1044–52. doi: 10.1038/leu.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia-Sanz R, Orfao A, Gonzalez M, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics. Blood. 1999;93:1032–7. [PubMed] [Google Scholar]
- 9.San Miguel JF, Gonzalez M, Gascon A, et al. Immunophenotypic heterogeneity of multiple myeloma: influence on the biology and clinical course of the disease. Castellano-Leones (Spain) Cooperative Group for the Study of Monoclonal Gammopathies. Br J Haematol. 1991;77:185–90. doi: 10.1111/j.1365-2141.1991.tb07975.x. [DOI] [PubMed] [Google Scholar]
- 10.Hayman SR, Fonseca R. Plasma cell leukemia. Curr Treat Options Oncol. 2001;2:205–16. doi: 10.1007/s11864-001-0034-4. [DOI] [PubMed] [Google Scholar]
- 11.Pellat-Deceunynck C, Bataille R, Robillard N, et al. Expression of CD28 and CD40 in human myeloma cells: a comparative study with normal plasma cells. Blood. 1994;84:2597–603. [PubMed] [Google Scholar]
- 12.Avet-Loiseau H, Daviet A, Brigaudeau C, et al. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: a study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myelome and the Groupe Francais de Cytogenetique Hematologique. Blood. 2001;97:822–5. doi: 10.1182/blood.v97.3.822. [DOI] [PubMed] [Google Scholar]
- 13.Taniwaki M, Nishida K, Ueda Y, Takashima T. Non-random chromosomal rearrangements and their implications in clinical features and outcome of multiple myeloma and plasma cell leukemia. Leuk Lymphoma. 1996;21:25–30. doi: 10.3109/10428199609067575. [DOI] [PubMed] [Google Scholar]
- 14.Fonseca R, Barlogie B, Bataille R, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64:1546–58. doi: 10.1158/0008-5472.can-03-2876. [DOI] [PubMed] [Google Scholar]
- 15.Chang H, Qi XY, Samiee S, et al. Genetic risk identifies multiple myeloma patients who do not benefit from autologous stem cell transplantation. Bone Marrow Transplant. 2005;36:793–6. doi: 10.1038/sj.bmt.1705131. [DOI] [PubMed] [Google Scholar]
- 16.Hanamura I, Stewart JP, Huang Y, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108:1724–32. doi: 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chang H, Qi X, Trieu Y, et al. Multiple myeloma patients with CKS1B gene amplification have a shorter progression-free survival post-autologous stem cell transplantation. Br J Haematol. 2006;135:486–91. doi: 10.1111/j.1365-2141.2006.06325.x. [DOI] [PubMed] [Google Scholar]
- 18.Fonseca R, Van Wier SA, Chng WJ, et al. Prognostic value of chromosome 1q21 gain by fluorescent in situ hybridization and increase CKS1B expression in myeloma. Leukemia. 2006;20:2034–40. doi: 10.1038/sj.leu.2404403. [DOI] [PubMed] [Google Scholar]
- 19.Shaughnessy JD, Jr, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–84. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 20.Chang H, Ning Y, Qi X, Yeung J, Xu W. Chromosome 1p21 deletion is a novel prognostic marker in patients with multiple myeloma. Br J Haematol. 2007;139:51–4. doi: 10.1111/j.1365-2141.2007.06750.x. [DOI] [PubMed] [Google Scholar]
- 21.Avet-Loiseau H, Attal M, Moreau P, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myelome. Blood. 2007;109:3489–95. doi: 10.1182/blood-2006-08-040410. [DOI] [PubMed] [Google Scholar]
- 22.Chang H, Qi X, Yeung J, Reece D, Xu W, Patterson B. Genetic aberrations including chromosome 1 abnormalities and clinical features of plasma cell leukemia. Leuk Res. 2009;33:259–62. doi: 10.1016/j.leukres.2008.06.027. [DOI] [PubMed] [Google Scholar]
- 23.Fonseca R, Blood E, Rue M, et al. Clinical and biologic implications of recurrent genomic aberrations in myeloma. Blood. 2003;101:4569–75. doi: 10.1182/blood-2002-10-3017. [DOI] [PubMed] [Google Scholar]
- 24.Chng WJ, Price-Troska T, Gonzalez-Paz N, et al. Clinical significance of TP53 mutation in myeloma. Leukemia. 2007;21:582–4. doi: 10.1038/sj.leu.2404524. [DOI] [PubMed] [Google Scholar]
- 25.Lloveras E, Granada I, Zamora L, et al. Cytogenetic and fluorescence in situ hybridization studies in 60 patients with multiple myeloma and plasma cell leukemia. Cancer Genet Cytogenet. 2004;148:71–6. doi: 10.1016/s0165-4608(03)00233-4. [DOI] [PubMed] [Google Scholar]
- 26.Gertz MA, Lacy MQ, Dispenzieri A, et al. Clinical implications of t(11;14)(q13;q32), t(4;14)(p16.3;q32), and −17p13 in myeloma patients treated with high-dose therapy. Blood. 2005;106:2837–40. doi: 10.1182/blood-2005-04-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stott FJ, Bates S, James MC, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998;17:5001–14. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eymin B, Gazzeri S, Brambilla C, Brambilla E. Mdm2 overexpression and p14(ARF) inactivation are two mutually exclusive events in primary human lung tumors. Oncogene. 2002;21:2750–61. doi: 10.1038/sj.onc.1205359. [DOI] [PubMed] [Google Scholar]
- 29.Esteller M, Cordon-Cardo C, Corn PG, et al. p14ARF silencing by promoter hypermethylation mediates abnormal intracellular localization of MDM2. Cancer Res. 2001;61:2816–21. [PubMed] [Google Scholar]
- 30.Magdinier F, Wolffe AP. Selective association of the methyl-CpG binding protein MBD2 with the silent p14/p16 locus in human neoplasia. Proc Natl Acad Sci USA. 2001;98:4990–5. doi: 10.1073/pnas.101617298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahuja HG, Foti A, Bar-Eli M, Cline MJ. The pattern of mutational involvement of RAS genes in human hematologic malignancies determined by DNA amplification and direct sequencing. Blood. 1990;75:1684–90. [PubMed] [Google Scholar]
- 32.Bezieau S, Devilder MC, Avet-Loiseau H, et al. High incidence of N and K-Ras activating mutations in multiple myeloma and primary plasma cell leukemia at diagnosis. Hum Mutat. 2001;18:212–24. doi: 10.1002/humu.1177. [DOI] [PubMed] [Google Scholar]
- 33.Ortega MM, Faria RM, Shitara ES, et al. N-RAS and K-RAS gene mutations in Brazilian patients with multiple myeloma. Leuk Lymphoma. 2006;47:285–9. doi: 10.1080/10428190500300969. [DOI] [PubMed] [Google Scholar]
- 34.Liang DC, Shih LY, Fu JF, et al. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer. 2006;106:950–6. doi: 10.1002/cncr.21687. [DOI] [PubMed] [Google Scholar]
- 35.Fonseca R, Witzig TE, Gertz MA, et al. Multiple myeloma and the translocation t(11;14)(q13;q32): a report on 13 cases. Br J Haematol. 1998;101:296–301. doi: 10.1046/j.1365-2141.1998.00700.x. [DOI] [PubMed] [Google Scholar]
- 36.Lai JL, Michaux L, Dastugue N, et al. Cytogenetics in multiple myeloma: a multicenter study of 24 patients with t(11;14)(q13;q32) or its variant. Cancer Genet Cytogenet. 1998;104:133–8. doi: 10.1016/s0165-4608(97)00469-x. [DOI] [PubMed] [Google Scholar]
- 37.Dimopoulos MA, Palumbo A, Delasalle KB, Alexanian R. Primary plasma cell leukaemia. Br J Haematol. 1994;88:754–9. doi: 10.1111/j.1365-2141.1994.tb05114.x. [DOI] [PubMed] [Google Scholar]
- 38.Musto P, Rossini F, Gay F, et al. Efficacy and safety of bortezomib in patients with plasma cell leukemia. Cancer. 2007;109:2285–90. doi: 10.1002/cncr.22700. [DOI] [PubMed] [Google Scholar]
- 39.Benson DM, Jr, Smith MK. Effectiveness of lenalidomide (Revlimid) for the treatment of plasma cell leukemia. Leuk Lymphoma. 2007;48:1423–5. doi: 10.1080/10428190701361851. [DOI] [PubMed] [Google Scholar]
- 40.Musto P, Pietrantuono G, Guariglia R, et al. Salvage therapy with lenalidomide and dexamethasone in relapsed primary plasma cell leukemia. Leuk Res. 2008;32:1637–8. doi: 10.1016/j.leukres.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 41.Johnston RE, Abdalla SH. Thalidomide in low doses is effective for the treatment of resistant or relapsed multiple myeloma and for plasma cell leukaemia. Leuk Lymphoma. 2002;43:351–4. doi: 10.1080/10428190290006143. [DOI] [PubMed] [Google Scholar]
- 42.Tsiara S, Chaidos A, Kapsali H, Tzouvara E, Bourantas KL. Thalidomide administration for the treatment of resistant plasma cell leukemia. Acta Haematol. 2003;109:153–5. doi: 10.1159/000069284. [DOI] [PubMed] [Google Scholar]
- 43.Wohrer S, Ackermann J, Baldia C, et al. Effective treatment of primary plasma cell leukemia with thalidomide and dexamethasone - a case report. Hematol J. 2004;5:361–3. doi: 10.1038/sj.thj.6200375. [DOI] [PubMed] [Google Scholar]
- 44.Bauduer F. Efficacy of thalidomide in the treatment of VAD-refractory plasma cell leukaemia appearing after autologous stem cell transplantation for multiple myeloma. Br J Haematol. 2002;117:996–7. doi: 10.1046/j.1365-2141.2002.03537_4.x. [DOI] [PubMed] [Google Scholar]
- 45.Guglielmelli T, Merlini R, Giugliano E, Saglio G. Lenalidomide, melphalan, and prednisone association is an effective salvage therapy in relapsed plasma cell leukaemia. J Oncol. 2009;2009:867380. doi: 10.1155/2009/867380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esparis-Ogando A, Alegre A, Aguado B, et al. Bortezomib is an efficient agent in plasma cell leukemias. Int J Cancer. 2005;114:665–7. doi: 10.1002/ijc.20793. [DOI] [PubMed] [Google Scholar]
- 47.Finnegan DP, Kettle P, Drake M, et al. Bortezomib is effective in primary plasma cell leukemia. Leuk Lymphoma. 2006;47:1670–3. doi: 10.1080/10428190500520954. [DOI] [PubMed] [Google Scholar]
- 48.Grassinger J, Sudhoff T, Andreesen R, Hennemann B. Complete remission and successful stem cell mobilization after treatment of refractory plasma cell leukemia with bortezomib. Ann Hematol. 2006;85:132–3. doi: 10.1007/s00277-005-0027-7. [DOI] [PubMed] [Google Scholar]
- 49.Katodritou E, Verrou E, Gastari V, Hadjiaggelidou C, Terpos E, Zervas K. Response of primary plasma cell leukemia to the combination of bortezomib and dexamethasone: do specific cytogenetic and immunophenotypic characteristics influence treatment outcome? Leuk Res. 2008;32:1153–6. doi: 10.1016/j.leukres.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 50.Al-Nawakil C, Tamburini J, Bardet V, et al. Bortezomib, doxorubicin and dexamethasone association is an effective option for plasma cell leukemia induction therapy. Leuk Lymphoma. 2008;49:2012–4. doi: 10.1080/10428190802290660. [DOI] [PubMed] [Google Scholar]
- 51.Chan SM, George T, Cherry AM, Medeiros BC. Complete remission of primary plasma cell leukemia with bortezomib, doxorubicin, and dexamethasone: a case report. Cases J. 2009;2:121. doi: 10.1186/1757-1626-2-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saccaro S, Fonseca R, Veillon DM, et al. Primary plasma cell leukemia: report of 17 new cases treated with autologous or allogeneic stem-cell transplantation and review of the literature. Am J Hematol. 2005;78:288–94. doi: 10.1002/ajh.20272. [DOI] [PubMed] [Google Scholar]
- 53.Mahindra A, Vesole D, Kalaycio M, et al. Autologous Hematopoietic Stem Cell Transplantation (HCT) is a Safe and Effective Treatment for Primary Plasma Cell Leukemia: The CIBMTR Experience. [abstract 532] Blood. 2009;114:22. ASH Annual Meeting Abstracts. [Google Scholar]