

Abstract
The Gene Ontology (GO) is a structured controlled vocabulary developed to describe the roles and locations of gene products in a consistent fashion, in a way that can be shared across organisms. The unicellular fungus Candida albicans is similar in many ways to the model organism Saccharomyces cerevisiae, but as both a commensal and a pathogen of humans, differs greatly in its lifestyle. With an expanding at-risk population of immunosuppressed patients, increased use of invasive medical procedures, the increasing prevalence of drug resistance, and the emergence of additional Candida species as serious pathogens, it has never been more critical to improve our understanding of Candida biology to guide the development of better treatments. In this brief review, we examine the importance of GO in the annotation of C. albicans gene products, with a focus on those involved in pathogenesis. We also discuss how sequence information combined with GO facilitates the transfer of knowledge across related species, and the challenges and opportunities that such an approach presents.
Introducing Candida albicans, a pathogen of increasing relevance in the present day
Candida albicans, the best studied of the human fungal pathogens, is a frequent commensal of humans and is responsible for mucosal infections, such as vaginitis or oral thrush, in otherwise healthy individuals. However, systemic Candida infection is a serious threat to severely ill or immunocompromised patients, including low-birthweight infants, transplant recipients, chemotherapy patients, burn victims, and HIV-infected patients [1, 2]. In addition, C. albicans can readily colonize catheters and other medical devices, where it forms biofilms that are significantly more resistant to drug treatment than are planktonic (individual free-floating) cells [3, 4]. For these reasons, C. albicans has become the third or fourth most common cause of nosocomial bloodstream infections, and mortality rates from systemic Candida infection are high [5, 6]. Emerging resistance to the existing arsenal of antifungal drugs is also of increasing concern, and azole resistance is now common among isolates from HIV-positive patients [7]. Additionally, other fungal species are emerging as pathogens of increasing prevalence and concern [8]. Thus, the need for new and more effective strategies to combat these infections, as well as those caused by other fungal pathogens for which C. albicans can serve as a model, has driven recent studies designed to elucidate the molecular basis of C. albicans virulence. Genomic-scale experiments are powerful tools that can provide insight into pathogenesis and other processes at a molecular level, but which require a well-annotated gene catalog. The Gene Ontology (GO) vocabulary is a classification system of particular utility for this purpose (see: http://www.geneontology.org).
Introducing the Gene Ontology
GO comprises three hierarchical controlled vocabularies and a set of evidence codes. These three vocabularies contain terms to describe the location of a gene product within the cell (Cellular Component), the activity that it carries out (Molecular Function), and the broader role that it fulfills (Biological Process). Each term is related to both the less-specific “parental terms” that are placed above it in the hierarchical tree, as well as to the more specific “child” terms that it encompasses. The evidence codes describe the type of inference that was used to make each GO annotation to a gene product and, as such, indicate a level of confidence. A gene product may be annotated with any number of terms from any of the three vocabularies, depending on the available data. Direct annotation to a particular term also implies indirect annotation to all terms from which that term is descended. For example, a protein annotated to the Cellular Component term “mitochondrial inner membrane” is also implicitly annotated to “mitochondrial membrane” and “mitochondrion”, which are more general, parental terms of “mitochondrial inner membrane” in the Cellular Component vocabulary.
Candida albicans biology and pathogenic strategies
Genetic diversity is crucial to adaptation and survival in the diverse and often hostile environments within the host. C. albicans usually exists in a diploid or near-diploid state, and has never been observed to undergo meiosis. However, diploids that become homozygous at the mating-type-like (MTL) locus can mate to form tetraploids in a tightly regulated process that requires morphological switching from the nonmating “white” cell type to the mating-competent “opaque” cell type [9-12] (Figure 1a). Genetic recombination occurs in resulting tetraploids, which then shed chromosomes to return to diploidy in a process termed the parasexual cycle [13, 14]. Another mechanism leading to genetic diversity is the frequent occurrence of aneuploidy and gross chromosomal rearrangements (such as isochromosome formation, during which one chromosome arm is duplicated and the other arm is lost), for which C. albicans exhibits a remarkable tolerance. Such events can confer selective advantages with respect to drug resistance and capacity for metabolism of specific nutrients [15-17].
Figure 1.

Life cycle and morphology of Candida albicans. (a) C. albicans parasexual cycle. Diploid yeast-form cells that have become homozygous (‘a/a’ or ‘α/α’) at the mating-type-like locus (MTL) may undergo a phenotypic switch from "white" to mating-competent, "opaque" cells. The mating of opaque cells of opposite mating types produces a tetraploid ‘aa/αα’ cell that returns to the diploid state via chromosome loss. (b) Different shapes of C. albicans. Depending on the environmental conditions, C. albicans can grow in distinct morphological forms: round, budding yeast-like cells easily separate from each other, whereas elongated, filament-forming true hyphal and pseudohyphal cells remain attached; the constriction between cells is a distinguishing feature of pseudohyphae. Figure adapted from reference [36].
C. albicans grows in several morphological forms, including: (i) a budding yeast form that can switch from the smooth-walled white phase to the rough-walled, mating-competent opaque phase, (ii) pseudohyphal filaments, and (iii) filaments with a true hyphal morphology (Figure 1b) [18, 19]. The ability to switch morphology is essential for its pathogenicity, as different growth forms exhibit differential success in the various ecological niches encountered in the host [20, 21]. Yeast-form cells are more amenable to dispersal in the bloodstream, while hyphae exhibit adhesive and mechanical properties that allow them to penetrate host cell membranes [22, 23]. Engulfment of yeast-form cells by host macrophages induces a stress response to the low-glucose conditions therein and triggers a switch to hyphal growth, which facilitates physical escape from this hostile environment [24], as well as other changes in gene expression that can affect the properties of macrophages to benefit the pathogen [25]. White- and opaque-phase cells are adapted to growth in different niches, and white-opaque switching is an infrequent event under most conditions studied. However, this switching is greatly induced in the WO-1 strain in the mammalian gastrointestinal tract, though other tested strains did not show such an induction, suggesting that there is genetic variation in control of this response [20].
The capacity to grow in a multicellular community known as a biofilm is an important component of C. albicans pathogenicity, allowing it to grow on catheters and other medical devices, and dental prostheses. Compared to planktonic cells, cells in biofilms exhibit a remarkable resistance to drugs, which complicates patient treatment [4, 26-28]. Biofilm formation depends on a means of cell-to-cell communication, and C. albicans exhibits signaling responses to cell density (quorum sensing) that regulate growth and probably facilitate its survival in the host [29, 30].
The acquisition of drug resistance is another important component of C. albicans virulence in the clinical setting. It can readily develop resistance to antifungal drug treatment via multiple mechanisms, including regulation of drug efflux transporters (e.g., Cdr1p, Cdr2p, and Mdr1p [31]), mutation of drug targets (e.g., Erg11p, lanosterol 14-alpha-demethylase, the target of azole antifungals [32]), mutation of regulatory factors (e.g., Tac1p, a transcriptional regulator of drug efflux [15]), and chromosomal rearrangements [15, 17, 33].
Candida albicans in the “genomic age”
The genomic sequence of C. albicans strain SC5314 was published in 2004 as an assembly comprising 266 contiguous segments across a haploid reference genome [34]. Subsequently, using these contigs as a starting point, a chromosome-level assembly was constructed [35]. Publication of the sequence served as a springboard to the genomic age [36, 37], and numerous genomic resources have been developed for Candida [38, 39], several of which have been made widely available to the research community (e.g., CandidaDB: http://genodb.pasteur.fr/cgi-bin/WebObjects/CandidaDB, Broad Fungal Genome Initiative: http://www.broad.mit.edu/node/304, Sanger Institute: http://www.sanger.ac.uk/Projects/Fungi/, Genolevures: http://www.genolevures.org/yeastgenomes.html; also see http://www.candidagenome.org/CommunityNews.shtml). The recent availability of genome sequences for related pathogenic and non-pathogenic fungi has enabled the use of comparative genomics approaches to refine the ORF catalog and gene annotation, as well as to examine gene families that have expanded in the pathogenic fungi.
The genome-scale experimental approaches (such as microarrays) that are now available for research into Candida pathogenesis mechanisms can generate large volumes of data, including long lists of genes of interest, providing a significant challenge to make sense of the results. It is therefore vital that gene products are annotated consistently, not only across all gene products within an organism, but also across similar gene products in other (often closely related) organisms. With such annotation, experimental data from many organisms can be leveraged to enable more comprehensive analyses of these large-scale datasets, and to provide biological relevance above and beyond simple phenomenological descriptions. The most comprehensive and most widely used system for organization and classification of gene products that satisfies this criterion is the Gene Ontology (GO) [40].
Gene Ontology at Candida Genome Database
GO annotation provides an at-a-glance summary of the most salient features of a gene product. Applied to the annotation of a large gene set, GO acts as a handle to rapidly retrieve lists of genes that share functions or roles, or whose products reside together in a common cellular location. For example, a Quick Search for “virulence” at Candida Genome Database (CGD, http://www.candidagenome.org/ [41]) retrieves the GO term “pathogenesis” from the Biological Process branch of the ontology, since “virulence” is included in GO as a synonym of “pathogenesis”. The GO Term page at CGD displays a graphical view of the term’s placement in the hierarchy, with its parental and child terms (if it has children), and also lists all of the genes that have been directly annotated with the term, along with the corresponding evidence codes, references from which the information was curated, and links to each Locus Summary page. An example GO Term Page is shown in Figure 2.
Figure 2.

Example GO term page for the Biological Process term “hyphal growth” from the CGD web site (http://www.candidagenome.org/cgi-bin/GO/go.pl?goid=30448, as seen on January 13, 2009). The page displays the definition of the term, with a graphic showing its location within the hierarchy. The page also contains a table that shows the number of annotations within various summary categories, including annotations made from high-throughput and computationally based experiments. A list of all of the annotated genes, along with references and evidence codes, is displayed at the bottom of the page. AmiGO is a web-based tool for searching and browsing the GO, which is provided by the Gene Ontology Consortium and is available at http://amigo.geneontology.org/cgi-bin/amigo/go.cgi. The evidence code IMP indicates that the annotation is “Inferred from Mutant Phenotype.”
The more comprehensive the annotation of an organism’s gene catalog, the easier it is to discover patterns and biological relevance in groups of genes that exhibit common properties in large-scale studies. The concise, structured, and standardized descriptions offered by GO annotation greatly facilitate the computational analysis of lists of gene products of interest. For example, a list of several dozen genes that are determined to be co-regulated in a microarray experiment, or whose products are co-localized, may include genes with seemingly disparate roles, or with completely unknown roles. Analysis of the GO annotations of such a group of genes may reveal significant enrichment of particular biological processes and can therefore generate testable hypotheses, by suggesting that the uncharacterized gene products may also participate in one or more of the enriched processes. Many tools have been created to facilitate grouping and statistical analysis of gene lists based on the GO terms assigned to the genes (e.g., GO::TermFinder [42]; GO Slim Mapper, http://db.yeastgenome.org/cgi-bin/GO/goSlimMapper.pl; and numerous others listed at http://www.geneontology.org/GO.tools.shtml).
Manual GO annotation at CGD
At CGD, GO annotation of C. albicans gene products is manually extracted from the scientific literature by CGD curators and added to the database, using the appropriate evidence code and reference for each annotation. The scientific literature does not typically indicate particular GO terms; rather, experimental data in a publication are used by a curator to determine the appropriate GO terms to use in annotating a gene product. Manual curation therefore requires biological expertise, as well as a working knowledge of GO itself, and the consistent application of a set of curation criteria. As of April 2009, CGD includes 5,860 manually curated GO annotations, collected from 1,176 distinct published references.
Early development of the GO focused on model organisms that are by and large nonpathogenic, such as Saccharomyces cerevisiae and Drosophila melanogaster. Beginning in 2004, a group of researchers formed an interest group called the Plant-Associated Microbe Gene Ontology group (PAMGO), to improve the parts of the GO that relate to interactions between microbes and the hosts that may be beneficial, neutral, or detrimental to either of the partner organisms (http://pamgo.vbi.vt.edu/ [43, 44]). These improvements to the GO have been used heavily in the annotation of C. albicans genes. For example, in CGD, over 200 genes are currently annotated to the term “symbiosis, encompassing mutualism through parasitism” and its child terms in this branch of the Biological Process ontology, which the PAMGO collaborators developed.
The C. albicans gene catalog contains a number of gene families implicated in pathogenicity, for example, agglutinin-like sequence (ALS) genes involved in adhesion to host cells, and genes coding for ferric reductases, secreted aspartyl proteases and secreted lipases [45-48]. The expansion of these gene families has likely been driven by evolutionary pressure to diversify the genes responsible for carrying out functions important for survival in the host. For example, host organisms strictly limit the amount of iron that is available to any pathogen, as an innate defense strategy against infection [49]. The microbial ferric reductases are an important component of the iron acquisition pathways that are essential for survival under the iron-limiting environment of the host [50]. As an apparent consequence of this evolutionary arms race between pathogen and host, it has been observed that the C. albicans genome contains an expanded complement of ferric reductase genes compared to its nonpathogenic cousin, S. cerevisiae [48]. As of January 2009, a total of 15 genes have been demonstrated or predicted to encode ferric reductases in C. albicans, but only 8 in S. cerevisiae (see http://www.candidagenome.org/cgi-bin/GO/go.pl?goid=293 and http://www.yeastgenome.org/cgi-bin/GO/goTerm.pl?goid=293, respectively). Of these 15 C. albicans genes, only two have been functionally characterized by direct experimentation [51-53]; the remaining gene functions have been inferred by similarity-based computational methods, and the experimental follow-up on these predictions is an opportunity for further study in the laboratory. As an example, the similarity of the gene orf19.7077 to ferric reductase genes has been noted, but the actual function and role of this gene has not been explored experimentally (http://www.candidagenome.org/cgi-bin/locus.pl?locus=orf19.7077, accessed on 6 April 2009).
Characterization of the genes within these families has demonstrated that individual family members can fill specialized roles and that different family members show differing regulation of expression under varying conditions [54]. For example, individual members of the family of cell surface glycoproteins encoded by the ALS genes preferentially mediate adherence to distinct ranges of host cell and extracellular matrix substrates, and even the products of a pair of allelic genes have been observed to show differential association with host cells, by virtue of differential regulation or protein sequence [55].
However, in general, not all the members of these important gene families have been experimentally characterized, and it is therefore often inferred from the family membership that all of the closely related members share the same function and role. (At this time, we curate such inferences only as they are published in the scientific literature; we do not make them de novo. In the future, we plan to incorporate predictions that are based on characterized protein domains.) GO annotation provides a concise way to express such inferences, by assigning a shared term that indicates the common function, and then using the GO evidence codes to clearly indicate whether the inference was based on experimental characterization or sequence similarity. However, these predictions based on family membership should be considered to be testable hypotheses about the relatively uncharacterized gene products within a gene family, as sequence similarity does not guarantee identical behavior; indeed, the cases where family members show differences are interesting biologically, and of particular relevance to C. albicans’ pathogenesis.
GO term transfer from S. cerevisiae to C. albicans
Because GO assignment is such a useful “handle” for genes in an annotated genome, and because only a subset of C. albicans genes have been characterized directly, it is both advantageous and necessary to predict functions of the uncharacterized or less-characterized genes. Predictions can be made using GO annotations in conjunction with a set of criteria for transitive term assignment, and with the use of appropriate evidence codes to provide the CGD user community with an at-a-glance indication of how these assignments were made.
The lack of a meiotic cycle, a haploid phase, and stable low-copy episomes in C. albicans make it more laborious to conduct genetic screens and phenotypic analyses than in S. cerevisiae. Direct experimental characterization of C. albicans gene products has therefore been concentrated in areas of virulence-related properties such as morphological switching, infectivity, and drug resistance. In contrast, S. cerevisiae is probably the best characterized model organism, and much is known about many cellular processes that are unlikely to ever be studied directly in C. albicans. In addition, annotation of the S. cerevisiae genome and curation of its scientific literature have been conducted for many years by the Saccharomyces Genome Database (SGD); indeed the Gene Ontology curation at SGD is considered the “gold standard” in the field, and 90% of known S. cerevisiae gene products have at least one manually assigned GO annotation (excluding annotations to "unknown" terms). Thus, there is great utility in leveraging this body of annotation, by interspecies GO term transfer from S. cerevisiae, to round out the annotation of the C. albicans gene catalog (see Figure 3).
Figure 3.
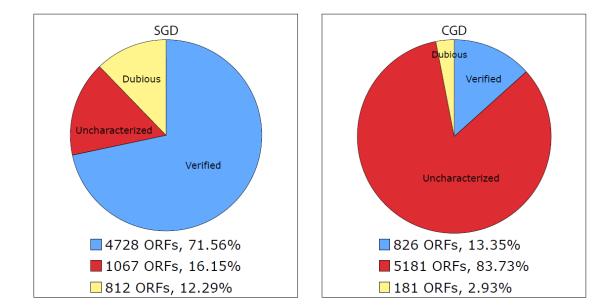
Comparison of C. albicans and S. cerevisiae genome annotation. ORFs are classified as ‘Verified’ if there is experimental evidence for a functional gene product. ‘Uncharacterized’ ORFs are predicted based on sequence analysis, but currently lack experimental characterization. ORFs labeled as ‘Dubious’ have no experimental characterization and appear indistinguishable from random non-coding sequence.
To conduct transitive annotation at CGD, we first determined orthology relationships between C. albicans and S. cerevisiae genes using the InParanoid algorithm, which identifies reciprocal best BLAST hits between species [56]. Candidate GO annotations for transfer from S. cerevisiae to C. albicans were only those with experimental evidence in SGD, with evidence codes of either Inferred from Direct Assay (IDA), Inferred from Physical Interaction (IPI), Inferred from Genetic Interaction (IGI) or Inferred from Mutant Phenotype (IMP), as these are deemed to be the most reliable annotations. Any annotations that were themselves inferred in S. cerevisiae, either based on sequence similarity or by some other algorithm, were excluded from this group in order to avoid propagating predictions. From this candidate set of S. cerevisiae annotations, those that were either redundant with existing manually curated GO annotations in CGD, or redundant with other SGD annotations that were candidates for transfer, were removed. For example, if an annotation to “kinase activity” and an annotation to “protein kinase activity” were candidates for transfer for a particular gene product, only the more specific annotation to “protein kinase activity” was transferred, as the less specific annotation (“kinase activity”) can be inferred from the annotation to its child term (“protein kinase activity”). All annotations that were transferred to CGD by this procedure were given the evidence code IEA (Inferred from Electronic Annotation), and thus identified as being computationally derived. In total, 11,191 new annotations were transferred in April 2008, affecting 3113 genes. An overview of the GO annotations in CGD, highlighting the annotations derived from S. cerevisiae annotations at SGD, is shown in Figure 4. This process is repeated periodically, to take advantage of new GO annotations at SGD, so that C. albicans researchers can continue to derive the maximum benefit from this transitive transfer.
Figure 4.

Statistics of GO term transfer to C. albicans based on S. cerevisiae annotation. Red bars represent the number of C. albicans genes at CGD annotated to the selected GO terms (annotated directly to each term itself, or annotated to one of its more specific child terms) by transferring annotations from orthologous characterized S. cerevisiae genes at SGD. Blue bars represent genes annotated at CGD based on published literature.
This method of transitive annotation is not without intrinsic biases. Firstly, it is important to consider that the pool of annotations available for transfer will exhibit dependence on the organism from which term transfer is initiated, and that the types of processes that are typically studied in any given model organism will affect the well-roundedness of any set of transferred terms. In addition, some organisms simply lack genes or even entire processes that may be of great importance in others, even in the case of reasonably closely related organisms. For example, S. cerevisiae entirely lacks both the NADH dehydrogenase subunits of respiratory complex I and the alternative oxidase (AOX), which are present in C. albicans and other fungi, suggesting that C. albicans is more representative of other fungi in this respect [57-61]. As S. cerevisiae is nonpathogenic under ordinary circumstances, GO term assignments related to virulence processes are not well represented in this organism. For instance, S. cerevisiae completely lacks orthologs of the genes encoding the secreted lipases of C. albicans, a gene family for which possible roles in host colonization and in virulence have been suggested, but not yet conclusively demonstrated [47, 62, 63].
Another caveat to consider is that the accuracy of transfer of these annotations relies on correct orthology designations. We used only sequences from C. albicans and S. cerevisiae to infer orthology (with Caenorhabditis elegans used as an outgroup, to eliminate some false positive predictions), whereas a more comprehensive orthology mapping might use sequences from many related organisms, such as is done in the TreeFam project [64], which is used to build trees of animal genes families. In the future, we will extend our procedure to predict GO term assignments based on orthology to the fission yeast Schizosaccharomyces pombe and more distantly related eukaryotes for which a thorough experimental evidence-based set of GO annotation has been curated. Above all, orthology-based transfer is predicated on the conservation of functions and roles of orthologs since their last common ancestor. In many, if not most cases, this assumption is likely to be correct, especially when the organisms diverged relatively recently. However, we stress that these orthology-based transferred annotations are predictions, and that subsequent experimental analysis is necessary to make a conclusive statement. Experimental follow-up on sequence-based predictions can yield exciting and surprising results, some of which are discussed in detail below.
Rewiring during evolution
C. albicans is very similar to S. cerevisiae in many ways, and early investigations into C. albicans molecular biology often emphasized the commonalities: many C. albicans genes were first cloned by complementation of S. cerevisiae mutations or by cross-hybridization with S. cerevisiae genes. More recent work has demonstrated, though, that C. albicans is not a baker’s yeast that happens to possess extra abilities allowing it to establish commensal and pathogenic interactions with mammalian hosts. Rather, the molecular details of some of the most basic and essential processes have diverged between the two organisms in the approximately 140-850 million years since their evolutionary separation [36]. The elucidation of several cases of transcriptional network “rewiring” has highlighted the capacity of transcriptional networks to evolve rapidly. “Rewiring”, or “reprogramming”, consists of changes in membership among groups of coregulated genes, achieved through addition or removal of upstream transcription factor binding motifs, and/or substitutions in the identities of the transcription factors involved in regulation of a particular process. Thus, two different species may exhibit a similar response to a particular environmental stimulus, but the molecular cast of characters used to achieve the response may be quite different [65]. Because of the phenomenon of rewiring, it can be argued that genetic nomenclature guidelines for C. albicans should be revised (Box 1).
Box 1. Gene naming conventions and functional divergence.
The phenomenon of rewiring may suggest that genetic nomenclature guidelines for C. albicans should be revised. The general rule has been that C. albicans genes should be named for their S. cerevisiae orthologs. However, such names can be misleading where rewiring has occurred: for example, whereas the GAL4 gene in S. cerevisiae is involved in galactose metabolism, C. albicans GAL4) is not involved in galactose metabolism [70]. The Candida research community will need to decide whether priority should be placed on orthology or on biological role, when choosing gene names.
The first name used for each gene in the published literature is entered as its standard gene name in CGD (see http://www.candidagenome.org/Nomenclature.shtml). However, standard gene names can be changed; gene name requests may be initiated by contacting CGD, at candida-curator@genome.stanford.edu. The procedure for gene name change is as follows: CGD will contact each of the other research groups who have published papers concerning the gene, let them know about the name change proposal, and give everyone a chance to voice any objections or concerns. If there is consensus in support of the change, the CGD standard name will be changed; the old name will become an alias that remains permanently associated with the gene.
In perhaps the most dramatic example, the transcriptional regulatory network for genes encoding ribosomal proteins has been significantly rewired in S. cerevisiae and/or C. albicans since their last common ancestor, which is surprising given the essential nature of ribosomal protein expression. In S. cerevisiae, Rap1p, in concert with cofactors that provide specificity, binds upstream of ribosomal protein genes and activates their transcription. In contrast, the C. albicans ortholog of S. cerevisiae Rap1p is not involved in this process; rather, both in C. albicans and in many other Ascomycetes, this task is performed by the ortholog of the S. cerevisiae protein Tbf1p, which is likely the ancestral state [66]. In this case, both the transcription factors involved and the regulatory motifs upstream of the set of regulated genes have diverged in the S. cerevisiae lineage. In another remarkable example, the genes under mating-type control are regulated in opposite ways in the two species, but with the same outcome in terms of gene expression in the ‘a’ and ‘alpha’ mating types; furthermore, C. albicans employs a transcriptional activator in this process, Mtla2p, that has been entirely lost from the S. cerevisiae lineage [67]. Several other instances of divergence in regulatory circuits, and differing roles of orthologous transcription factors, have been uncovered [68-72].
Transcriptional regulation is not the only area in which rewiring has occurred. It has long been an apparent paradox that although the C. albicans genome includes orthologs of a number of S. cerevisiae meiotic genes [73], meiosis has never been observed. Recent work has established that genetic recombination occurs during the parasexual cycle and is mediated by C. albicans Spo11p, whose S. cerevisiae ortholog functions exclusively in meiotic recombination [14]. This raises the possibility that other C. albicans orthologs of meiotic genes may also have roles in the generation of genetic diversity via recombination in the parasexual cycle [14].
While these examples of rewiring do not detract from the overall utility of GO annotation transfer between species, they illustrate some limitations to the approach. The strategy depends on the presence of identifiable orthologs in both species, so it is only applicable to the common set of genes; if an ortholog has been lost from one of the species, for example MTLA2, which is not present in S. cerevisiae, then GO annotation transfer is not possible. In addition, since rewiring is more likely to change the role of a gene product than its biochemical activity, transferred Biological Process annotations may be less reliable than Molecular Function or Cellular Component annotations. Like any GO annotations made on the basis of automated analysis, they should be treated as predictions that can be used to formulate testable hypotheses to be pursued in the laboratory.
Concluding remarks and future directions
Characterization of the complete gene catalog of the fungal pathogen C. albicans, although challenging, is a goal that is likely to bring exciting new insights about fungal biology and pathogenesis. Interspecies transfer of GO annotations presents an opportunity to make a large number of inferences quickly. Being able to round out the catalog of annotated genes, and, in particular, to infer annotations for genes that are unlikely ever to see experimental characterization in the organism of interest, is greatly helpful in analysis of large-scale experiments. It is important to bear in mind that GO annotations transferred on the basis of orthology are predictions and need to be further subject to experimental validation. However, in cases where experimental results do not correspond to sequence-based predictions, there is interesting biology to be explored. Comparison to S. cerevisiae genes is only the first step: there are likely to be as-yet-unrecognized C. albicans genes, which may be either not yet annotated as open reading frames, or annotated but without similarity to any currently characterized genes, that will be revealed as the genome sequence is refined and as the genes of other organisms, particularly those of other fungal pathogens, are experimentally characterized [74]. In addition, recent microarray and transcriptome sequencing studies have revealed on the order of a thousand previously unknown transcripts in S. cerevisiae [75-78], about which almost nothing is known, and many of which show little or no conservation with the corresponding syntenic regions in closely related species [76, 79]. It is expected that such studies will reveal novel genes in C. albicans, many of which are likely on the forefront of C. albicans’ evolutionary arms race with its host, and intimately involved in pathogenesis. The ongoing annotation of the C. albicans genome affords opportunities to facilitate laboratory research into fungal pathogenesis (Box 2), with the exciting potential that new discoveries may unlock molecular secrets that will allow us to better tame the organism’s pathogenic ways to benefit human patients suffering from Candida disease.
Box. Questions for future research.
What can comparative genomics tell us about Candida pathogenesis genes and possible antifungal targets?
Can we prevent the acquisition of antifungal drug resistance by Candida during infection of human patients?
Can the fungus be “locked” into a commensal, avirulent state in the host?
What are the explicit biological requirements for clinical Candida virulence?
How can the host response be made more effective against systemic Candida infection?
How has the relationship between Candida and its hosts developed over the course of evolution?
Acknowledgements
The Candida Genome Database staff thanks our close collaborators, Mike Cherry and the other members of the Saccharomyces Genome Database Group. CGD is supported by NIH grant R01 DE015873 from the NIDCR at the NIH.
References
- 1.Pfaller MA, Diekema DJ. Epidemiology of invasive candidiasis: a persistent public health problem. Clinical microbiology reviews. 2007;20:133–163. doi: 10.1128/CMR.00029-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schelenz S. Management of candidiasis in the intensive care unit. The Journal of antimicrobial chemotherapy. 2008;61(Suppl 1):i31–34. doi: 10.1093/jac/dkm430. [DOI] [PubMed] [Google Scholar]
- 3.d'Enfert C. Biofilms and their role in the resistance of pathogenic Candida to antifungal agents. Current drug targets. 2006;7:465–470. doi: 10.2174/138945006776359458. [DOI] [PubMed] [Google Scholar]
- 4.Lewis K. Multidrug tolerance of biofilms and persister cells. Current topics in microbiology and immunology. 2008;322:107–131. doi: 10.1007/978-3-540-75418-3_6. [DOI] [PubMed] [Google Scholar]
- 5.Mean M, et al. Bench-to-bedside review: Candida infections in the intensive care unit. Critical care (London, England) 2008;12:204. doi: 10.1186/cc6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wisplinghoff H, et al. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. 2004;39:309–317. doi: 10.1086/421946. [DOI] [PubMed] [Google Scholar]
- 7.Traeder C, et al. Candida infection in HIV positive patients 1985-2007. Mycoses. 2008;51(Suppl 2):58–61. doi: 10.1111/j.1439-0507.2008.01574.x. [DOI] [PubMed] [Google Scholar]
- 8.Lai CC, et al. Current challenges in the management of invasive fungal infections. J Infect Chemother. 2008;14:77–85. doi: 10.1007/s10156-007-0595-7. [DOI] [PubMed] [Google Scholar]
- 9.Bennett RJ, Johnson AD. Mating in Candida albicans and the search for a sexual cycle. Annual review of microbiology. 2005;59:233–255. doi: 10.1146/annurev.micro.59.030804.121310. [DOI] [PubMed] [Google Scholar]
- 10.Magee PT, Magee BB. Through a glass opaquely: the biological significance of mating in Candida albicans. Current opinion in microbiology. 2004;7:661–665. doi: 10.1016/j.mib.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen K, Heitman J. Sex and virulence of human pathogenic fungi. Advances in genetics. 2007;57:143–173. doi: 10.1016/S0065-2660(06)57004-X. [DOI] [PubMed] [Google Scholar]
- 12.Soll DR. Mating-type locus homozygosis, phenotypic switching and mating: a unique sequence of dependencies in Candida albicans. Bioessays. 2004;26:10–20. doi: 10.1002/bies.10379. [DOI] [PubMed] [Google Scholar]
- 13.Bennett RJ, Johnson AD. Completion of a parasexual cycle in Candida albicans by induced chromosome loss in tetraploid strains. The EMBO journal. 2003;22:2505–2515. doi: 10.1093/emboj/cdg235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forche A, et al. The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS biology. 2008;6:e110. doi: 10.1371/journal.pbio.0060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coste A, et al. Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryotic cell. 2007;6:1889–1904. doi: 10.1128/EC.00151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rustchenko E. Chromosome instability in Candida albicans. FEMS yeast research. 2007;7:2–11. doi: 10.1111/j.1567-1364.2006.00150.x. [DOI] [PubMed] [Google Scholar]
- 17.Selmecki A, et al. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science (New York, N.Y. 2006;313:367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sudbery P, et al. The distinct morphogenic states of Candida albicans. Trends in microbiology. 2004;12:317–324. doi: 10.1016/j.tim.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 19.Whiteway M, Bachewich C. Morphogenesis in Candida albicans. Annual review of microbiology. 2007;61:529–553. doi: 10.1146/annurev.micro.61.080706.093341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramirez-Zavala B, et al. Environmental induction of white-opaque switching in Candida albicans. PLoS pathogens. 2008;4:e1000089. doi: 10.1371/journal.ppat.1000089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saville SP, et al. Use of a genetically engineered strain to evaluate the pathogenic potential of yeast cell and filamentous forms during Candida albicans systemic infection in immunodeficient mice. Infection and immunity. 2008;76:97–102. doi: 10.1128/IAI.00982-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumamoto CA, Vinces MD. Contributions of hyphae and hypha-co-regulated genes to Candida albicans virulence. Cellular microbiology. 2005;7:1546–1554. doi: 10.1111/j.1462-5822.2005.00616.x. [DOI] [PubMed] [Google Scholar]
- 23.Whiteway M, Oberholzer U. Candida morphogenesis and host-pathogen interactions. Current opinion in microbiology. 2004;7:350–357. doi: 10.1016/j.mib.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 24.Lorenz MC, et al. Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryotic cell. 2004;3:1076–1087. doi: 10.1128/EC.3.5.1076-1087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcil A, et al. Analysis of PRA1 and its relationship to Candida albicans-macrophage interactions. Infection and immunity. 2008;76:4345–4358. doi: 10.1128/IAI.00588-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blankenship JR, Mitchell AP. How to build a biofilm: a fungal perspective. Current opinion in microbiology. 2006;9:588–594. doi: 10.1016/j.mib.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Mukherjee PK, et al. Candida biofilm: a well-designed protected environment. Med Mycol. 2005;43:191–208. doi: 10.1080/13693780500107554. [DOI] [PubMed] [Google Scholar]
- 28.Nobile CJ, Mitchell AP. Genetics and genomics of Candida albicans biofilm formation. Cellular microbiology. 2006;8:1382–1391. doi: 10.1111/j.1462-5822.2006.00761.x. [DOI] [PubMed] [Google Scholar]
- 29.Hogan DA. Talking to themselves: autoregulation and quorum sensing in fungi. Eukaryotic cell. 2006;5:613–619. doi: 10.1128/EC.5.4.613-619.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nickerson KW, et al. Quorum sensing in dimorphic fungi: farnesol and beyond. Applied and environmental microbiology. 2006;72:3805–3813. doi: 10.1128/AEM.02765-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akins RA. An update on antifungal targets and mechanisms of resistance in Candida albicans. Med Mycol. 2005;43:285–318. doi: 10.1080/13693780500138971. [DOI] [PubMed] [Google Scholar]
- 32.Lupetti A, et al. Molecular basis of resistance to azole antifungals. Trends in molecular medicine. 2002;8:76–81. doi: 10.1016/s1471-4914(02)02280-3. [DOI] [PubMed] [Google Scholar]
- 33.Selmecki A, et al. An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Molecular microbiology. 2008;68:624–641. doi: 10.1111/j.1365-2958.2008.06176.x. [DOI] [PubMed] [Google Scholar]
- 34.Jones T, et al. The diploid genome sequence of Candida albicans. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7329–7334. doi: 10.1073/pnas.0401648101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van het Hoog M, et al. Assembly of the Candida albicans genome into sixteen supercontigs aligned on the eight chromosomes. Genome biology. 2007;8:R52. doi: 10.1186/gb-2007-8-4-r52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berman J, Sudbery PE. Candida albicans: a molecular revolution built on lessons from budding yeast. Nature reviews. 2002;3:918–930. doi: 10.1038/nrg948. [DOI] [PubMed] [Google Scholar]
- 37.Odds FC, et al. Candida albicans genome sequence: a platform for genomics in the absence of genetics. Genome biology. 2004;5:230. doi: 10.1186/gb-2004-5-7-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryotic cell. 2005;4:298–309. doi: 10.1128/EC.4.2.298-309.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Noble SM, Johnson AD. Genetics of Candida albicans, a diploid human fungal pathogen. Annual review of genetics. 2007;41:193–211. doi: 10.1146/annurev.genet.41.042007.170146. [DOI] [PubMed] [Google Scholar]
- 40.The Gene Ontology Consortium The Gene Ontology project in 2008. Nucleic acids research. 2008;36:D440–444. doi: 10.1093/nar/gkm883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnaud MB, et al. Sequence resources at the Candida Genome Database. Nucleic acids research. 2007;35:D452–456. doi: 10.1093/nar/gkl899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boyle EI, et al. GO::TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics (Oxford, England) 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Collmer CW, et al. Gene Ontology (GO) for Microbe–Host Interactions and Its Use in Ongoing Annotation of Three Pseudomonas syringae Genomes via the Pseudomonas –Plant Interaction (PPI) Web Site. In: Fatmi MB, et al., editors. In Pseudomonas syringae Pathovars and Related Pathogens – Identification, Epidemiology and Genomics. Springer; 2008. [Google Scholar]
- 44.The Gene Ontology Consortium The Gene Ontology (GO) project in 2006. Nucleic acids research. 2006;34:D322–326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoyer LL, et al. Discovering the secrets of the Candida albicans agglutinin-like sequence (ALS) gene family--a sticky pursuit. Med Mycol. 2008;46:1–15. doi: 10.1080/13693780701435317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Naglik J, et al. Candida albicans proteinases and host/pathogen interactions. Cellular microbiology. 2004;6:915–926. doi: 10.1111/j.1462-5822.2004.00439.x. [DOI] [PubMed] [Google Scholar]
- 47.Schaller M, et al. Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses. 2005;48:365–377. doi: 10.1111/j.1439-0507.2005.01165.x. [DOI] [PubMed] [Google Scholar]
- 48.Baek YU, et al. Candida albicans ferric reductases are differentially regulated in response to distinct forms of iron limitation by the Rim101 and CBF transcription factors. Eukaryotic cell. 2008;7:1168–1179. doi: 10.1128/EC.00108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bullen JJ, et al. Natural resistance, iron and infection: a challenge for clinical medicine. Journal of medical microbiology. 2006;55:251–258. doi: 10.1099/jmm.0.46386-0. [DOI] [PubMed] [Google Scholar]
- 50.Howard DH. Iron gathering by zoopathogenic fungi. FEMS immunology and medical microbiology. 2004;40:95–100. doi: 10.1016/S0928-8244(03)00301-8. [DOI] [PubMed] [Google Scholar]
- 51.Hammacott JE, et al. Candida albicans CFL1 encodes a functional ferric reductase activity that can rescue a Saccharomyces cerevisiae fre1 mutant. Microbiology (Reading, England) 2000;146:869–876. doi: 10.1099/00221287-146-4-869. Pt 4. [DOI] [PubMed] [Google Scholar]
- 52.Knight SA, et al. Reductive iron uptake by Candida albicans: role of copper, iron and the TUP1 regulator. Microbiology (Reading, England) 2002;148:29–40. doi: 10.1099/00221287-148-1-29. [DOI] [PubMed] [Google Scholar]
- 53.Knight SA, et al. Iron acquisition from transferrin by Candida albicans depends on the reductive pathway. Infection and immunity. 2005;73:5482–5492. doi: 10.1128/IAI.73.9.5482-5492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumamoto CA. Niche-specific gene expression during C. albicans infection. Current opinion in microbiology. 2008;11:325–330. doi: 10.1016/j.mib.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao X, et al. Unequal contribution of ALS9 alleles to adhesion between Candida albicans and human vascular endothelial cells. Microbiology (Reading, England) 2007;153:2342–2350. doi: 10.1099/mic.0.2006/005017-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O'Brien KP, et al. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic acids research. 2005;33:D476–480. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helmerhorst EJ, et al. Characterization of the mitochondrial respiratory pathways in Candida albicans. Biochimica et biophysica acta. 2002;1556:73–80. doi: 10.1016/s0005-2728(02)00308-0. [DOI] [PubMed] [Google Scholar]
- 58.Huh WK, Kang SO. Molecular cloning and functional expression of alternative oxidase from Candida albicans. Journal of bacteriology. 1999;181:4098–4102. doi: 10.1128/jb.181.13.4098-4102.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huh WK, Kang SO. Characterization of the gene family encoding alternative oxidase from Candida albicans. The Biochemical journal. 2001;356:595–604. doi: 10.1042/0264-6021:3560595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McDonough JA, et al. Involvement of Candida albicans NADH dehydrogenase complex I in filamentation. Fungal Genet Biol. 2002;36:117–127. doi: 10.1016/S1087-1845(02)00007-5. [DOI] [PubMed] [Google Scholar]
- 61.Nosek J, Fukuhara H. NADH dehydrogenase subunit genes in the mitochondrial DNA of yeasts. Journal of bacteriology. 1994;176:5622–5630. doi: 10.1128/jb.176.18.5622-5630.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hube B, et al. Secreted lipases of Candida albicans: cloning, characterisation and expression analysis of a new gene family with at least ten members. Archives of microbiology. 2000;174:362–374. doi: 10.1007/s002030000218. [DOI] [PubMed] [Google Scholar]
- 63.Schofield DA, et al. Differential Candida albicans lipase gene expression during alimentary tract colonization and infection. FEMS microbiology letters. 2005;244:359–365. doi: 10.1016/j.femsle.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 64.Li H, et al. TreeFam: a curated database of phylogenetic trees of animal gene families. Nucleic acids research. 2006;34:D572–580. doi: 10.1093/nar/gkj118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scannell DR, Wolfe K. Rewiring the transcriptional regulatory circuits of cells. Genome biology. 2004;5:206. doi: 10.1186/gb-2004-5-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hogues H, et al. Transcription factor substitution during the evolution of fungal ribosome regulation. Molecular cell. 2008;29:552–562. doi: 10.1016/j.molcel.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tsong AE, et al. Evolution of alternative transcriptional circuits with identical logic. Nature. 2006;443:415–420. doi: 10.1038/nature05099. [DOI] [PubMed] [Google Scholar]
- 68.Ihmels J, et al. Rewiring of the yeast transcriptional network through the evolution of motif usage. Science (New York, N.Y. 2005;309:938–940. doi: 10.1126/science.1113833. [DOI] [PubMed] [Google Scholar]
- 69.Kadosh D, Johnson AD. Rfg1, a protein related to the Saccharomyces cerevisiae hypoxic regulator Rox1, controls filamentous growth and virulence in Candida albicans. Molecular and cellular biology. 2001;21:2496–2505. doi: 10.1128/MCB.21.7.2496-2505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martchenko M, et al. Transcriptional rewiring of fungal galactose-metabolism circuitry. Curr Biol. 2007;17:1007–1013. doi: 10.1016/j.cub.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nicholls S, et al. Msn2- and Msn4-like transcription factors play no obvious roles in the stress responses of the fungal pathogen Candida albicans. Eukaryotic cell. 2004;3:1111–1123. doi: 10.1128/EC.3.5.1111-1123.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ramsdale M, et al. MNL1 regulates weak acid-induced stress responses of the fungal pathogen Candida albicans. Molecular biology of the cell. 2008;19:4393–4403. doi: 10.1091/mbc.E07-09-0946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tzung KW, et al. Genomic evidence for a complete sexual cycle in Candida albicans. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3249–3253. doi: 10.1073/pnas.061628798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Butler G, et al. Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature. 2009 doi: 10.1038/nature08064. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.David L, et al. A high-resolution map of transcription in the yeast genome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5320–5325. doi: 10.1073/pnas.0601091103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee A, et al. Novel low abundance and transient RNAs in yeast revealed by tiling microarrays and ultra high-throughput sequencing are not conserved across closely related yeast species. PLoS Genet. 2008;4:e1000299. doi: 10.1371/journal.pgen.1000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miura F, et al. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:17846–17851. doi: 10.1073/pnas.0605645103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nagalakshmi U, et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science (New York, N.Y. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li QR, et al. Revisiting the Saccharomyces cerevisiae predicted ORFeome. Genome research. 2008;18:1294–1303. doi: 10.1101/gr.076661.108. [DOI] [PMC free article] [PubMed] [Google Scholar]