

Abstract
The localization and signaling of S-palmitoylated peripheral membrane proteins is sustained by an acylation cycle in which acyl protein thioesterases (APTs) depalmitoylate mislocalized palmitoylated proteins on endomembranes. However, the APTs are themselves reversibly S-palmitoylated, which localizes thioesterase activity to the site of the antagonistc palmitoylation activity on the Golgi. Here, we resolve this conundrum by showing that palmitoylation of APTs is labile due to autodepalmitoylation, creating two interconverting thioesterase pools: palmitoylated APT on the Golgi and depalmitoylated APT in the cytoplasm, with distinct functionality. By imaging APT-substrate catalytic intermediates, we show that it is the depalmitoylated soluble APT pool that depalmitoylates substrates on all membranes in the cell, thereby establishing its function as release factor of mislocalized palmitoylated proteins in the acylation cycle. The autodepalmitoylating activity on the Golgi constitutes a homeostatic regulation mechanism of APT levels at the Golgi that ensures robust partitioning of APT substrates between the plasma membrane and the Golgi.
Introduction
The acylation cycle is a dynamic reaction-diffusion system that maintains the spatial organization of palmitoylated peripheral membrane proteins, which include proto-oncogene products of the Ras and Src families as well as signal transducers of the heterotrimeric G-protein family (1). In addition to reversible palmitoylation, these proteins typically also obtain an irreversible lipid modification such as S-prenylation (2) or N-myristoylation (3). Therefore, the depalmitoylated forms of these proteins still possess a weak membrane affinity that allows them to rapidly equilibrate to all membranes in the cell. This equilibration is facilitated by interaction with GDI-like solubilizing factors (GSFs) (4). S-palmitoylation of membrane-proximal cysteine residues leads to an increase in the hydrophobicity and hence the membrane affinity of peripheral membrane proteins. The cytoplasmic face of the Golgi apparatus is known to possess S-palmitoyltransferase activity, thus enriching palmitoylatable peripheral membrane proteins on this membrane compartment by kinetic trapping (5,6). Vesicular transport along the secretory pathway directs this protein enrichment to the plasma membrane (PM), thereby transferring the nonequilibrium Golgi enrichment to the PM. Palmitoylated PM-associated proteins may spontaneously dissociate or be redistributed to the endomembrane system due to membrane fission processes such as endocytosis. Thus, S-palmitoylated proteins enriched at the PM will eventually equilibrate to a random distribution on all membranes in the cell that is indistinguishable from their nonpalmitoylated forms (1,7). However, widespread thioesterase activity in the cell depalmitoylates these proteins on endomembranes, thereby converting slowly diffusing, randomly distributed (mislocalized) palmitoylated proteins to their rapidly diffusing unpalmitoylated forms, allowing them to get efficiently (re-)trapped and concentrated at the Golgi apparatus by repalmitoylation (1,4,7). It is therefore the palmitoylation trap at the Golgi that maintains the out-of-equilibrium distribution of palmitoylated peripheral membrane proteins. The acylation cycle has been shown to be a constitutive and general correction mechanism that counters equilibration (mislocalization) over all endomembranes and generates the dynamic PM/Golgi localization of several palmitoylated peripheral membrane proteins (1). Although additional nuances in spatial localization can be conferred by additional modifications or domains in these proteins, perturbation of the acylation cycle typically results in disruption of the native localization of palmitoylated peripheral membrane proteins. For example, inhibition of the depalmitoylation activity restricts these proteins to a randomized aspecific distribution over endomembranes in their palmitoylated state (7). This prevents enrichment on the Golgi, causing subsequent loss of specific PM localization and suppression of oncogenic signaling by palmitoylated Ras family proteins. This critical depalmitoylation has been shown to be performed by the thioesterases acyl protein thioesterase 1 (APT1) and its homolog, APT2, in cells (7–10), but little is known about how thioesterase activity is regulated to maintain the localization of palmitoylated substrates on the PM. APT activity must be tightly regulated in the cell; too much activity will cause depalmitoylated substrates to equilibrate to all endomembranes, whereas too little will cause the same randomized distribution of palmitoylated substrates. Although alternative regulatory mechanisms based on inactivation of APTs by dimerization (11) have been suggested, biochemical labeling experiments have shown APT1/2 to be palmitoylated (12). Recently, it was also reported that APT1/2 are palmitoylated at Cys-2, and that APT1 is capable of depalmitoylating itself as well as APT2 (13). This raises the possibility that an acylation cycle on APTs could also affect thioesterase activity within the cell. We therefore investigated the dynamic mechanisms that regulate thioesterase activity in cellular space, and what role these mechanisms play in maintaining palmitoylated peripheral membrane proteins in specific membrane compartments.
Materials and Methods
For a description of the molecular and cell biology procedures used in this work, see Supporting Material.
Confocal imaging
Confocal laser scanning microscopy was performed on a Leica TCS SP5 DMI6000 equipped with an HCX PL APO 63× 1.4-.6 NA Blau CS objective and an environment-control chamber maintained at 37°C and 5% CO2.
mCerulean, mCitrine, and mCherry fluorescence was excited using 458 nm and 514 nm argon laser lines and a 561 nm HeNe laser, respectively. Detection of fluorescence emission was restricted with an Acousto-Optical Beam Splitter (AOBS) as follows: mCerulean, 468–504 nm; mCitrine, 524–551 nm; and mCherry, 571–650 nm. Scanning was performed in line-by-line sequential mode with 2× line averaging. Confocality was controlled by limiting the pinhole size between 1.0 and 1.5 Airy units.
Fluorescence recovery after photobleaching imaging and quantification
Fluorescence recovery after photobleaching (FRAP) experiments were carried out at 37°C on a Leica SP5 confocal microscope with settings similar to those described above, typically with a 63× oil immersion objective. Cells were allowed to equilibrate in the environment-control chamber on the microscope. A custom routine was set up that included the three steps of FRAP analysis: 1), prebleach imaging; 2), bleaching; and 3), postbleach imaging.
For a detailed description of the FRAP experiments and analysis, see Supporting Material.
Förster resonance energy transfer/fluorescence-lifetime imaging microscopy-based enzyme-substrate imaging of APT activity
Fluorescence lifetime imaging microscopy (FLIM) experiments were carried out at 37°C on a Leica SP5 or Olympus FV1000 confocal microscope equipped with Picoquant FLIM modules (LSM Upgrade Kit/SMD Module; Picoquant). On the Leica SP5, mCitrine fluorescence was excited at 514 nm with a pulsed supercontinuum laser with a repetition rate of 79.2 MHz. Fluorescence signal was collected as described above. On the Olympus FV1000, a pulsed 470 nm diode laser (LDH 470; Picoquant) with a repetition rate of 40 MHz was used. Fluorescence signal was collected through an oil immersion objective (×60/1.35 UPlanSApo (Olympus) or HCX PL APO 63x 1.4-.6 NA Blau CS (Leica)) and spectrally filtered using a narrow-band emission filter (HQ 525/15; Chroma).
Photons from the mCitrine channel were detected using avalanche photodiodes (APDs) and a time-correlated single-photon counting (TCSPC) module (PicoHarp 300; Picoquant).
To obtain the molar fraction of enzyme substrate over total enzyme (ES/ET) of APT-mCitrine (Förster resonance energy transfer (FRET) donor) in complex with the substrate Gα (1-11)-mCherry or APT1/2-mCherry (FRET acceptors) (14), FLIM data sets were analyzed as previously described (15).
To estimate the extent of ES complexes on membranes, the fraction of pixels per cell with an ES/ET above a certain threshold was computed. This threshold was determined as the mean + 2 standard deviations (SDs) of ES/ET values in the APT substrate-free nucleus of the cell.
For a detailed description of the FLIM experiments and analysis, see Supporting Material.
Metabolic labeling, click chemistry, and western blotting to detect palmitoylation
To detect palmitoylation of APT proteins, we performed metabolic labeling with 15-hexadecynoic acid (15-HDYA) in MDCK cells expressing His-tagged APT wild-type (WT) or APTC2S. 15-HDYA is a palmitate analog with an ω-alkyne group. The alkyne group can be utilized for Huisgen azide-alkyne 1,3-dipolar cycloaddition with biotin azide, allowing covalent labeling of palmitoylated proteins with biotin.
For a more detailed description, see Supporting Material.
Results
APT palmitoylation generates a cytoplasmic and Golgi pool of thioesterase activity
Ectopically expressed, fluorescently tagged APT1-mCitrine and APT2-mCitrine showed a characteristic diffuse cytoplasmic localization (16). In addition to this large cytoplasmic fraction, both proteins also exhibited enrichment on the Golgi apparatus, reminiscent of the localization of their palmitoylated substrates (Fig. 1, A–C). This distribution indicates that APT1/2 are palmitoylated and may be substrates of their own depalmitoylating activity. Bioinformatics analysis with CSS-PALM 3.0 (17) showed that the conserved Cys residue at position 2 (Fig. S1) of both proteins has a high probability of being palmitoylated. In line with these observations, APTs were recently reported to be palmitoylated at the Cys residue at position 2 (13). To determine the connection between this conserved Cys residue and localization of APTs in the cell, we compared the localization of WT APT1/2-mCitrine and Cys2-to-Ser point-mutated APT1/2-C2S-mCitrine (Fig. 1, A–C). WT APTs localized to the Golgi as well as the cytosol, whereas the C2S mutation abolished Golgi enrichment. APTs do not possess any irreversible lipid modification such as prenylation or myristoylation, and therefore unpalmitoylated APTs are entirely cytosolic. Localization of WT APTs on the Golgi apparatus was also lost when cells were treated with 2-bromopalmitate, a general inhibitor of palmitoylation and palmitate synthesis (18), again leaving the APTs cytosolic. In contrast to their palmitoylated substrates, no enrichment of APTs on the PM was observed. However, specific inhibition of cellular thioesterase activity using Palmostatin B results in stabilization of APT palmitoylation and has been reported to result in a subsequent localization of APTs to the PM (13). This temporary and nonphysiological localization can arise from the stabilized palmitoylated state of APT, which interacts long enough with membranes to be transported from the Golgi via vesicular transport (Fig. 1 C; see also Fig. 3). However, prolonged thioesterase inhibition by Palmostatin B leads to stabilization of APT palmitoylation, resulting in an entropic equilibration to an aspecific redistribution of APTs to all cellular membranes, as is characteristic for palmitoylated proteins when the acylation cycle is interrupted (7). Some residual enrichment of APTs that remains at the Golgi apparatus may be attributed to residual cycling due to incomplete inhibition of thioesterase activity or weak interactions with Golgi-resident proteins. Using metabolic labeling with 15-hexadecynoic acid followed by click chemistry (19) to detect palmitoylation (Fig. 1 D), we confirmed that APTs, but not APTC2S mutants, are indeed palmitoylated. We therefore conclude that APT1/2 are palmitoylated at the Cys residue at position 2, and that this is the primary cause of APT enrichment on the Golgi. Changes in its localization upon thioesterase inhibitor or palmitoylation inhibitor treatment suggest that this palmitoylation is dynamic and APTs undergo an acylation cycle that is responsible for the enrichment of these proteins on the Golgi apparatus.
Figure 1.
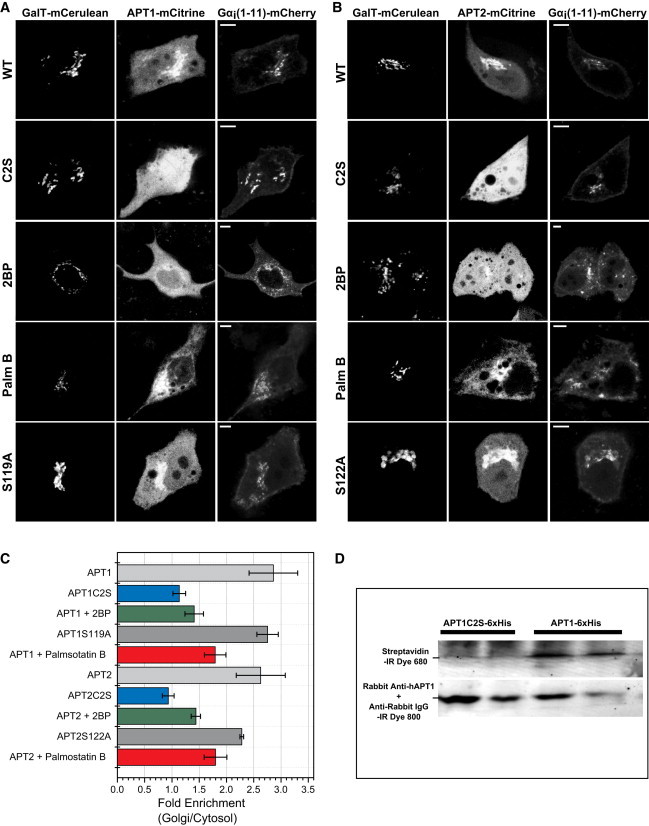
Localization of APT1-mCitrine and APT2-mCitrine in live cells shows a cytosolic pool and significant Golgi enrichment. GalT-mCerulean fluorescence marks the Golgi apparatus. Gα (1-11)-mCherry is an N-terminal 11-amino-acid fragment of Gαi1, a palmitoylated substrate of APTs. (A) Localization of WT APT1-mCitrine (WT, top row), Cys2-to-Ser mutant APT1C2S-mCitrine (C2S, second row), APT1-mCitrine under inhibition of palmitoylation with 2-bromopalmitate (2BP, third row), APT1-mCitrine under inhibition of thioesterase activity with Palmostatin B (Palm B, fourth row), and catalytically inactive Ser119-to-Ala mutant APT1S119A-mCitrine (S119A, bottom row). (B) Same as in A, with APT2-mCitrine and catalytically inactive Ser122-to-Ala APT2S122A-mCitrine mutant (S122A, bottom row). Scale bars: 10 μm. (C) Quantification of the ratio of intensities of APT1/2-mCitrine on the Golgi versus cytosol (n = 10 each) exemplified in A and B. (D) Western blot showing palmitoylation of His-tagged APT1, but not APT1C2S mutant, as detected by streptavidin binding to a biotinylated palmitate analog (upper row). APT1 was detected with an anti-hAPT1 antibody (lower row).
Figure 3.
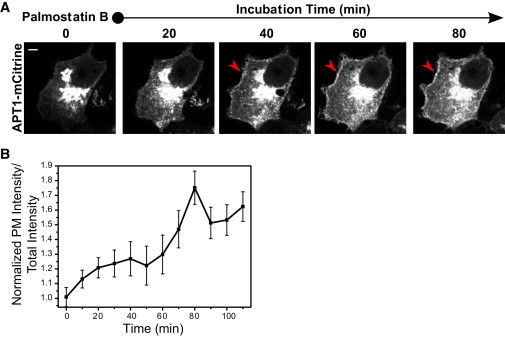
(A) APT localization depends on thioesterase activity. Time-lapse imaging of APT1-mCitrine localization upon Palmostatin B-mediated thioesterase inhibition. Red arrows show PM localization. (B) Graph shows quantification of fold change in APT1-mCitrine intensity at the PM over Palmostatin B incubation time (min). Error bars denote SEM for (n = 5). To see this figure in color, go online.
Golgi and cytoplasmic APT pools interconvert rapidly due to labile palmitoylation
To ascertain the reversibility of APT palmitoylation, we performed FRAP studies on APT1/2-mCitrine on the Golgi apparatus. APT1/2-mCitrine fluorescence intensity recovered rapidly on the Golgi apparatus after photobleaching (Fig. 2, A–E). APT localization to the Golgi is therefore dynamic, suggesting reversible S-palmitoylation as its cause. The fluorescence recovery times did not show any dependence on the total fluorescence intensity of ectopically expressed APT1-mCitrine or coexpressed APT1-mCherry, indicating that APT acylation dynamics on the Golgi apparatus are robust with respect to varying expression levels of total thioesterase activity (Fig. 2, D). However, since the APTs themselves are thioesterases, ectopic expression of WT APTs undoubtedly alters the gross thioesterase activity in the cell, and as such, our measurements may not reflect the fraction of palmitoylated protein as determined by the endogenous thioesterase activity. Therefore, catalytically inactive mutants APT1(S119A) and APT2(S122A) were expressed to serve as tracers for APT1 palmitoylation and localization without altering thioesterase activity in the cell.
Figure 2.
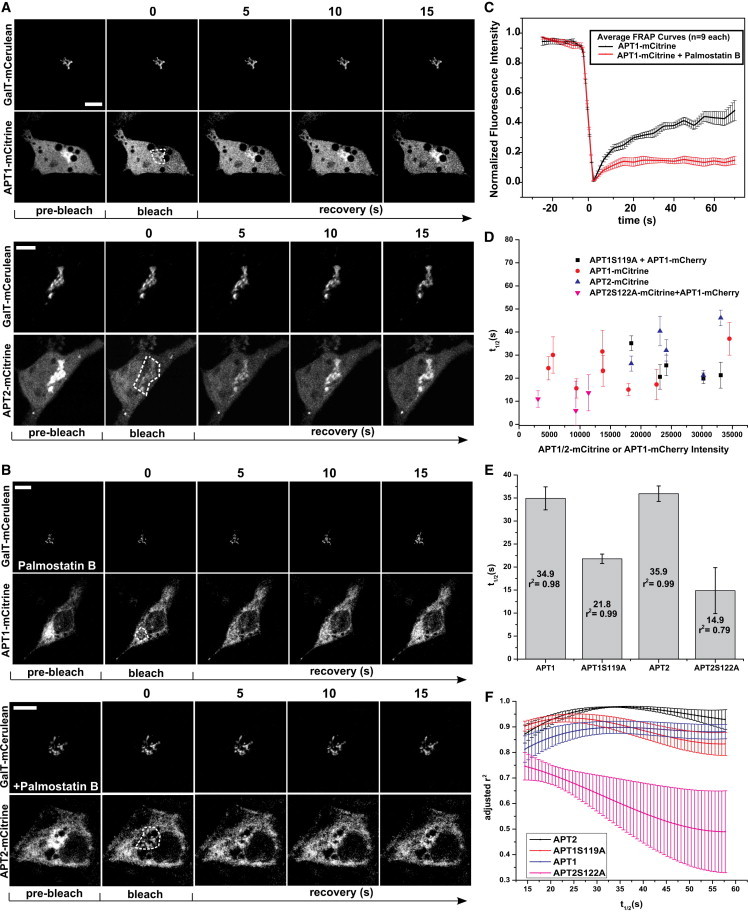
Dynamic maintenance of APTs on the Golgi. (A) Time-lapse sequence of APT1-mCitrine (top) and APT2-mCitrine (bottom) fluorescence recovery after photobleaching on the Golgi. GalT-mCerulean fluorescence marks the Golgi apparatus. Prebleach indicates the images acquired before bleaching. Bleach indicates the images acquired directly after photobleaching in the region of interest (white dashes). Time (s) after photobleaching is indicated above the images. (B) Same as in A, except under inhibition of depalmitoylation activity with 5 μM Palmostatin B. Scale bars: 10 μm. (C) Averaged Golgi FRAP recovery curves with SEM for APT1-mCitrine in cells with (n = 9) and without (n = 9) Palmostatin B treatment. (D) Half times (t1/2) of fluorescence recovery for APTs and their catalytically inactive mutants with respect to intensity of APT1/2-mCitrine or coexpressed APT1-mCherry. (E) Half times of fluorescence recovery on Golgi for APTs and their catalytically inactive mutants. Error bars are SDs as derived from searching error space in the global fit; r2 is the coefficient of determination indicating goodness-of-fit. (F) Rigorous error analysis of fits spanning a range of t1/2 (s) values for APT recovery on the Golgi. Error bars represent SD of the goodness-of-fit (r2) for multiple cells.
The localization of APT1(S119A) and APT2(S122A) was similar to that of WT APTs (Fig. 1, A and B). Therefore, changes in gross cellular thioesterase activity do not have a significant effect on APT localization. However, the catalytically inactive APTs showed significantly reduced fluorescence recovery times on the Golgi apparatus as compared to their WT counterparts (Fig. 2, E and F). Fluorescence recovery times on the Golgi are determined by the balance between palmitoylation and depalmitoylation activities that would be slowed by any autodepalmitoylating activity of Golgi-localized APTs. The enhanced thioesterase activity upon ectopic expression of WT APT1/2 thus leads to an apparent retardation of fluorescence recovery after photobleaching, and the faster recovery of catalytically inactive mutants indicates that APT1/2 autodepalmitoylate on the Golgi.
Consistent with these results, inhibition of APT catalytic activity by Palmostatin B blocks fluorescence recovery of APT1/2-mCitrine after photobleaching on the Golgi (Fig. 2, B and C). The redistribution of APTs to endomembranes after inhibition of thioesterase activity occurs over an hour (7), whereas the data presented here show that palmitate turnover on APTs is on the order of seconds in the presence of thioesterase activity. Therefore, the average residence time of APTs on membranes is too short to generate any substantial PM or endomembrane enrichment, in agreement with localization studies in which no such enrichment was detected in confocal micrographs of ectopically expressed APTs (Fig. 1). The nonphysiological PM localization can be transiently induced by inhibition of thioesterase activity using Palmostatin B to stabilize the palmitoylated state of APTs on membranes long enough for them to be transported from the Golgi via vesicular transport to the PM (Fig. 3, A and B). After prolonged thioesterase inhibition, the palmitoylated APTs eventually equilibrate to an aspecific distribution to all cellular membranes (Fig. 1, A and B), as is characteristic for palmitoylated proteins when the acylation cycle is interrupted (7).
The steady-state partitioning in the Golgi and cytosol of APTs is similar and seemingly unaffected by the levels of total thioesterase activity in the cell. Therefore, the rate of recruitment and rate of loss of APTs from the Golgi compartment must be maintained in proportion. The negative feedback on thioesterase activity due to autodepalmitoylation of APTs on the Golgi could be a mechanism to maintain such proportionality. In this scenario, the apparent rate of fluorescence recovery for a given APT species would appear to be independent of the total thioesterase activity in the cell but dependent on the Golgi localized thioesterase activity, consistent with our observations.
APTs autodepalmitoylate on the Golgi, generating a homeostatic negative feedback
To determine whether a negative feedback due to APT autodepalmitoylation on the Golgi occurs, we directly investigated the presence of ES complexes between APTs in live cells. FLIM (19) was performed to image the interaction between mCitrine- and mCherry-tagged APTs quantitatively. The application of FLIM to quantify the fraction of ES complexes that undergo FRET is referred to as ES imaging (14).
ES imaging was performed using combinations of APT1/2-mCitrine donor fusion constructs with APT1/2-mCherry acceptor fusion constructs. Interaction between APT1-APT1, APT2-APT2, and APT1-APT2 was detected on the Golgi apparatus, with no detectable interaction in the cytoplasm at steady state (Fig. 4). This high interacting fraction at the Golgi is maintained by the continuous replenishment of palmitoylated APTs at the Golgi due to the local palmitoyltransferase activity (1,20,21), and is lost upon inhibition of APTs with Palmostatin B, which reacts with the catalytic serine in its active site (7) (Fig. 4 A, second column, and B). Therefore, the interaction occurs at the active site of at least one of the APTs in the complex and confirms the formation of ES complexes. Correspondingly, the lack of any significant interaction in the cytoplasm confirms that cytoplasmic soluble APTs are unpalmitoylated and represent the depalmitoylated product of the thioesterase activity. We also demonstrate the formation of equivalent homotypic and heterotypic ES complexes between APT1/2, showing that both APTs can depalmitoylate themselves or their paralog with similar efficiency.
Figure 4.
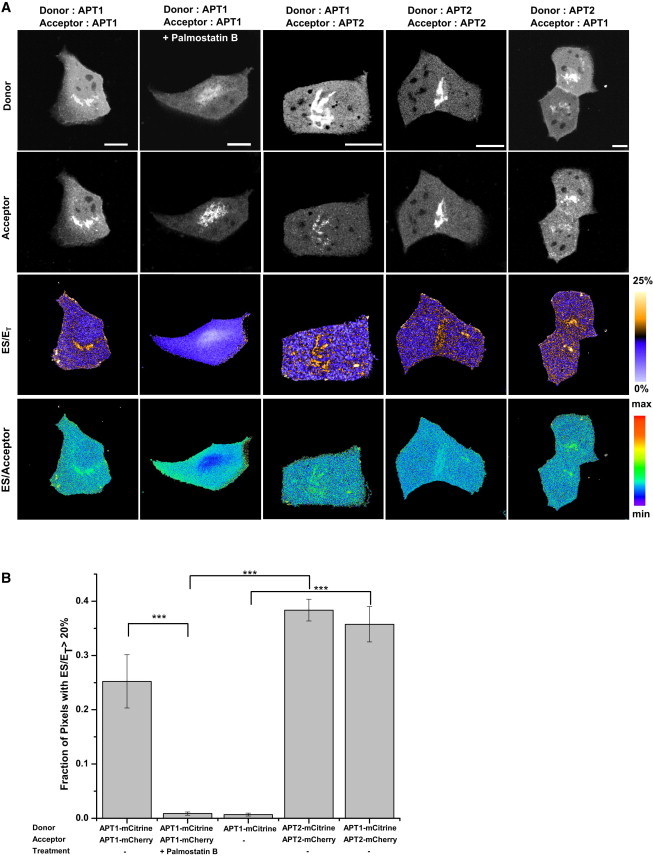
ES complexes of APTs. (A) Representative images of fluorescence intensities and the fraction of ES complexes (ES/ET, bottom row), with combinations of APT1-mCitrine or APT2-mCitrine as FRET donors (top row), and APT1-mCherry or APT2-mCherry as FRET acceptors (second row). The second column shows that ES interaction of APT1-mCitrine with APT1-mCherry is lost upon treatment of cells with Palmostatin B for 30 min, before redistribution of palmitoylated APT to all membranes. The color bar indicates the level of ES/ET. The fourth row shows ES/Acceptor, which is equivalent to the ratio of ES/ET over Acceptor/Donor, across all experiments. Scale bars: 10 μm. (B) Graph shows quantification of the fraction of pixels with ES/ET > 20% (20% is the (mean + 2 SD) ES/ET value in the membrane-free nuclear region representing a high-confidence threshold for identifying pixels showing interaction). Error bars denote SEM for (n = 5, 5, 5, 3, 4, respectively) from left to right. The number of asterisks indicates the significance in units of SEM.
To rule out the possibility of aspecific FRET between APTs, we generated spatial maps of the ratio of (ES/ET) over (acceptor/donor) intensity. This ratio is equivalent to the ES/acceptor ratio and indicates the dependence of the FRET signal on acceptor concentrations. The ES FRET signal did not show any dependence on acceptor intensity, indicating that the acceptor is present at saturating concentrations with respect to the APT-binding sites. Indeed, upon treatment with Palmostatin B, the ES/acceptor ratio in the Golgi apparatus was lower than that in the cytoplasm, in spite of the high acceptor concentrations present in the Golgi apparatus (Fig. 4 A, fourth row, second column). Additionally, a pixelwise correlation analysis of the FRET signal with respect to the acceptor intensity gave an average Pearson’s correlation of 0.09 for all conditions in the Golgi, and 0.0 in the PM (Fig. S2 A). The lack of a significant correlation between the ES/ET and acceptor/donor ratio affirms the conclusion that the FRET signal is specific and not a result of acceptor-concentration-dependent bystander FRET (22).
Soluble depalmitoylated APTs depalmitoylate mislocalized substrates on endomembranes
To test whether the APTs can access palmitoylated substrates on endomembranes, we performed ES imaging (14) to directly quantify the interaction between mCitrine-tagged APT enzymes and an mCherry-tagged palmitoylated substrate in live cells. For this purpose, we coexpressed APT1/2-mCitrine with Gα (1-11)-mCherry, an 11-amino acid N-terminal palmitoylated fragment of a substrate of APT1, and quantified the spatial distribution and extent of the ES interaction. Under normal conditions, APT1 and the substrate were found to interact extensively on the Golgi apparatus, detected as a high fraction of APT1/2-mCitrine exhibiting FRET with the substrate (Fig. 5, A and B). The fraction of APT that interacted with substrate was reduced after the cells were incubated with Palmostatin B, which inhibits APT activity by reacting with the catalytic serine in its active site. This confirms that the FLIM signal indeed reports on the binding of the substrate in the active site of APTs. The high molar fraction of APT-substrate complex that is detected on the Golgi is likely due to the continuous regeneration of palmitoylated substrate from depalmitoylated product by the local palmitoyltransferase activity. A substantial fraction of the soluble APTs that interacts with the membrane-bound substrate could also be measured on the PM due to the local enrichment of the palmitoylated Gα (1-11)-mCherry that is transported to the PM by the secretory pathway (Fig. 5 A). By contrast, the APT-substrate complex could not be detected on other endomembranes in the cytoplasm. This is likely due to endomembranes being virtually devoid of palmitoylated substrate due to depalmitoylation activity in the acylation cycle.
Figure 5.
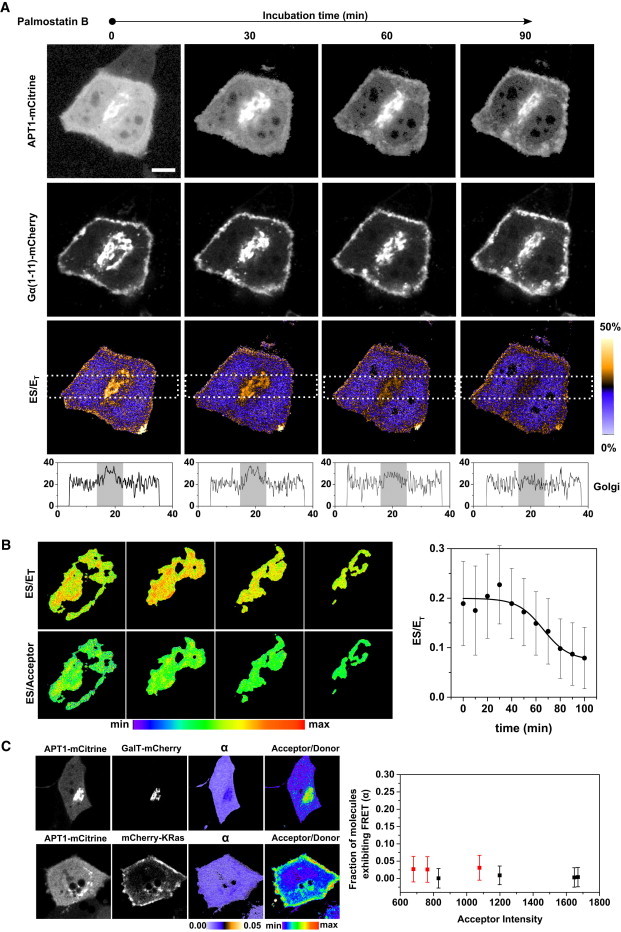
ES imaging of APT1 interacting with its palmitoylated substrate. (A) Representative image sequence of fluorescence intensity of APT1-mCitrine (top row), its palmitoylated substrate Gα (1-11)-mCherry (second row), and fraction of ES complexes (ES/ET, third row) during inhibition of APT activity by Palmostatin B. The acceptor/donor ratio on the Golgi remains invariant throughout the experiment. ES/ET represents the fraction of APT1-mCitrine in complex with the Gα (1-11)-mCherry substrate as determined from mCitrine FLIM decay profiles. The color bar indicates the level of ES/ET. Scale bar: 10 μm. Bottom profiles show average ES/ET levels along a rectangular region (white dashes) passing through the Golgi area (indicated in gray). (B) Magnified view of Golgi in A shows a spatial map of (ES/ET) and ES/Acceptor. ES/Acceptor is equivalent to the ratio of (ES/ET) over (Acceptor/Donor). Graph shows quantification of average ES/ET on the Golgi versus Palmostatin B incubation time (min). Error bars denote SEM for (n = 5). (C) Representative example of cells showing interaction of APT1-mCitrine with the nonpalmitoylated Golgi-resident protein GalT-mCherry (top row) and the nonpalmitoylated PM-resident protein mCherry-KRas (bottom row). Columns show (from left to right) APT1-mCitrine (donor) intensity, GalT-mCherry or mCherry-KRas (acceptor) intensity, and average fraction (α) of APT1-mCitrine molecules exhibiting FRET and Acceptor/Donor intensity ratio. Graph shows the average fraction of molecules exhibiting FRET (α, mean + SD) for cells (n = 7) expressing varying levels of mCherry-KRas (red) or GalT-mCherry (black) acceptors.
We confirmed the specificity of the FRET signal for the APT1-mCitrine (donor) and Gα (1-11)-mCherry (acceptor) pair by generating spatial maps of the ES/acceptor ratio on the Golgi (Fig. 5 B, left) and PM, and performing a correlation analysis as described above. PM and Golgi segmentation of the spatial maps showed that variance of the ES FRET signal does not depend on acceptor intensity. Thus, wherever the acceptor is localized, it is present at saturating concentrations with respect to the APT-binding sites. This was corroborated by the near absence of Pearson’s correlation between (ES/ET) and (acceptor/donor) intensity (ρ = −0.04 on Golgi and −0.07 on PM) throughout the Palmostatin B incubation (Fig. S2 B). FLIM measurements of the FRET between APT1-mCitrine and the unpalmitoylated Golgi-membrane-resident protein GalT-mCherry or the unpalmitoylated PM-resident protein KRas-mCherry (Fig. 5 C) showed an insignificant fraction α of APT1-mCitrine exhibiting FRET (α = 0.02 ± 0.03), over a 3-fold variation of acceptor intensity, confirming the absence of aspecific bystander FRET effects (22) in our experiments.
To show that soluble APT activity acts on palmitoylated substrates on endomembranes, the level of substrates on endomembranes must be increased by interfering with the acylation cycle. Ectopically expressed, palmitoylation-deficient but catalytically active APT-C2S mutants do not exhibit Golgi localization due to a lack of palmitoylation. These types of mutants represent the cytoplasmic pool of APTs that show a similar level of interaction with substrates on the Golgi and PM as WT APTs at steady state (Fig. 6 A, first column, B, and C). To increase the level of substrates on endomembranes, we incubated cells with Palmostatin B to inhibit APT activity, which leads to a redistribution of palmitoylated substrates to endomembranes. Under these conditions, the palmitoylation-deficient APT-C2S remains cytoplasmic but is inactive (Fig. 6 A, second column, B, and C). Removal of Palmostatin B from the cellular medium leads to a slow reactivation of the enzyme by hydrolysis of the Palmostatin B-APT-adduct (7), which precedes reinstatement of the PM-Golgi localization of the substrate. A substantial interaction of cytosolic, reactivated APTs with the delocalized substrate can be observed in this transient period (Fig. 6 A, third column, B, and C), which shows that depalmitoylated, cytoplasmic APTs can act on mislocalized substrates on endomembranes. This interaction eventually becomes undetectable as the reactivated depalmitoylation activity clears palmitoylated substrate from endomembranes and restores the steady-state substrate localization on the Golgi and PM (Fig. 6, B and C).
Figure 6.
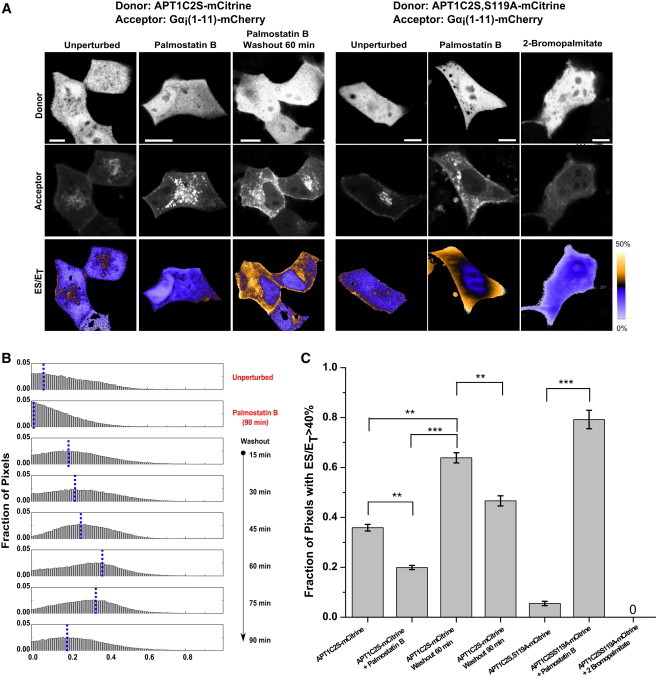
Cytosolic APTs depalmitoylate substrates on all cellular membranes. (A) Representative fluorescence images of APT1C2S-mCitrine and APT1C2S,S119A-mCitrine (top row) and palmitoylated substrate Gα (1-11)-mCherry (middle row). The fraction of ES complexes (ES/ET) as determined from mCitrine fluorescence decay profiles obtained by FLIM is shown in the third row. The first column shows the distribution of interaction between palmitoylation-deficient APT1C2S-mCitrine and Gα (1-11)-mCherry in the unperturbed state. The second column shows the loss of this interaction after interruption of the Gα (1-11)-mCherry acylation cycle with Palmostatin B-mediated APT inhibition. The third column shows the interaction between cytosolic APT1C2S-mCitrine and Gα (1-11)-mCherry on endomembranes after washout of Palmostatin B and reactivation of APT depalmitoylation activity. The fourth column shows the interaction of palmitoylation-deficient and catalytically inactive APT1C2S,S119A-mCitrine with Gα (1-11)-mCherry. The fifth column shows the increase in this interaction after inhibition of endogenous APTs with Palmostatin B. The sixth column shows the loss of this interaction in cells treated with 2-bromopalmitate. The color bar indicates the level of ES/ET. Scale bars: 10 μm. (B) Normalized ES/ET histograms of pixels pooled from experiments for APT1C2S-mCitrine shown in A. The blue line indicates the modal value of ES/ET for each condition (right). From top to bottom: unperturbed cells (n = 4), cells with Palmostatin B-mediated APT inhibition (n = 3), and cells at various time points following washout of Palmostatin B (n = 4, 3, 3, 3, 2, 5 respectively). (C) Spatial extent of ES interaction. Graph shows average fraction of pixels with ES/ET > 40% in cells corresponding to experiments described in A (40% is the (mean + 2 SD) ES/ET value in the membrane-free nuclear region representing a high-confidence threshold for identifying pixels showing interaction). Error bars denote SEM for (n = 4, 3, 3, 5, 5, 5, 2, respectively). The number of asterisks indicates the significance in units of SEM.
To further substantiate that cytoplasmic APTs indeed access their substrates on endomembranes, we studied the stable interaction of catalytically impaired, palmitoylation-deficient APT1-C2S,S119A-mCitrine with mislocalized palmitoylated substrates. Palmostatin B inhibits thioesterase activity by forming a stable covalent adduct with the catalytic serine residue in the active site of APT1/2, which leads to the redistribution of palmitoylated substrates on all membranes. The interaction of cytoplasmic APTs with mislocalized substrates can therefore be demonstrated with a catalytic serine-lacking mutant that cannot react with Palmostatin B but still forms stable, albeit unproductive, ES complexes (7,11). In untreated cells, the level of ES complexes with catalytically impaired APT1C2S,S119A-mCitrine was much lower than that for WT APT1/2-mCitrine (Fig. 6 A, fourth column, and C). The overall lower level of catalytically impaired ES complexes that was detected under these conditions is probably due to competitive binding of endogenous active APTs to the Gα (1-11)-mCherry substrate. However, upon inhibition of endogenous APT activity by Palmostatin B, cytosolic catalytically impaired APTs strongly interacted with the mislocalized palmitoylated Gα (1-11)-mCherry proteins on endomembranes (Fig. 6 A, fifth column, and C). Inhibiting palmitoylation of the substrate with 2-bromopalmitate abolished this interaction, confirming that APTs interact with only palmitoylated Gα (1-11)-mCherry proteins (Fig. 6 A, sixth column, and C). These experiments show that depalmitoylated, soluble APTs can readily access palmitoylated substrates bound to membranes, and that ubiquitous depalmitoylation of substrates by APTs occurs on all membranes.
Discussion
APT1/2 have been reported to be cytosolic enzymes based on the presence of APT1 in cytosolic fractions of yeast cells (8). Fractionation and immunohistochemical studies suggested that hemagglutinin (HA)-tagged mammalian APT1/2 are cytosolic and palmitoylated, and at least APT1 is capable of autodepalmitoylation (13). In particular, Kong et al. (13) reported that palmitoylation allows APTs to be enriched on the PM in a canonical acylation cycle, and thus enhances their reactivity toward their PM-bound substrates. However, this view does not take into account the spatial dynamics and stability of APT palmitoylation. Indeed, the enrichment of APT thioesterase activity on the PM is antagonistic to the enrichment of its palmitoylated substrates, and would impede the PM-localized functions of palmitoylated proteins of the Ras and Src family. We provide evidence for an alternative model wherein APTs do not localize to the PM. Rather, soluble unpalmitoylated APTs can depalmitoylate their substrates on all membranes in the cell such that they can be repalmitoylated and trapped on the Golgi apparatus. This model, where cytosolic APTs counter equilibration of their substrates over all membranes, is consistent with the known function of the acylation cycle to enrich palmitoylated proteins on the PM.
The data presented here show that in mammalian cells, a significant fraction of APTs are enriched on the Golgi apparatus due to dynamic S-palmitoylation, and that APTs can autodepalmitoylate in trans. In contrast to other proteins that undergo the acylation cycle, APTs possess only a single reversible lipidation site and lack irreversible lipidations such as S-prenylation or N-myristoylation. Further, palmitoylation of APTs on the Golgi counteracts itself as it concomitantly recruits and enriches the opposing thioesterase activity. The membrane attachment of APTs is therefore labile, and far too short-lived for APTs to be transported to the PM via the secretory pathway. Thus, the autocatalytic acylation cycle of APTs maintains its steady-state distribution between the cytosol and Golgi apparatus, in contrast to its substrates that are partitioned between the Golgi and the PM.
Palmitoylation of APTs could enhance their reactivity toward their membrane-bound substrates. Yet, palmitoylation localizes the depalmitoylating activity of APTs to the Golgi apparatus, a compartment where the opposing palmitoylation activity occurs. The absence of APTs on endomembranes where in fact depalmitoylation of mislocalized proteins should occur is a result of their labile palmitoylation. Here, we resolve this conundrum by demonstrating ES complexes of unpalmitoylated cytosolic APTs with membrane-bound palmitoylated substrates on endomembranes. We thus demonstrate the role of the depalmitoylated, cytoplasmic pool of APTs in the depalmitoylating activity that corrects mislocalization of palmitoylated proteins on endomembranes in the acylation cycle.
What, then, is the role of palmitoylation and the Golgi-localized pool of APTs? Several lines of evidence presented in this work show that dynamic S-palmitoylation creates a negative feedback of APT activity on the Golgi. This couples the rate of palmitoylation to the rate of depalmitoylation on Golgi membranes, thereby establishing a balance that controls the steady-state amounts of palmitoylated substrates on the Golgi. This renders the partitioning of substrates in the acylation cycle robust with respect to variations in cellular geometry (23), vesicular transport (24,25), organelle structure, palmitate metabolism, and other dynamic factors. Although the specificity and regulation of protein palmitoyltransferases are currently poorly understood, we previously reported that in mammalian cells, the partitioning of peripheral-membrane proteins remains relatively unaffected by an RNAi-mediated reduction in the palmitoylation activity of specific DHHC proteins (1). In this case, the negative feedback loop created by the autodepalmitoylating APT activity would compensate for the decreased palmitoylation by reducing the opposing local thioesterase activity. In concert with other recently described mechanisms that regulate access of APTs to their substrates (26), such a negative feedback would also render the acylation cycle resilient against perturbations of APT activity, thus maintaining substrate partitioning over membrane compartments (7,26). Simulation of APT palmitoylation dynamics on the Golgi supports this model, wherein the negative feedback loop imparts robustness to the acylation cycle of its substrates (Fig. 7). In this two-compartment model, substrate partitioning is defined as the ratio of substrate localized to the Golgi compartment to substrate localized in the cytosol compartment (see “Simulation of APT dynamics on the Golgi apparatus” in the Supporting Material). An instantaneous increase of the amount of substrate localized to the Golgi results in a rapid increase of the substrate partitioning, which recovers to the original steady state due to the negative feedback mediated by APT thioesterase activity on the Golgi. This recovery is absent when APTs cannot be palmitoylated and therefore are not localized to the Golgi. The simulation thus conceptually shows that the negative feedback in the system maintains a constant amount of APT on the Golgi apparatus at steady state, and ensures that variations in substrate concentrations or thioesterase concentrations do not alter substrate partitioning over cellular membranes. This implies that thioesterase inhibition must be persistent and nearly complete to affect the activities of palmitoylated substrate proteins, such as oncogenic HRas, in a therapeutic setting. Indeed, previous studies have required extended incubation at high thioesterase inhibitor concentrations (7) or chronic downregulation (27–29) to observe effects on cell physiology. On the other hand, palmitoyltransferases have been reported to have overlapping specificity, and such a negative feedback at the level of the thioesterase activity on the Golgi would allow the system to generate a variety of partitioning modes for different proteins depending on the relative affinities of these proteins to palmitoyltransferases and APTs, and the strength of the negative feedback. The autodepalmitoylating activity of APTs therefore adds a nested reaction cycle as a layer of regulation to the acylation cycle of APT substrates in maintaining specific spatial partitioning of palmitoylated peripheral-membrane proteins over cellular membranes.
Figure 7.
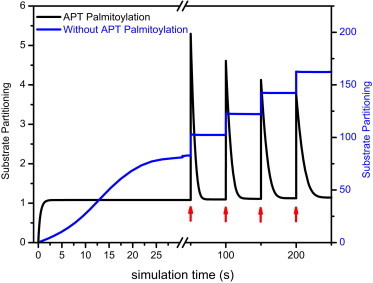
Substrate partitioning is robust when APTs are palmitoylated. Representative simulation of substrate dynamics between the Golgi and the cytosol with translocation between compartments coupled to palmitoylation state. Substrate partitioning is defined as the ratio of substrate concentration on the Golgi versus that in the cytosol. When APTs are palmitoylated, substrate partitioning recovers to steady state from a periodic and cumulative increase of substrate concentration on the Golgi (red arrows). This homeostatic behavior is completely lost when APTs are not palmitoylated. Initial conditions for this particular simulation run: SCyt = 5 μM, SGolgi = 0 μM, APTCyt = 5 μM, APTGolgi = 0 μM, and (S-palmitoyltransferase activity) PAT = 10 μM; parameters: kcatPAT = 1 s−1, kcatAPT = 1 s−1, kMPAT = 1 μM, and kMAPT = 0.1 μM.
Acknowledgments
We thank Astrid Kraemer for assistance in preparing the manuscript, and Marion Rusch and Michael Albers for assistance in the metabolic labeling experiments. N.V. and P.B. designed experiments and wrote the manuscript. N.V. performed localization, FRAP, bioinformatics, metabolic labeling, and simulation studies. N.V. and B.P. performed ES imaging studies, H.G. performed FLIM analysis. L.R. cloned APT1 and APT2 constructs. C.H. and H.W. provided initial insights and results showing that APTs can be palmitoylated.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Rocks O., Gerauer M., Bastiaens P.I. The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell. 2010;141:458–471. doi: 10.1016/j.cell.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 2.Hougland J.L., Fierke C.A. Getting a handle on protein prenylation. Nat. Chem. Biol. 2009;5:197–198. doi: 10.1038/nchembio0409-197. [DOI] [PubMed] [Google Scholar]
- 3.Martin D.D.O., Beauchamp E., Berthiaume L.G. Post-translational myristoylation: fat matters in cellular life and death. Biochimie. 2011;93:18–31. doi: 10.1016/j.biochi.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 4.Chandra A., Grecco H.E., Bastiaens P.I. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2012;14:148–158. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- 5.Schroeder H., Leventis R., Silvius J.R. S-Acylation and plasma membrane targeting of the farnesylated carboxyl-terminal peptide of N-ras in mammalian fibroblasts. Biochemistry. 1997;36:13102–13109. doi: 10.1021/bi9709497. [DOI] [PubMed] [Google Scholar]
- 6.Rocks O., Peyker A., Bastiaens P.I. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science. 2005;307:1746–1752. doi: 10.1126/science.1105654. [DOI] [PubMed] [Google Scholar]
- 7.Dekker F.J., Rocks O., Waldmann H. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat. Chem. Biol. 2010;6:449–456. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 8.Duncan J.A., Gilman A.G. A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein α subunits and p21(RAS) J. Biol. Chem. 1998;273:15830–15837. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 9.Görmer K., Bürger M., Waldmann H. Chemical-biological exploration of the limits of the Ras de- and repalmitoylating machinery. ChemBioChem. 2012;13:1017–1023. doi: 10.1002/cbic.201200078. [DOI] [PubMed] [Google Scholar]
- 10.Rusch M., Zimmermann T.J., Waldmann H. Identification of acyl protein thioesterases 1 and 2 as the cellular targets of the Ras-signaling modulators palmostatin B and M. Angew. Chem. Int. Ed. Engl. 2011;50:9838–9842. doi: 10.1002/anie.201102967. [DOI] [PubMed] [Google Scholar]
- 11.Devedjiev Y., Dauter Z., Derewenda Z.S. Crystal structure of the human acyl protein thioesterase I from a single X-ray data set to 1.5 A. Structure. 2000;8:1137–1146. doi: 10.1016/s0969-2126(00)00529-3. [DOI] [PubMed] [Google Scholar]
- 12.Yang W., Di Vizio D., Freeman M.R. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol. Cell. Proteomics. 2010;9:54–70. doi: 10.1074/mcp.M800448-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong E., Peng S., Mukherjee A.B. Dynamic palmitoylation links cytosol-membrane shuttling of acyl-protein thioesterase-1 and acyl-protein thioesterase-2 with that of proto-oncogene H-ras product and growth-associated protein-43. J. Biol. Chem. 2013;288:9112–9125. doi: 10.1074/jbc.M112.421073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yudushkin I.A., Schleifenbaum A., Bastiaens P.I. Live-cell imaging of enzyme-substrate interaction reveals spatial regulation of PTP1B. Science. 2007;315:115–119. doi: 10.1126/science.1134966. [DOI] [PubMed] [Google Scholar]
- 15.Grecco H.E., Roda-Navarro P., Verveer P.J. Global analysis of time correlated single photon counting FRET-FLIM data. Opt. Express. 2009;17:6493–6508. doi: 10.1364/oe.17.006493. [DOI] [PubMed] [Google Scholar]
- 16.Hirano T., Kishi M., Izumi T. Thioesterase activity and subcellular localization of acylprotein thioesterase 1/lysophospholipase 1. Biochim. Biophys. Acta. 2009;1791:797–805. doi: 10.1016/j.bbalip.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Ren J., Wen L., Yao X. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng. Des. Sel. 2008;21:639–644. doi: 10.1093/protein/gzn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coleman R.A., Rao P., Bardes E.S. 2-Bromopalmitoyl-CoA and 2-bromopalmitate: promiscuous inhibitors of membrane-bound enzymes. Biochim. Biophys. Acta. 1992;1125:203–209. doi: 10.1016/0005-2760(92)90046-x. [DOI] [PubMed] [Google Scholar]
- 19.Bastiaens P.I.H., Squire A. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell. Trends Cell Biol. 1999;9:48–52. doi: 10.1016/s0962-8924(98)01410-x. [DOI] [PubMed] [Google Scholar]
- 20.Hou H., John Peter A.T., Ungermann C. Analysis of DHHC acyltransferases implies overlapping substrate specificity and a two-step reaction mechanism. Traffic. 2009;10:1061–1073. doi: 10.1111/j.1600-0854.2009.00925.x. [DOI] [PubMed] [Google Scholar]
- 21.Ohno Y., Kihara A., Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim. Biophys. Acta. 2006;1761:474–483. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 22.Chiu V.K., Bivona T., Philips M.R. Ras signalling on the endoplasmic reticulum and the Golgi. Nat. Cell Biol. 2002;4:343–350. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- 23.Altschuler S.J., Wu L.F. Cellular heterogeneity: do differences make a difference? Cell. 2010;141:559–563. doi: 10.1016/j.cell.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Apolloni A., Prior I.A., Hancock J.F. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol. Cell. Biol. 2000;20:2475–2487. doi: 10.1128/mcb.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watson R.T., Furukawa M., Pessin J.E. The exocytotic trafficking of TC10 occurs through both classical and nonclassical secretory transport pathways in 3T3L1 adipocytes. Mol. Cell. Biol. 2003;23:961–974. doi: 10.1128/MCB.23.3.961-974.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahearn I.M., Tsai F.D., Philips M.R. FKBP12 binds to acylated H-ras and promotes depalmitoylation. Mol. Cell. 2011;41:173–185. doi: 10.1016/j.molcel.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eskildsen T., Taipaleenmäki H., Kassem M. MicroRNA-138 regulates osteogenic differentiation of human stromal (mesenchymal) stem cells in vivo. Proc. Natl. Acad. Sci. USA. 2011;108:6139–6144. doi: 10.1073/pnas.1016758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X., Jiang L., Zhou X. MicroRNA-138 suppresses invasion and promotes apoptosis in head and neck squamous cell carcinoma cell lines. Cancer Lett. 2009;286:217–222. doi: 10.1016/j.canlet.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Siegel G., Obernosterer G., Schratt G.M. A functional screen implicates microRNA-138-dependent regulation of the depalmitoylation enzyme APT1 in dendritic spine morphogenesis. Nat. Cell Biol. 2009;11:705–716. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Currie L.A. Limits for qualitative detection and quantitative determination. Application to radiochemistry. Anal. Chem. 1968;40:586–593. [Google Scholar]
- 31.McNally J.G. Quantitative FRAP in analysis of molecular binding dynamics in vivo. Methods Cell Biol. 2008;85:329–351. doi: 10.1016/S0091-679X(08)85014-5. [DOI] [PubMed] [Google Scholar]
- 32.Sprague B.L., Pego R.L., McNally J.G. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smisdom N., Braeckmans K., Ameloot M. Fluorescence recovery after photobleaching on the confocal laser-scanning microscope: generalized model without restriction on the size of the photobleached disk. J. Biomed. Opt. 2011;16:046021. doi: 10.1117/1.3569620. [DOI] [PubMed] [Google Scholar]
- 34.Tannert A., Tannert S., Schaefer M. Convolution-based one and two component FRAP analysis: theory and application. Eur. Biophys. J. 2009;38:649–661. doi: 10.1007/s00249-009-0422-4. [DOI] [PubMed] [Google Scholar]
- 35.Martin B.R., Wang C., Cravatt B.F. Global profiling of dynamic protein palmitoylation. Nat. Methods. 2012;9:84–89. doi: 10.1038/nmeth.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rostovtsev V.V., Green L.G., Sharpless K.B. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 37.Jennings B.C., Linder M.E. DHHC protein S-acyltransferases use similar ping-pong kinetic mechanisms but display different acyl-CoA specificities. J. Biol. Chem. 2012;287:7236–7245. doi: 10.1074/jbc.M111.337246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satou M., Nishi Y., Sugimoto H. Identification and characterization of acyl-protein thioesterase 1/lysophospholipase I as a ghrelin deacylation/lysophospholipid hydrolyzing enzyme in fetal bovine serum and conditioned medium. Endocrinology. 2010;151:4765–4775. doi: 10.1210/en.2010-0412. [DOI] [PubMed] [Google Scholar]
- 39.Yeh D.C., Duncan J.A., Michel T. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. J. Biol. Chem. 1999;274:33148–33154. doi: 10.1074/jbc.274.46.33148. [DOI] [PubMed] [Google Scholar]
- 40.UniProt Consortium The Universal Protein Resource (UniProt) 2009. Nucleic Acids Res. 2009;37(Database issue):D169–D174. doi: 10.1093/nar/gkn664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waterhouse A.M., Procter J.B., Barton G.J. Jalview version 2—a multiple sequence alignment analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.