

Abstract
p53 is a tumor suppressor gene with well-characterized roles in cell cycle regulation, apoptosis and the maintenance of genome stability. Recent evidence suggests that p53 may also contribute to the regulation of migration and invasion. Epithelial cell adhesion molecule (EpCAM) is a transmembrane glycoprotein that is overexpressed in the majority of human epithelial carcinomas, including breast and colorectal carcinomas. We demonstrate by chromatin immunoprecipitation assays that p53 interacts with a candidate p53 binding site within the EpCAM gene. p53-mediated transcriptional repression of EpCAM was confirmed in gain-of-function, and loss-of-function experimental systems. Induction of wildtype p53 was associated with a significant dose-dependent decrease in EpCAM expression; conversely, specific ablation of p53 was associated with a significant increase in EpCAM expression. At the functional level, specific ablation of p53 expression is associated with increased breast cancer invasion, and this effect is abrogated by concomitant specific ablation of EpCAM expression. Taken together, these biochemical and functional data are the first demonstration that (1) wildtype p53 protein binds to a response element within the EpCAM gene and negatively regulates EpCAM expression, and (2) transcriptional repression of EpCAM contributes to p53 control of breast cancer invasion.
Keywords: p53, EpCAM, breast cancer, invasion
INTRODUCTION
p53 is a tumor suppressor gene that is frequently mutated in human cancers, including breast cancer (1). The p53 gene encodes a transcription factor that plays a central role in the activation of DNA repair, cell cycle arrest, initiation of apoptosis, and maintenance of genome stability. In normal cells p53 is typically inactive; upon activation, the p53 protein is transiently stabilized and accumulates in the nucleus where it functions in part to induce or repress the transcription of downstream target genes. Identification of p53-regulated genes has provided critical insights into understanding the biologic function of the p53 protein.
Epithelial cell adhesion molecule (EpCAM) is a 40-kD cell-surface glycoprotein that is expressed at the basolateral membrane of the majority of epithelial tissues. EpCAM is overexpressed in the majority of human epithelial cancers (2), and has long been considered to be a candidate for molecular therapy (3). Of note, EpCAM expression in primary breast cancers has been associated with poor clinical outcome (4–6). In these studies of primary breast cancer specimens from more than 2300 patients, EpCAM expression was independently associated with prognosis. Recent in vitro studies confirm the potential functional role of EPCAM in breast cancer; specific ablation of EpCAM expression using RNA interference results in a dramatic decrease in the invasive potential of breast cancer cell lines (7).
Despite recent interest in the functional biology of EpCAM, the transcriptional regulation of EpCAM in breast and other epithelial cancers remains to be elucidated. There is experimental evidence to suggest that p53 may regulate the methylation status and amplification of the EpCAM gene (8). However, recent analyses of primary breast cancer specimens demonstrate no significant correlation between promoter methylation and EpCAM expression, suggesting that alternate mechanisms may be involved in the regulation of EpCAM expression (9). Since p53 loss or mutation is a common event in human breast cancer, we investigated the potential role of wildtype p53 in the regulation of EpCAM expression.
MATERIALS AND METHODS
Cell culture
Human breast epithelial and cancer cell lines MCF10A and MCF10CA1a (CA1a) were obtained from Dr. Fred Miller at Wayne State University (Detroit, MI). MCF-7 cells were obtained from ATCC (Manassas, VA). Wildtype and p53-deficient HCT116 cells were obtained from Dr. Bert Vogelstein at Johns Hopkins University (Baltimore, MD).
Plasmids and luciferase reporter assay
A luciferase reporter corresponding to the p53 response element EpCAM-RE1 was generated by cloning two copies of EpCAM-RE1 into pGL3-Basic (Promega, Madison, WI). pNeo, p53, p53-V143A, and p53-R273H were obtained from Dr. Simon Powell (Washington University in Saint Louis). p53-R280K was cloned from the MDA-MB-231 cell line. These constructs were transiently transfected with 20ng-TK-Renilla, 400ng of the EpCAM-RE1 luciferase reporter, and 200ng of pNEo or the indicated p53 expression constructs into CA1a or MCF-7 cells. Relative light units (RLU) reflect the percent mean change in luciferase activity in experimental cells compared to cells co-transfected with empty pGL3-Basic and pcDNA3.1 vectors.
Lentiviral RNA interference
The lentiviral construct pSicoR was obtained from Dr. Tyler Jacks (Massachusetts Institute of Technology, Boston, MA) (10). shRNA target sequences specific for p53 (GACTCCAGTGGTAATCTAC), EpCAM (CTGGCTTTACCAATCTTGA ) and a scrambled control (TTCTCCGAACGTGTCACGT) were cloned into the pSicoR vector. shRNA constructs were transfected into HEK293T cells with VSVG and Δ8.9 and viral supernatants were collected to transduce the indicated cell lines at 10 MOI.
Chromatin immunoprecipitation (ChIP) assay
MCF-7 cells were exposed to a minimal dose of UV radiation (10-J/M2) to induce p53 expression and processed for ChIP assay at 8 h using the Millipore ChIP kit (Billerica, MA). Briefly, confluent MCF-7 cells were fixed with 1% formaldehyde and lysed. After sonication, p53 bound DNA was immunoprecipitated using proteinA-DO-1 antibody, washed and eluted in 1% SDS, 0.1M NaHCO3. Reverse-crosslinked DNA was purified using DNA columns and analyzed by PCR analysis using EpCAM and p21 specific primer sequences.
Immunoblotting
Cells were lysed in RIPA buffer, and the protein concentration was measured using the BCA protein assay (Pierce, Rockford, IL). Cell extracts were subjected to 12% SDS-PAGE, and electrophoretically transferred to a PVDF membrane. Membranes were incubated with the indicated primary antibodies, and secondary antibodies conjugated with peroxidase (Santa Cruz Biotechnology, Santa Cruz, CA). Signal was then detected using the ECL chemiluminescent immunodetection system (Applied Biosystems, Foster City, CA).
Flow cytometry
EpCAM expression levels were measured by flow cytometry using PE-labeled EpCAM antibody using a FACScan flow cytometer (BD Biosciences, San Jose, CA).
cDNA synthesis and real-time PCR analysis
Total RNA was purified from cell lines using RNAeasy (Qiagen). Two micrograms of RNA was reverse transcribed using a cDNA synthesis kit (Ambion, Austin, TX). Quantitative mRNA expression was measured using SYBR green chemistry and an ABI Prism 7700 Sequence Detector (Applied Biosystems). Reaction conditions and primer sequences are available upon request.
Invasion and Adhesion assays
For invasion assays, stably transduced CA1a cells (4×104) were added into transwell matrigel invasion chambers (BD Biosciences) and incubated for 48–72 h. Cells invading through the matrigel membrane were counted by a laboratory technician blinded to the experimental conditions. For the adhesion assay, 1×105 stably transduced CA1a cells were plated in triplicate on 96-well fibronectin coated plates (BD Biosciences). After 30 minutes of incubation, the plates were placed on a shaker for 2 minutes, washed 2 times with PBS, stained with crystal-violet, lysed with 2% SDS and read at 550nm on an ELISA reader.
RESULTS
Identification of candidate p53 binding sites in the EpCAM gene, and in vivo confirmation of wildtype p53 binding to the highest scoring binding site
Candidate p53 binding sites in the EpCAM gene were identified using the p53MH computer algorithm (11). The p53MH computer algorithm identified 10 candidate p53 binding sites in the EpCAM genomic sequence (Figure 1A), including two candidate binding sites, RE1 and RE2, located in introns within the EpCAM gene with a score > 90%. ChIP assays confirmed p53 binding to the highest scoring candidate binding site, EpCAM-RE1, and to p21, a gene that is bound and regulated by p53 (Figure 1B). EpCAM-RE1 and EpCAM-RE2 reporter constructs were then transiently transfected into CA1a cells and wildtype p53 was induced by transient transfection, UV radiation or camptothecin (CPT) treatment. Wildtype p53 expression was associated with a dose-dependent increase in EpCAM-RE1, but not EpCAM-RE2 luciferase activity (Figure 1C, 1D). No luciferase activity was observed following transient transfection with the p53 mutants tested (Figure 1C), suggesting that only wildtype p53 binds the EpCAM-RE1 binding site.
Figure 1. Identification of a p53 binding site in the human EpCAM gene.
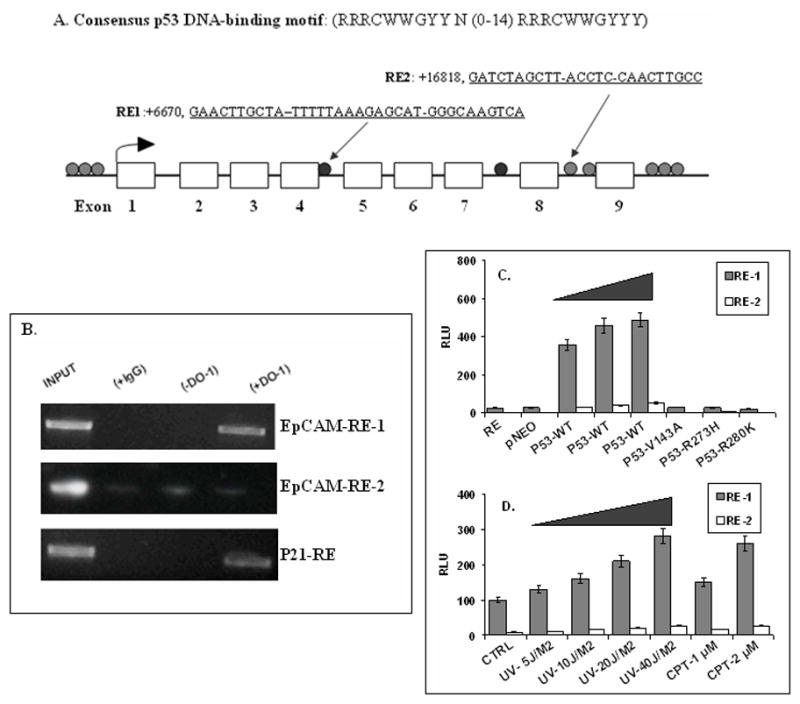
(A) Ten candidate p53 binding sites in the human EpCAM gene were identified. The two candidate binding sites with scores > 90% are indicated. (B) ChIP assay confirms an interaction between p53 and EpCAM-RE1, but not EpCAM-RE2. A known p53 binding site in the p21 gene served as a positive control. INPUT: no immunoprecipitation prior to PCR; (+IgG): immunoprecipitation with isotype control antibody; (−DO-1): immunoprecipitation in the absence of p53-specific primary antibody; (+DO-1): immunoprecipitation with p53-specific primary antibody. (C, D) Dose-dependent association between p53 expression and EpCAM-RE1 reporter activity.
Wildtype p53 negatively regulates EpCAM expression in breast cancer cells
In gain-of-function experiments, induction of p53 expression was associated with a dose-dependent decrease in EpCAM expression (Figure 2A, 2B). No change in EpCAM expression was observed following transient transfection with the mutant p53 constructs (Figure 2C). In loss-of-function experiments, specific ablation of p53 expression resulted in a significant reduction (>90%) in p53 mRNA and protein expression (Figure 3A, 3B), a significant decrease in p21 expression (data not shown), and a significant increase in EpCAM expression (Figure 3B, 3C).
Figure 2. p53 expression is associated with a dose-dependent decrease in EpCAM expression.
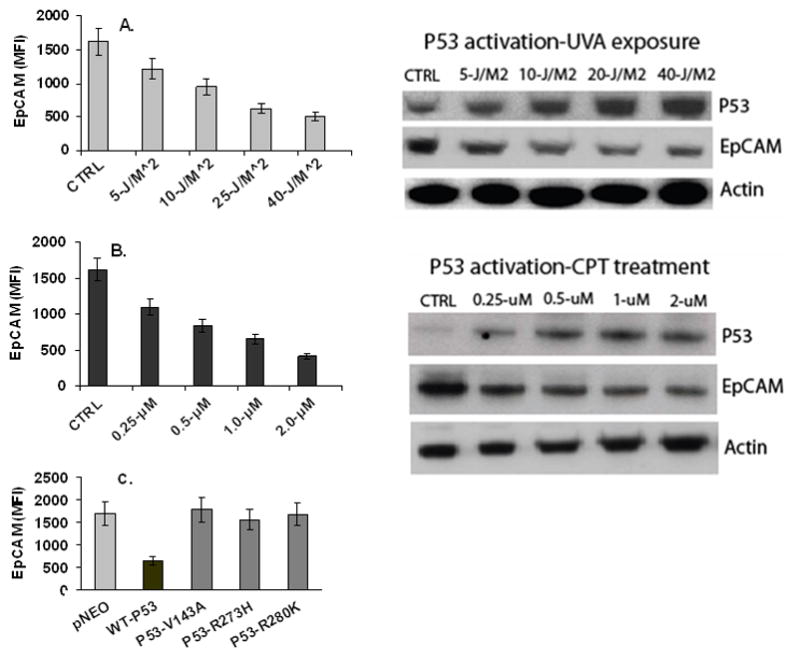
(A, B) MCF-7 cells were treated with UV radiation or CPT as indicated. EpCAM protein expression was measured by flow cytometry in the left panel, and immunoblot in the right panel. (C) Transient transfection of MCF-7 cells with wildtype and p53 mutants demonstrate that wildtype, but not mutant p53, is associated with decreased EpCAM expression.
Figure 3. Specific ablation of p53 expression is associated with increased EpCAM expression.
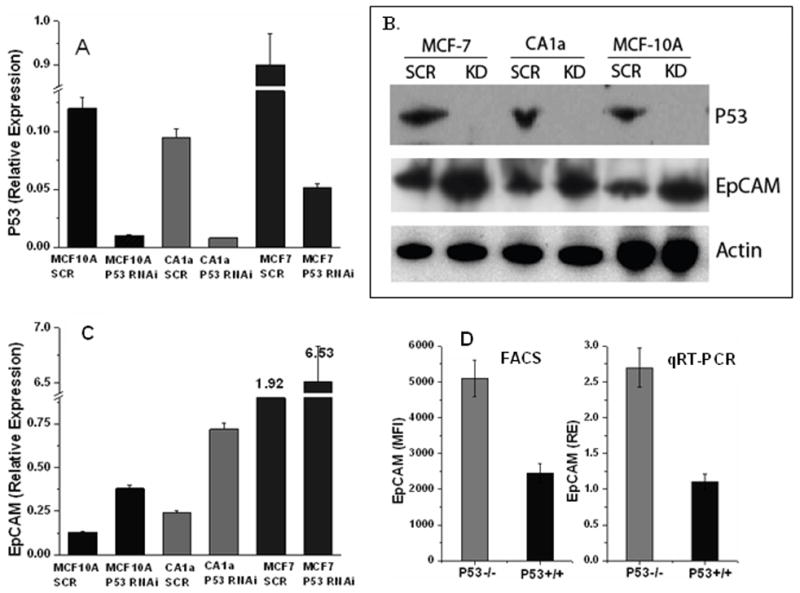
Lentiviral shRNA constructs specific for p53 were used to specifically ablate p53. (A, B) p53 expression is decreased as demonstrated by qRT-PCR and immunoblot. (B, C) EpCAM expression is increased as demonstrated by qRT-PCR and immunoblot. (D) EpCAM expression is increased in p53-deficient HCT116 cells as measured by flow cytometry and qRT-PCR.
We also assessed EpCAM expression in wildtype and p53-deficient HCT116 cells, demonstrating increased EpCAM mRNA and protein expression in p53-deficient HCT116 cells (Figure 3D). As an important control, we specifically ablated p53 expression with RNA interference prior to treatment of MCF-7 breast cancer cells with UV or camptothecin. Under these conditions, no change in EpCAM expression was observed following the UV or camptothecin treatment, confirming that the change in EpCAM expression observed is mediated by p53 (data not shown).
Transcriptional repression of EpCAM contributes to p53 control of breast cancer invasion
To determine the functional relevance of p53-mediated transcriptional repression of EpCAM, we assessed the impact of concomitant specific ablation of p53 and EpCAM expression on breast cancer invasion. CA1a cells were stably transduced with lentiviral constructs containing p53, EpCAM, or control shRNA constructs. Transduction with EpCAM shRNA constructs resulted in a significant decrease in EpCAM expression (Figure 4A), and a decrease in breast cancer invasion as measured in a transwell invasion assay (Figure 4C). Conversely, transduction with p53 shRNA constructs resulted in a significant increase in EpCAM expression (Figure 4A), and an increase in breast cancer invasion (Figure 4C). When used in combination with p53 shRNA constructs, EpCAM shRNA constructs were able to successfully abrogate the increase in EpCAM expression and breast cancer invasion observed following specific ablation of p53 expression (Figure 4A, 4C). In parallel adhesion experiments we observed similar results; (Figure 4 D). These data strongly suggest that transcriptional repression of EpCAM contributes to p53 control of breast cancer invasion.
Figure 4. Transcriptional repression of EpCAM contributes to p53 control of breast cancer invasion.
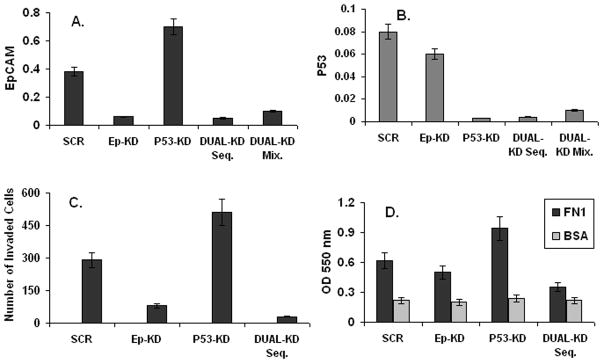
Lentiviral shRNA constructs targeting p53, EpCAM or control sequences were used to specifically ablate p53 expression, EpCAM expression, or expression of both genes. (A, B) Decreased p53 and EpCAM expression measured by qRT-PCR following transduction with the indicated constructs. (C) Invasion was measured in a matrigel transwell assay following stable transduction with the indicated shRNA sequences. (D) Adhesion was measured by assessing adherence to fibronectin-coated plates.
DISCUSSION
Identification of p53-responsive genes has provided critical insights into the role of p53 as a tumor suppressor protein. Although genome-wide expression profiling suggest that there may be over 1500 p53-responsive genes, the majority of these genes are believed to be indirect targets (12). In 2002, it was estimated that there were only 20 genes that had confirmed p53 binding sites, were known to bind p53, and were clearly regulated by p53 (11). In this study, we confirmed p53 binding/regulation of the EpCAM gene in vivo, and identified a biological function due to p53 binding of the EpCAM gene. Taken together, these studies suggest that EpCAM may be a key transcriptional target of p53.
Regulation of migration and invasion is a recently described tumor suppressor function of p53 (13). Although the mechanisms of p53 control of migration and invasion remain to be defined, there is experimental evidence to suggest that p53 is capable of regulating the function of the Rho family of small GTPases, including the prototypic family members RhoA, Rac1 and Cdc42, (14, 15). In these studies, phosphoinositide 3-kinase (PI3K) was demonstrated to be a key intermediary between p53 and the Rho GTPases (14). In this study we confirm our previous findings that EpCAM expression is associated with breast cancer invasion (7). Although the mechanism(s) of EpCAM control of breast cancer invasion remain to be defined, EpCAM directly interacts with PI3K in normal epithelial cells (16), and the introduction of EpCAM in normal epithelial cells is associated with cytoskeletal rearrangements (17). These previous studies, and the demonstration that EpCAM contributes to p53 control of breast cancer invasion in this study, strongly suggest that EpCAM may be an intermediary between p53, PI3K and the Rho GTPases.
In contrast to the well-studied mechanisms of p53 transcriptional activation, the mechanisms underlying p53-mediated transcriptional repression are less well characterized. Several potential mechanisms for p53-mediated transcriptional repression have been proposed (18): (1) interference with DNA-binding transcriptional activators, (2) nonspecific interference with basal transcriptional machinery, (3) alteration of downstream target gene chromatin structure by recruitment of histone deacetylases (HDAC), and (4) recruitment of DNA (cytosine-5) methyltransferase (DNMT) and concomitant promoter methylation. Recruitment of HDAC to the EpCAM gene with concomitant repression of EpCAM expression has recently been described (19). In this study, Tai et al. also demonstrated that promoter methylation contributes to the repression of EpCAM gene expression in lung cancer cells. It is possible that p53 is actively involved in the recruitment of an HDAC-DNMT1-p53 complex to the EpCAM gene, and that this complex mediates gene repression by promoter methylation as has been recently demonstrated for the survivin gene (20).
In the present study, we took a step-wise approach to demonstrate that p53 represses the transcription of EpCAM. These data imply that the loss or mutation of wildtype p53 in breast and other epithelial cancers may contribute to EpCAM overexpression. Further study will be necessary to define the mechanisms by which p53 represses EpCAM expression, to determine if evidence of this interaction can be demonstrated in human cancer specimens, and to explore the functional implications in more detail.
Acknowledgments
Funding provided by Susan G. Komen for the Cure
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Went PT, Lugli A, Meier S, et al. Frequent EpCam protein expression in human carcinomas. Hum Pathol. 2004;35:122–8. doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong A, Eck SL. EpCAM: A new therapeutic target for an old cancer antigen. Cancer Biol Ther. 2003;2:320–6. doi: 10.4161/cbt.2.4.451. [DOI] [PubMed] [Google Scholar]
- 4.Spizzo G, Went P, Dirnhofer S, et al. High Ep-CAM expression is associated with poor prognosis in node-positive breast cancer. Breast Cancer Res Treat. 2004;86:207–13. doi: 10.1023/B:BREA.0000036787.59816.01. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt M, Hasenclever D, Schaeffer M, et al. Prognostic effect of epithelial cell adhesion molecule overexpression in untreated node-negative breast cancer. Clin Cancer Res. 2008;14:5849–55. doi: 10.1158/1078-0432.CCR-08-0669. [DOI] [PubMed] [Google Scholar]
- 6.Spizzo G, Obrist P, Ensinger C, et al. Prognostic significance of Ep-CAM AND Her-2/neu overexpression in invasive breast cancer. Int J Cancer. 2002;98:883–8. doi: 10.1002/ijc.10270. [DOI] [PubMed] [Google Scholar]
- 7.Osta WA, Chen Y, Mikhitarian K, et al. EpCAM is overexpressed in breast cancer and is a potential target for breast cancer gene therapy. Cancer research. 2004;64:5818–24. doi: 10.1158/0008-5472.CAN-04-0754. [DOI] [PubMed] [Google Scholar]
- 8.Nasr AF, Nutini M, Palombo B, Guerra E, Alberti S. Mutations of TP53 induce loss of DNA methylation and amplification of the TROP1 gene. Oncogene. 2003;22:1668–77. doi: 10.1038/sj.onc.1206248. [DOI] [PubMed] [Google Scholar]
- 9.Spizzo G, Gastl G, Obrist P, et al. Methylation status of the Ep-CAM promoter region in human breast cancer cell lines and breast cancer tissue. Cancer Lett. 2007;246:253–61. doi: 10.1016/j.canlet.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Ventura A, Meissner A, Dillon CP, et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A. 2004;101:10380–5. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002;99:8467–72. doi: 10.1073/pnas.132268899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mirza A, Wu Q, Wang L, et al. Global transcriptional program of p53 target genes during the process of apoptosis and cell cycle progression. Oncogene. 2003;22:3645–54. doi: 10.1038/sj.onc.1206477. [DOI] [PubMed] [Google Scholar]
- 13.Roger L, Gadea G, Roux P. Control of cell migration: a tumour suppressor function for p53? Biology of the cell/under the auspices of the European Cell Biology Organization. 2006;98:141–52. doi: 10.1042/BC20050058. [DOI] [PubMed] [Google Scholar]
- 14.Guo F, Gao Y, Wang L, Zheng Y. p19Arf-p53 tumor suppressor pathway regulates cell motility by suppression of phosphoinositide 3-kinase and Rac1 GTPase activities. The Journal of biological chemistry. 2003;278:14414–9. doi: 10.1074/jbc.M300341200. [DOI] [PubMed] [Google Scholar]
- 15.Guo F, Zheng Y. Rho family GTPases cooperate with p53 deletion to promote primary mouse embryonic fibroblast cell invasion. Oncogene. 2004;23:5577–85. doi: 10.1038/sj.onc.1207752. [DOI] [PubMed] [Google Scholar]
- 16.Winter MJ, Cirulli V, Briaire-de Bruijn IH, Litvinov SV. Cadherins are regulated by Ep-CAM via phosphaditylinositol-3 kinase. Molecular and cellular biochemistry. 2007;302:19–26. doi: 10.1007/s11010-007-9420-y. [DOI] [PubMed] [Google Scholar]
- 17.Winter MJ, Nagelkerken B, Mertens AE, Rees-Bakker HA, Briaire-de Bruijn IH, Litvinov SV. Expression of Ep-CAM shifts the state of cadherin-mediated adhesions from strong to weak. Exp Cell Res. 2003;285:50–8. doi: 10.1016/s0014-4827(02)00045-9. [DOI] [PubMed] [Google Scholar]
- 18.Ho J, Benchimol S. Transcriptional repression mediated by the p53 tumour suppressor. Cell Death Differ. 2003;10:404–8. doi: 10.1038/sj.cdd.4401191. [DOI] [PubMed] [Google Scholar]
- 19.Tai KY, Shiah SG, Shieh YS, et al. DNA methylation and histone modification regulate silencing of epithelial cell adhesion molecule for tumor invasion and progression. Oncogene. 2007;26:3989–97. doi: 10.1038/sj.onc.1210176. [DOI] [PubMed] [Google Scholar]
- 20.Esteve PO, Chin HG, Pradhan S. Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci U S A. 2005;102:1000–5. doi: 10.1073/pnas.0407729102. [DOI] [PMC free article] [PubMed] [Google Scholar]