

Abstract
Researchers have used transmission electron microscopy (TEM) to make contributions to cell biology for well over 50 years, and TEM continues to be an important technology in our field. We briefly present for the neophyte the components of a TEM-based study, beginning with sample preparation through imaging of the samples. We point out the limitations of TEM and issues to be considered during experimental design. Advanced electron microscopy techniques are listed as well. Finally, we point potential new users of TEM to resources to help launch their project.
Transmission electron microscopy (TEM) has been an important technology in cell biology ever since it was first used in the early 1940s. The most frequently used TEM application in cell biology entails imaging stained thin sections of plastic-embedded cells by passage of an electron beam through the sample such that the beam will be absorbed and scattered, producing contrast and an image (see Table 1 for a definition of terms). Because of the short wavelength of the electron beam (100,000-fold shorter than photons in visible light), TEM can achieve subnanometer resolution—well below that of even the highest-resolution light microscopes, ∼20 nm. Similar to our longer-wavelength light-microscopy brethren, electron microscopists are faced with a growing array of instruments for specimen preparation and imaging. The emerging technologies focus on preserving the native ultrastructure of samples by vitrification, achievement of higher resolution, three-dimensional ultrastructure by electron tomography, and precise correlation of specific cell structures using immunofluorescence microscopy and TEM (correlative light and electron microscopy [CLEM]). The combination of these techniques is being used to cover the complete spectrum of structure, from molecular to whole cell. Here we discuss conventional TEM of thin sections for morphology and immunolocalization. Even when the EM is done with the assistance of a core facility, we suggest that it is worthwhile for cell biologists who propose to use TEM in their studies to be well enough versed in the specimen preparation and imaging technologies to design appropriate experiments and be able to participate in trouble shooting when necessary.
TABLE 1:
Talk EM Like a Pro.
| Term | Definition |
|---|---|
| Beem capsule | Plastic forms that hold samples in resin during polymerization |
| Blocks (bullets) | Polymerized samples in plastic removed from the Beem capsule and ready to section |
| Block face | Small surface trimmed on a block before sectioning |
| Boat | Water reservoir in which sections float after being cut by a knife |
| CLEM | Correlative light and electron microscopy |
| Dehydration | Removal of water from a sample by replacement with solvent |
| Electron tomography (ET) | A method to image thick sections (200–300 nm) and produce three-dimensional images |
| Embedding | Process of infiltrating the sample with resin |
| Fixation | Sample preservation with low temperature and/or chemicals to maintain sample integrity |
| Grid | Small metal support that holds the sections for viewing in the electron microscope |
| HPF/FS | High-pressure freezing/freeze substitution sample preparation technique |
| Immuno-EM | Detection of proteins in EM samples using antibodies |
| In-FXXKing credible!!!! | Actual user quote in response to particularly beautiful sample. You may embellish with your own words. |
| Knife | A very sharp edge, either glass or diamond, used to slice off resin-embedded samples into sections |
| Pre-embedding labeling | Application of antibodies before fixation and embedding |
| Post-embedding labeling | Application of antibodies to sections on the grid |
| Poststaining | Staining with heavy metals of sections on a grid |
| Resin | Liquid form of the plastics used for embedding |
| Ribbon | Collection of serial sections placed on the grid |
| Serials sections | One-after-the-other thin sections in a ribbon |
| TEM | Transmission electron microscopy |
| Thin sections | The 60- to 70-nm sections cut from the samples in blocks |
| Trimming | Process of cutting away excess resin to create a block face |
| Ultramicrotome | Instrument used to cut sections |
| Vitrification/vitreous ice | Unordered ice in which samples can be viewed without fix or stain |
TEM has proven valuable in the analysis of nearly every cellular component, including the cytoskeleton, membrane systems, organelles, and cilia, as well as specialized structures in differentiated cells, such as microvilli and the synaptonemal complex. There is simply no way to visualize the complexity of cells and see cellular structures without TEM. Despite its power, the use of TEM does have limitations. Among the limitations are the relatively small data set of cells that can be imaged in detail, the obligate use of fixed—therefore deceased—cells, and the ever-present potential for fixation and staining artifacts. However, many of these artifacts are well known and have been catalogued (e.g., Bozzola and Russell, 1999; Maunsbach and Afzelius, 1999).
A typical TEM experiment consists of two phases: the live-cell experiment, in which a cell type, possibly a mutant, is grown under given conditions for analysis, followed by preparation of the specimen and imaging by TEM. Specimen preparation for conventional TEM is comprehensively reviewed in Hayat (1970) and briefly described here (Figure 1).
FIGURE 1:
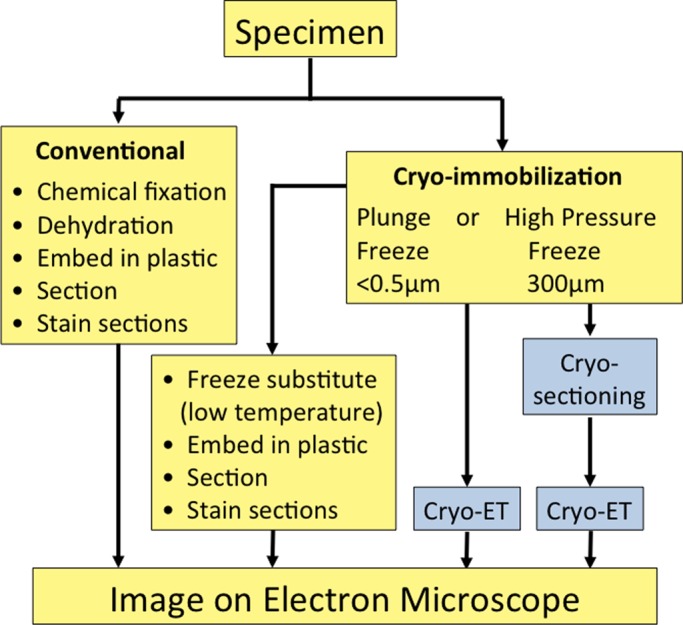
A brief flowchart showing the work to be done with different types of sample preparation for conventional electron microscopy (yellow background). The advanced cryo-EM techniques are shown with a blue background. For immuno-EM, the samples can be stained before embedding (pre-embedding staining) or the sections can be stained (post-embedding staining).
SAMPLE PREPARATION
Traditionally, cells are chemically fixed at room temperature using glutaraldehyde, followed by osmium tetroxide. Glutaraldehyde primarily cross-links proteins. Osmium tetroxide reacts strongly with membrane lipids and also with proteins (and is dangerous). Osmium is a heavy enough metal to confer some electron density to the membranes and other cellular structures to which it binds. Because this method depends on the diffusion of the fixative into the cell, concerns include slow infiltration of the fixative and extraction of cellular contents. Both of these issues can lead to fixation artifacts, such as distorted cellular membranes or organelles, and to loss of material, making the cell appear less dense than it is in reality. This is especially a concern with specimens that contain natural barriers to diffusion, such as a cell wall or cuticle. Nonetheless, the relative speed and simplicity of these protocols has led to their continued use for routine analysis and for a rapid first look at new samples destined for more-advanced approaches. Furthermore, many selective fixation and staining protocols and reagents exist that have been optimized for various cellular structures or compartments. An excellent resource for protocols and procedures for EM sample preparation can be found in the Practical Methods for Electron Microscopy series (e.g., Glauert and Reid, 1975).
After fixation with aqueous fixatives, samples are dehydrated in increasing concentrations of a solvent. Common solvents used are acetone or ethanol, often followed by propylene oxide. These reagents all have the necessary capacity to extract cellular water and later serve as solvents for the embedding resin.
Significant advances in sample preparation have been achieved by rapid freezing of cells, tissue samples, or small organisms under high pressure (Gilkey and Staehelin, 1986; Moor, 1987; Dahl and Staehelin, 1989; McDonald, 1999, 2007). Other methods for cryofixation of biological samples exist, including plunge freezing, slam freezing, and propane jet freezing. However, only plunge freezing in propane or ethane is in wide use, primarily for subcellular samples and monolayers of tissue culture cells. Freezing under high pressure suppresses the formation of very damaging ice crystals as the sample is rapidly cooled (∼200°C in 10 ms). The advantages of high pressure freezing (HPF) are rapid cessation of cellular activity, more faithful preservation of ultrastructure, and better retention of antigenicity for subsequent immunolabeling. Disadvantages of HPF include the high cost of the equipment, the relatively small size of sample that can be vitrified, and, in many cases, the necessity to use some reagents to further suppress the formation of ice crystals (cryoprotectants). There are excellent reviews on the principles, practice, and utility of HPF sample preparation (Moor, 1987; McDonald et al., 2007).
HPF samples destined for conventional TEM are dehydrated at low temperature by a process called freeze substitution (FS). Basically, the frozen water within the sample dissolves (as opposed to melts) in a solvent, usually acetone or methanol, at about −80 to −90°C. The FS cocktail generally contains one or more of the same fixatives used for aqueous, ambient-temperature fixations. Most FS protocols include a gradual warming to temperatures that permit fixation chemistry to occur at a reasonable rate.
Embedding is the process of infiltrating the sample with resins that can be polymerized into a hard plastic suitable for thin sectioning. The process is similar for samples produced by traditional protocols or by freeze substitution. A variety of embedding resins are available. Epoxy resins such as Epon are among the easiest to section and allow for excellent poststaining. Epoxy resins are normally polymerized at 60–70°C and are not conducive to immunolabeling. Methacrylate resins such as the Lowicryls can infiltrate into dehydrated specimens and be polymerized at low temperature by ultraviolet light. Combined with high-pressure freezing and freeze substitution, embedding in these resins retains antigenicity, making HPF/FS samples suitable for post-embedding immunolabeling as described later. In any event, embedding and curing in any resin should yield a hard “block” with the sample in it: congratulations, you've created a fossil.
SECTIONING AND STAINING
To view the sample in the EM, thin sections (∼60–80 nm) must be cut from the block. There is real art to sectioning. The face of the block must be trimmed with a razor blade or glass microtome knife to a neat trapezoid, generally <1 mm on a side. Cutting very small sections allows the loading of dozens of serial sections on an EM specimen support called a grid. Once trimmed, the block is mounted in an ultramicrotome—a specialized machine that cuts sections of the material by slowly advancing the block face by small, precisely controlled increments over a diamond or glass knife edge to produce sections of a given thickness selected by the operator.
Sections are too small and fragile to be directly manipulated with forceps or other tools. Sections float off the ultramicrotome knife edge onto a small, water-filled reservoir built into the knife, and the sections must be carefully transferred onto metal grids. When done well, the adjacent sections will adhere to each other, creating a ribbon of serial sections, which are invaluable to tracking structures within individual cells. Ribbons are transferred to grids by a variety of methods, all of which are nerve wracking. Grids come with various mesh patterns or open slots through which the sections can be imaged. To support the sections over these holes, grids are coated with a thin plastic (Formvar is popular) that can be strengthened with carbon coating. The final step in sample preparation before imaging is postsectioning staining. This is often done with uranyl acetate, followed by lead citrate to enhance contrast, and is easily done by floating the grids, sections side down, on droplets of stain, followed by water rinses.
In addition to the broad-spectrum stains that lend contrast to all cell constituents, there are methods for selectively marking target molecules with antibodies (Griffiths et al., 1993). Pre-embedding labeling involves using primary and gold-labeled secondary antibodies on samples before fixing and embedding the material. This technique can yield strong staining signal but sometimes at a cost to preservation quality because the staining protocol is prior to fixation. Post-embedding immuno–electron microscopy protocols typically consist of placing the electron microscope grid–containing sections on a drop of primary antibody, often in a buffer containing bovine serum albumin, gelatin, or milk, and rinsing, followed by exposure to secondary antibody conjugated to colloidal gold particles in the 5- to 20-nm-diameter range. To retain antigenicity of the fixed material used for postsectioning immuno–electron microscopy, osmium tetroxide is usually eliminated, and the concentration of glutaraldehyde is reduced or eliminated. As mentioned, methacrylate resins are often used for immuno-EM samples. Often, more-hydrophilic embedding resins such as Lowicryl K4M and LR White yield stronger immunolabeling than the more-hydrophobic varieties such as Lowicryl HM20. In either case, the target antigen may be a native protein or one that has been tagged with a marker peptide sequence. Green fluorescent protein is a particularly useful epitope tag, as its fluorescence can be exploited to follow the localization of the marked protein in living cells. When necessary, the correlation can be made between fluorescence and electron micrographs of the same cell, before and after specimen preparation, a technique referred to as CLEM (Nixon et al., 2009; Kukulski et al., 2011; Muller-Reichert and Verkade, 2012). Finally, reporting of immuno-EM staining results should be quantified by compiling data from a number of stained structures in various cells and samples, followed by checking for background signal with secondary antibody–only controls. A fine example of this approach can be found in Rout et al. (2000).
IMAGING
The stained sections on grids are now ready for examination in the electron microscope. A grid is inserted into the column of the electron microscope using a holder that places the grid in the electron beam. Modern electron microscopes have extensive, user-friendly computer interfaces, making it relatively simple to learn how to safely operate the instruments. Digital cameras not only record the images but are also used for locating areas of interest, focusing, and correcting astigmatism and have almost universally replaced traditional film.
A general strategy for examining a sample is to begin at low magnification (in the 100–1000× range) to find the sections on the grid and perhaps choose cells to examine at higher magnification. Newer EMs have software tools that allow the user to map the grid and remember specific locations that can then be revisited automatically, a feature that is particularly useful for tracking a given cell through serial sections. In most cases, the best strategy is to take plenty of digital images, sort through them after the microscopy session, and perhaps share them with colleagues knowledgeable about EM and about the cellular ultrastructure of interest. Figure 2 shows examples of the complexity of cell structure as viewed in the TEM from samples prepared by conventional chemical fixation, such as the elaborate cytoskeletal arrays in cultured myocytes (Figure 2A) to the detailed ultrastructure of cellular organelles (Figure 2B). Excellent ultrastructural preservation, particularly of membrane organelles such as the Golgi, can be obtained using HPF/FS samples (Figure 2C).
FIGURE 2:

Cell structure as visualized by transmission electron microscopy. (A) Actin-myosin cytoskeleton revealed in a cultured cardiomyocyte prepared by conventional chemical fixation. Bar, 1 µm (B) Cytoplasmic organelles in a mouse macrophage prepared by conventional chemical fixation. Bar, 700 nm (C) Golgi membranes in a cultured 3T3 cell prepared by high-pressure freezing and freeze substitution. Bar, 200 nm. (D) Three-dimensional tomographic model of a forming mitotic spindle from budding yeast. Bar, 200 nm.
ADVANCED PROCEDURES
These brief comments concern conventional TEM using thin sections of plastic-embedded material. There are a number of advanced procedures aimed at better sample preservation or more specialized imaging. Electron tomography is an approach that produces three-dimensional image data from a series of images taken as the specimen is tilted at 1 or 2° increments over a ±60–70° range. Software interfaces capable of automating the tilt series collection and assembling tomograms are available from TEM manufacturers and as freeware (Kremer et al., 1996; Mastronarde, 1997). Tomography is often done with thick sections (200–300 nm imaged on an intermediate voltage electron microscope), making it possible to image complex structures in a single section (McIntosh et al., 2005). Three-dimensional models of cellular structures can then be created from the volume data, such as the forming yeast spindle shown in Figure 2D. An emerging technology for the preservation of unaltered molecular structure is to prepare samples in vitreous (unordered) ice without fixation or stain. To image such samples in situ, most cells must be vitrified and then sectioned with cryo-ultramicrotomes to produce frozen sections, a technique referred to as vitreous sectioning (Al-Amoudi et al., 2004; Bouchet-Marquis and Hoenger, 2011). Frozen sections are imaged using specialized cryo-specimen holders in the TEM. The idea is to image the cell in a manner close to its native state. Because the cells have not been fixed or stained, contrast is extremely low, and overall cellular structure can be difficult or impossible to discern. Thus many studies begin with the type of conventional TEM described here and progress to electron tomography, cryo-EM, or cryo–electron tomography to push the resolution and refine the accuracy of a particular structure. In many cases several types of data contribute to the understanding of the structure of interest.
HOW TO GET STARTED?
TEM is unrivaled in its ability to let one see inside the cell and appreciate the complexity of a chosen structure or organelle. The techniques are demanding and expensive and may require the expertise of a specialist to accomplish successfully. Experimental design is therefore key; the question to address must be narrowly defined, such that it can be clearly answered with a reasonably sized data set, which in an EM experiment is likely to be small (tens of cells). Investigators new to TEM may choose to work with their local EM core facility, or they can establish a collaboration with a group that has expertise in TEM analysis of the cell type, organism, or structure in question. In any case, there are significant advantages to having the scientist responsible for the experiment be directly involved with the imaging. Familiarity with the system and the experiment will facilitate the search for relevant data, and the process of searching for the best images imparts significantly more information than is captured in images. Lucky investigators will find that their core facility has appropriate cell type/organism-specific experience, and the facility will be able to help the investigator plan and execute the EM work. Barring such an experienced core facility, it is advisable to contact an investigator or core facility that is experienced with one's sample type. Their advice and help with be invaluable and greatly accelerate one's work. In fact, some core facilities offer sample preparation for outside users and will ship the blocks for sectioning and imaging to the user's local facility. However you get it done, TEM is a powerful and simply fun way to do cell biology.
Acknowledgments
Tomography was performed in the Boulder Laboratory for 3D Electron Microscopy of Cells under Grant 8P41GM103431 from the National Institute of General Medical Sciences to Andreas Hoenger. M.W.’s EM work has been supported by grants GM51312 and GM074746 from National Institute of General Medical Sciences.
Footnotes
REFERENCES
- Al-Amoudi A, Chang JJ, Leforestier A, McDowall A, Salamin LM, Norlén LP, Richter K, Blanc NS, Studer D, Dubochet J. Cryo-electron microscopy of vitreous sections. EMBO J. 2004;23:3583–3588. doi: 10.1038/sj.emboj.7600366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet-Marquis C, Hoenger A. Cryo-electron tomography on vitrified sections: a critical analysis of benefits and limitations for structural cell biology. Micron. 2011;42:152–162. doi: 10.1016/j.micron.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Bozzola JJ, Russell LD. Electron Biology Principles and Techniques for Biologists. Sudbury, MA: Jones & Bartlett Learning; 1999. [Google Scholar]
- Dahl R, Staehelin LA. High-pressure freezing for the preservation of biological structure: theory and practice. J Electron Microsc Tech. 1989;13:165–174. doi: 10.1002/jemt.1060130305. [DOI] [PubMed] [Google Scholar]
- Gilkey JC, Staehelin LA. Advances in ultrarapid freezing for the preservation of cellular ultrastructure. J Electron Microsc Tech. 1986;3:177–210. [Google Scholar]
- Glauert AM, Reid N. Fixation, Dehydration and Embedding of Biological Specimens. New York: Elsevier; 1975. [Google Scholar]
- Griffiths G, Burke B, Lucocq J. Fine Structure Immunocytochemistry. New York: Springer-Verlag; 1993. [Google Scholar]
- Hayat MA. Principles and Techniques of Electron Microscopy, Biological Applications, Vol. 1. New York: Van Nostrand-Reinhold; 1970. [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kukulski W, Schorb M, Welsch S, Picco A, Kaksonen M, Briggs JA. Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision. J Cell Biol. 2011;192:111–119. doi: 10.1083/jcb.201009037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN. Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol. 1997;120:343–352. doi: 10.1006/jsbi.1997.3919. [DOI] [PubMed] [Google Scholar]
- Maunsbach AB, Afzelius BA. Biomedical Electron Microscopy: Illustrated Methods and Presentations. San Diego: CA: Academic Press; 1999. [Google Scholar]
- McDonald KL. High-pressure freezing for preservation of high-resolution fine structure and antigenicity for immunolabeling. Methods Mol Biol. 1999;117:77–97. doi: 10.1385/1-59259-201-5:77. [DOI] [PubMed] [Google Scholar]
- McDonald K. Cryopreparation methods for electron microscopy of selected model systems. Methods Cell Biol. 2007;79:24–52. doi: 10.1016/S0091-679X(06)79002-1. [DOI] [PubMed] [Google Scholar]
- McDonald KL, Morphew M, Verkade P, Mueller-Reichert T. Recent advances in high-pressure freezing: equipment and specimen loading methods. Methods Mol Biol. 2007;369:143–173. doi: 10.1007/978-1-59745-294-6_8. [DOI] [PubMed] [Google Scholar]
- McIntosh JR, Nicastro D, Mastronarde D. New views of cells in 3D: an introduction to electron tomography. Trends Cell Biol. 2005;15:43–51. doi: 10.1016/j.tcb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Moor H. Theory and practice of high pressure freezing. In: Steinbrecht RA, Zierold K, editors. In: Cryotechniques in Biological Electron Microscopy. Berlin: Springer; 1987. pp. 175–191. [Google Scholar]
- Muller-Reichert T, Verkade P. Introduction to Correlative Light and Electron Microscopy. New York: Elsevier; 2012. [DOI] [PubMed] [Google Scholar]
- Nixon SJ, Webb RI, Floetenmeyer M., Schieber N, Lo HP, Parton RG. A single method for cryofixation and correlative light, electron microscopy and tomography of zebrafish embryos. Traffic. 2009;10:131–136. doi: 10.1111/j.1600-0854.2008.00859.x. [DOI] [PubMed] [Google Scholar]
- Rout MP, Aitchison JD, Suprapto A, Hjertaas K, Zhao Y, Chait BT. The yeast nuclear pore complex: composition, architecture, and transport mechanism. J Cell Biol. 2000;148:635–651. doi: 10.1083/jcb.148.4.635. [DOI] [PMC free article] [PubMed] [Google Scholar]