

Real-time recordings of ER ATP fluxes in single cells using an ER-targeted, genetically encoded ATP sensor within the lumen of the ER reveal a local Ca2+-controlled ATP signal that is restored during energy stress. The data highlight a novel Ca2+-controlled process that supplies the ER with additional energy upon cell stimulation.
Abstract
Multiple functions of the endoplasmic reticulum (ER) essentially depend on ATP within this organelle. However, little is known about ER ATP dynamics and the regulation of ER ATP import. Here we describe real-time recordings of ER ATP fluxes in single cells using an ER-targeted, genetically encoded ATP sensor. In vitro experiments prove that the ATP sensor is both Ca2+ and redox insensitive, which makes it possible to monitor Ca2+-coupled ER ATP dynamics specifically. The approach uncovers a cell type–specific regulation of ER ATP homeostasis in different cell types. Moreover, we show that intracellular Ca2+ release is coupled to an increase of ATP within the ER. The Ca2+-coupled ER ATP increase is independent of the mode of Ca2+ mobilization and controlled by the rate of ATP biosynthesis. Furthermore, the energy stress sensor, AMP-activated protein kinase, is essential for the ATP increase that occurs in response to Ca2+ depletion of the organelle. Our data highlight a novel Ca2+-controlled process that supplies the ER with additional energy upon cell stimulation.
INTRODUCTION
The endoplasmic reticulum (ER) is a complex organelle within eukaryotic cells that is central to synthesis (Groenendyk et al., 2010), glycosylation (Banerjee, 2012), folding and assembly (Naidoo, 2009), degradation (Merulla et al., 2013), and secretion of proteins (Zanetti et al., 2012). These processes are essential, and any defect or delay would have detrimental or even lethal effects on cells and the whole organism (Rasheva and Domingos, 2009). For all of them, energy in the form of ATP is required (Fang et al., 2010). In the ER, ATP is either consumed to run these processes or acts as a messenger or cofactor to initiate and maintain them (Bukau et al., 2006). Several steps during protein folding in the ER, such as the formation of disulfide bonds, need a constant supply of ATP to fulfill their energetic demands (Chia et al., 2012). It has been shown that the activity of ER chaperones is modulated by binding and hydrolysis of ATP (Braakman and Bulleid, 2011), whereas under conditions of ATP depletion, protein processing within the ER is impaired (Kapoor and Sanyal, 2009). Although these findings indicate that ATP transport into the ER is essential for protein folding, it is not clear whether the ER protein-folding machinery is indeed controlled by ER ATP transport. In addition, the ER reacts to a number of different cellular stress conditions by activating the unfolded protein response (UPR; Ron and Walter, 2007; Korennykh and Walter, 2012). The UPR is an early stress response, which activates a number of pathways to reestablish homeostasis and minimize cell damage in a first instance, but if that fails, UPR triggers programmed cell death (Rasheva and Domingos, 2009; Han et al., 2013). ATP within the ER has been found to control the dissociation of the glucose-regulated protein 78, an ER-resident chaperone, from the inositol response element 1, which is one of the main initiators of the UPR (Oikawa et al., 2009). These findings implicate the importance of ATP transport into the ER to maintain protein folding and control the UPR. In addition, many other processes, such as protein phosphorylation (Ron and Harding, 2012), glycosylation (Mohorko et al., 2011), and sterol biosynthesis (Blom et al., 2011) within the ER, require ATP. However, little is known about the regulation and identity of ER ATP transporters.
Cellular ATP is generated during glycolysis in the cytosol and more efficiently within mitochondria by oxidative phosphorylation (OXPHOS). Because the ER is incapable of generating ATP autonomously, it is assumed that ATP is transported into this organelle (Hirschberg et al., 1998). However, there are conflicting reports regarding the need for an active ER ATP transport, as passive diffusion of ATP across the rather leaky ER membrane was suggested (Le et al., 2004). So far, identification of ER ATP carrier(s) of mammals has not been made, but ATP translocation was characterized in ER-derived vesicles and proteoliposomes in vitro (Shin et al., 2000). Of note, a protein referred to as ER-ANT1 was identified as an ATP/ADP transporter of the ER in the plant Arabidopsis thaliana (Leroch et al., 2008), whereas Sac1p was shown to play an important role for the transport of ATP into the lumen of the ER in Saccharomyces sp. (Kochendörfer et al., 1999). Such findings point to the existence of regulated ER ATP transport machineries that might contribute to the regulation of vital processes in the lumen of this organelle.
Owing to the lack of suitable tools to monitor local ATP levels within intact cells, little is known about ER ATP fluxes in living cells. To overcome these limitations, we targeted a genetically encoded ATP probe to the lumen of the ER, which is based on Förster resonance energy transfer (FRET) between fluorescent proteins. Genetically encoded fluorescent ATP sensors, referred to as ATeams, were used to measure ATP levels in the cytosol, nucleus, and mitochondria of HeLa cells (Imamura et al., 2009). In this study we show that this approach can also be used for real-time monitoring of ATP dynamics within the lumen of the ER on the single-cell level. Using the ER ATP probe, we reveal ER ATP signals that are controlled by ER Ca2+ release. Our data point to high ER ATP dynamics that is primarily controlled by the ER Ca2+ content ([Ca2+]ER]) in an inverse manner. We identify the AMP-activated protein kinase (AMPK), a central sensor of energy stress (Hardie et al., 2012), as an essential determinant of Ca2+-coupled ER ATP increase. Eventually, we provide an original avenue to probe how the ATP content of the ER is altered in living cells under physiological and pathological conditions.
RESULTS
Targeting of a genetically encoded ATP probe to the ER
To monitor ATP within the lumen of the ER on the single-cell level, we constructed an ER-targeted, genetically encoded, FRET-based ATP probe, ERAT4.01, analogous to a previously reported ATP sensor (Imamura et al., 2009). The ATP probe reversibly binds ATP at the ε-subunit of the F0F1-ATP synthase from Bacillus subtilis. ATP binding induces a conformational rearrangement within the ATP sensor, which narrows the distance between the N- and C-terminal cyan and yellow fluorescent proteins (CFP and YFP), respectively, yielding increased FRET (Figure 1A). For ER targeting, the calreticulin signal sequence was fused to the N-terminus and the KDEL sequence, a classic ER-retention signal (Pelham, 1990; Clairmont et al., 1992), was added to the C-terminus of the ATP probe (Figure 1A). The ER-targeted ATP probe localized to the ER as expected (Figure 1B). Use of high-resolution array confocal microscopy revealed that most cells displayed correct ER localization of the ATP probe, whereas in 9–38% of the cells tested, ERAT4.01 was mistargeted, depending on the cell type (Supplemental Figure S1). Of note, in all imaging experiments, cells with correct targeting of ERAT4.01 were selected and analyzed exclusively.
FIGURE 1:

Targeting of a FRET-based genetically encoded ATP Probe to the ER. (A) Schematic representation of the ER-targeted ATP probe ERAT4.01. Binding of ATP to the ε-subunit of the bacterial FoF1-ATP synthase induces a conformational change within ERAT4.01, which is detected by a change of the FRET between the terminal enhanced CFP and the YFP variant citrine. ER targeting is achieved by adding both the calreticulin signal domain and the KDEL sequence. (B) EA.hy926 cells expressing both ERAT4.01 (left, green) and an ER-targeted RFP (middle, red), which colocalizes with the ERAT4.01 fluorescence signal (merge image, right).
ERAT4.01 senses ER ATP depletions in real time
First we tested the ER-targeted ATP probe in HeLa cells, which are known to generate ATP primarily via anaerobic glycolysis (Lu et al., 2002). Inhibition of glycolysis with the d-glucose analogue 2-deoxy-d-glucose (2-DG) gradually reduced the FRET ratio of ERAT4.01 (Figure 2, A and B). This drop in the ratio under these conditions was based on a decreased fluorescence intensity in the FRET channel accompanied by a significant increase of the donor (CFP) signal (Supplemental Figure S2A), indicating that the probe senses changes of the ATP concentration in the ER ([ATP]ER) in a ratiometric manner. The gradual and steady decline in the FRET ratio started 2.50 ± 0.18 min (average ± SEM, n = 10) after 2-DG addition, and the signal reached a minimum within 6.47 ± 0.48 min (average ± SEM, n = 10). Subsequent replacement of 2-DG by d-glucose in the medium partially restored the FRET signal in ∼20% of the HeLa cells tested (Figure 2, A and B), indicating that the probe senses ATP in the lumen of the ER in a dynamic and reversible manner. However, cells expressing AT1.03, an analogous genetically encoded cytosolic ATP sensor, showed a delayed decrease in the AT1.03 signal in response to 2-DG (Supplemental Figure S2, B–E). The reliability of the ER-targeted ATP probe to sense cellular ATP depletion was examined using four different cell types. In all of them the inhibition of ATP generation by a mixture of 2-DG and the mitochondrial ATP synthase inhibitor oligomycin A significantly reduced the FRET signal by ERAT4.01 over time. However, the kinetics of the decline varied among the individual cell types (Figure 2C). These findings indicate that ATP is differently transferred into and/or consumed in the ER under resting conditions, depending on the cell type. The basal FRET ratio, reflecting ER ATP levels under resting conditions, also varied in the different cell types tested (Figure 2D). These findings point to cell type–specific ER ATP levels and homeostasis.
FIGURE 2:

ERAT4.01 senses ER ATP on inhibition of ATP-generating processes in different cell types. (A) Representative pseudocolored ratio (F535/F480) images over time of two HeLa cells expressing ERAT4.01 that were treated with 10 mM 2-DG. Red pixels indicate high ratio values (>2), and blue pixels indicate low ratio values (<2). (B) Nonnormalized ratios of ERAT4.01 signals expressed in the two HeLa cells presented in A in response to 10 mM 2-DG. Dotted lines represent individual bleaching functions (R0) of the ratios, which were used for normalization. (C) Representative average curves of ratio ERAT4.01 signals over time in four different cell types in response to 10 mM 2-DG and 2 μM oligomycin A. Curves were fitted using the Boltzmann sigmoid function in Prism software 5.01 (GraphPad, La Jolla, CA). (D) Columns represent basal ratio values of ERAT4.01 in four different cell types: EA.hy926 (green, n = 17), INS-1 832/13 (red, n = 61), HEK-293 (blue, n = 25), and HeLa cells (purple, n = 23). *p < 0.05 vs. basal ratio value of ERAT4.01 expressed in EA.hy926 cells. (E) Comparison of the reduction of normalized ratio values over time between the mitochondria-targeted ATP probe mtAT1.03 (red dotted line) and ERAT4.01 (green continuous line) in response to 10 mM 2-DG using HeLa cells (left). Middle, statistical data of the maximal drop of normalized signal ratios in response to 2-DG of the mitochondrially targeted ATP probe (red column, n = 13) and the ER ATP sensor (green column, n = 10). Left, statistical analysis of the respective maximal slope of the 2-DG–induced reduction of normalized ratios of mtAT1.03 and ERAT4.01 signals in HeLa cells. *p < 0.05 vs. respective data from mtAT1.03 signals. (F) Representative normalized ratio time course of mtAT1.03 (red dotted line) and ERAT4.01 (green continuous line) in response to 2 μM oligomycin A in HeLa cells. Columns represent respective statistical data regarding the maximal reduction (middle) and the maximal slope (right) of the mitochondrial ATP signal (red columns, n = 5) and the ER ATP signal (green columns, n = 8) shown on the left. *p < 0.05 vs. respective data extracted from mtAT1.03 signals. (G) Representative normalized ratio time course of mtAT1.03 (red dotted line) and ERAT4.01 (green continuous line) in response to 2 μM oligomycin A in INS-1 832/13 cells. For comparison respective curves in HeLa cells from H are added in gray. Bottom, statistical data regarding the maximal reduction (left bottom) and the maximal slope (right bottom) of the mitochondrial ATP signal (red columns, n = 31) and the respective ER ATP signal (green columns, n = 39). *p < 0.05 vs. respective data extracted from mtAT1.03 signals.
The comparison between FRET signals of the mitochondria-targeted ATP probe mitAT1.03 and those of the ER ATP sensor showed that the mitochondrial ATP concentration ([ATP]mito) is affected before and more strongly than [ATP]ER by inhibition of glycolysis in HeLa cells (Figure 2E). However, inhibition of the mitochondrial ATP synthase in HeLa cells only minimally and transiently reduced the FRET ratio of mitAT1.03 and had almost no effect on respective FRET signals of ERAT4.01 (Figure 2F). This observation is in agreement with low levels of OXPHOS in HeLa cells (Supplemental Figure S2F) and previous reports showing that many cancer cells have reduced OXPHOS rates while generating ATP primarily by anaerobic glycolysis (Lu et al., 2002; Mathupala et al., 2010). However, in the clonal pancreatic beta cell line INS-1 832/13, which, in contrast to HeLa cells, shows a much higher rate of OXPHOS and less anaerobic glycolysis (Supplemental Figure S2F), an inhibition of the mitochondrial ATP-synthase reduced rapidly and considerably the ERAT4.01 signal (Figure 2G). Moreover, there was a delay (1.12 ± 0.17 min, n = 18) in the drop of the ratio of the ER-targeted ATP probe as compared with mitAT1.03 in response to oligomycin A (Figure 2G). These results point to an organelle- and cell type–specific ATP turnover and validate ERAT4.01 as a suitable probe to assess [ATP]ER in single living cells.
ER Ca2+ mobilization leads to an increase of ATP within the lumen of the ER
The ER plays a central role in cell signaling by storing and releasing Ca2+ ions (Berridge, 2002). Physiological Ca2+ mobilization from the ER is accomplished by various inositol 1,4,5-trisphosphate (IP3)–generating agonists (Miyazaki, 1993). We speculated that during such cell stimulations the ATP demand within the ER might be altered. Hence we investigated whether changes of [Ca2+]ER correlate with fluctuations of [ATP]ER in response to IP3-generating agonists. In both the glycolytic HeLa cells (Figure 3A) and the OXPHOS-dependent INS-1 832/13 cells (Figure 3B), IP3-mediated [Ca2+]ER depletion was coupled to a distinct elevation of the ERAT4.01 FRET signal. Removal of the IP3-generating agonists and Ca2+ addition to the medium restored [Ca2+]ER and [ATP]ER levels (Figure 3, A and B). Use of carbachol (CCh) as an IP3-generating agonist in INS-1 832/13 cells revealed a half-maximal effective concentration (EC50) of 6.62 (3.82–11.45) μM (n = 10–17) to trigger an increase in the ERAT4.01 FRET signal (Figure 3C), whereas the respective EC50 in HeLa cells, using ATP as an IP3-generating agonist, was 21.77 (10.25–46.21) μM (n = 5–14; Figure 3D). A correlation between the genetically encoded ER Ca2+ sensor D1ER (Palmer et al., 2004) and ERAT4.01 signals showed that the increase in [ATP]ER lagged slightly behind the [Ca2+]ER reduction (Figure 3E). There was a clear linear correlation between the maximal degree of [Ca2+]ER depletion and the increase of [ATP]ER over a concentration range from 1 to 100 μM of the IP3-generating agonist (Figure 3F). This constant proportionality between [Ca2+]ER and [ATP]ER indicates that the drop of Ca2+ within the ER is directly coupled to an elevation of [ATP]ER. In one given HeLa cell a consecutive treatment with a concentration lower than the EC50 followed by a maximal concentration of the IP3-generating agonist evoked a small and a large transient increase in [ATP]ER, respectively (Figure 3G). This finding indicates that the ER ATP signal can be evoked repetitively in an ascending order of concentration of an IP3-generating agonist.
FIGURE 3:

ERAT4.01 reveals an ER ATP increase on Ca2+ mobilization from the ER. (A) Individual responses of HeLa cells expressing the ER-targeted ATP sensor ERAT4.01 (top) or the ER Ca2+ probe D1ER (bottom) in response to cell treatment with 100 μM histamine and 100 μM ATP in the absence of Ca2+ in the medium (top). As indicated, 2 mM Ca2+ was readded upon washout of the IP3-generating agonists. Thick black lines represent respective average curves. (B) ER ATP (top) and ER Ca2+ signals (bottom) in INS-1 832/13 cells expressing either ERAT4.01 or D1ER. Cells were treated with a mixture of 100 μM carbachol (CCh) and 100 μM ATP in Ca2+-free medium before 2 mM Ca2+ was readded. (C) Concentration response curve of carbachol triggering an [ATP]ER increase in INS-1 832/13 cells (n = 10–17). Experiments were performed in the absence of Ca2+ in the medium. (D) Concentration response curve of ATP to trigger an ER ATP increase in HeLa cells (n = 5–14). Experiments were performed in the absence of Ca2+ in the bathing medium. (E) Correlation between the ER Ca2+ content (y-axis) and the [ATP]ER increase (x-axis). Data were extracted from experiments shown in B. Respective maximal delta effects were defined as 100%. (F) Linear correlation between [Ca2+]ER (n = 18–38) and [ATP]ER (n = 10–17) in INS-1 832/13. As indicated, cells were treated with different concentration of CCh in the absence of Ca2+. (G) HeLa cells expressing the ER ATP probe were treated consecutively with different concentrations of ATP. Representative curve is given as percentage of the maximal effect; the delta maximal effect in response to 300 μM ATP was defined as 100%.
To determine whether the Ca2+-induced increase of ATP within the ER requires the IP3 signaling pathway, we depleted the ER Ca2+ store in IP3-independent ways. First we treated cells with 2,5-di-tert-butylhydroquinone (BHQ), an inhibitor of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). Addition of BHQ slowly reduced [Ca2+]ER in Ca2+-free medium and gradually enhanced ER ATP levels (Supplemental Figure S3A). Subsequent stimulation with IP3-generating agonists further decreased [Ca2+]ER and increased [ATP]ER. This observation further confirms that the increase of [ATP]ER depends on the degree of [Ca2+]ER depletion. Thapsigargin, a more potent, irreversible, and selective SERCA inhibitor, also evoked a distinct increase of the ERAT4.01 FRET signal (Supplemental Figure S3B). Moreover, we used the Ca2+ ionophore ionomycin in Ca2+-free medium to deplete the ER Ca2+ store in a SERCA- and IP3-independent manner. Treatment of cells with ionomycin rapidly lowered [Ca2+]ER and caused a respective increase of the ERAT4.01 FRET signal (Supplemental Figure S3, C and D). These results indicate that, independent of its mode, ER Ca2+ mobilization causes a significant elevation of ER ATP level.
Ca2+-coupled ER ATP increase represents MgATP elevation and is neither due to direct effect of Ca2+ on the probe nor related to changes of ER redox
We next performed a series of in vitro experiments in which the Ca2+ sensitivity of the genetically encoded ATP probe was tested (Figure 4). Both the ratio signals of the ER ATP probe in permeabilized cells (Figure 4A) and the spectral changes of the purified ATP probe in vitro remained unaffected by Ca2+ addition in the absence of MgATP (Figure 4, B and E). These data show that the sensor used to study ER ATP dynamics is per se Ca2+ insensitive and indicate that the inverse correlation between [Ca2+]ER and the ERAT4.01 FRET signal in intact cells is not caused by a direct effect of Ca2+ on the sensor. As expected, addition of MgATP significantly increased the ratio signal in a ratiometric manner of ERAT4.01 in permeabilized cells (Figure 4F and Supplemental Figure S4A) and the purified ATP probe in vitro (Figure 4, C–E). However, in the presence of MgATP, addition of Ca2+ (100–500 or 2000 μM) slightly reduced the increased ratio signals of the ATP probe in vitro (Figure 4, C–E) and permeabilized cells (Figures 4G and Supplemental Figure S4B). The effect of Ca2+ on the ratio signal in vitro and permeabilized cells is likely to be caused by a competition between CaATP and MgATP. CaATP is not sensed by the genetically encoded ATP probe (Figure 4H). Accordingly, the Ca2+-coupled increase of the FRET signal of the ER-targeted ATP probe in intact cells (Figure 3) represents an increase in MgATP within the ER.
FIGURE 4:

Characterization of the effect of Ca2+ on the ATP probe in permeabilized cells and in vitro. (A) The effect of Ca2+ on ERAT4.01 signals was evaluated in cells permeabilized with 10 μM ionomycin in the presence of 10 μM digitonin. In permeabilized cells multiple Ca2+elevations (2 mM Ca2+) in the absence of ATP do not affect the FRET signal of ERAT4.01 (solid curve). [Ca2+]ER was detected with D1ER (dotted curve). (B) Fluorescence spectra of the purified ER ATP probe without MgATP and in the presence of different Ca2+ concentrations as indicated. (C) Fluorescence spectra of the purified ER ATP probe in the presence of 4 mM MgATP and in the presence of different Ca2+ concentrations as indicated in B. (D) Fluorescence spectra of the purified ER ATP probe in the presence of 8 mM MgATP and in the presence of different Ca2+ concentrations as indicated in B. (E) ATP-dependent changes of the emission ratio of the purified ATP probe in vitro vs. the Ca2+ concentration. (F) Normalized FRET ratio changes of ERAT4.01 in permeabilized cells upon the addition of 1 mM Mg ATP (white column, n = 12) and 10 mM Mg ATP (black column, n = 20). Respective changes of the fluorescence of the donor and FRET channel are presented in Supplemental Figure S4A. (G) Representative curve representing normalized ratio signals of ERAT4.01 in permeabilized HeLa cells in response to 10 mM MgATP and the subsequent addition and removal of 2 mM Ca2+. For statistical data see Supplemental Figure S4B. (H) Effect of a switch from 10 mM MgATP to 10 mM CaATP in permeabilized HeLa cells. The average curve was normalized to the maximal ratio signal, which was obtained by the addition of 10 mM MgATP (n = 4).
With an ER-targeted, genetically encoded thiol redox sensor it was shown recently that ER Ca2+ depletion induces a significant reductive shift within the organelle (Avezov et al., 2013). Hence we carefully verified whether the inverse correlation between [Ca2+]ER and the ERAT4.01 FRET signal might be due to Ca2+-coupled fluctuations of the ER thiol redox. For this purpose we generated and tested additional genetically encoded, ER-targeted ATP probes that differ in terms of donor and acceptor fluorescent proteins (FPs) and contain mutations within the ε-subunit (Supplemental Figure S4, C–J). Two to three cysteines (Cs), which might form transient reducible disulfide bounds, are present at different positions of all FPs used (two Cs in enhanced CFP, citrine, cpv [circularly permuted venus], and enhanced GFP [EGFP]; three Cs in Tag–red fluorescent protein [RFP]; see Supplemental Figure S4, C–E and G–I) but not within the ATP-binding ε-subunit of the ATP probes. However, a putative glycosylation site, asparagine (N) in position 7 within the ε-subunit, which if glycosylated might cause interactions between the ATP sensor and chaperones within the ER (Helenius and Aebi, 2004), was detected and, hence, mutated to glutamine (Q), yielding respective N7Q mutants of ER-targeted ATP probes (Supplemental Figure S4, D, E, H, and I). In addition CFP/YFP-based (Supplemental Figure S4E) and respective GFP/RFP-based (Supplemental Figure S4I), red-shifted, ER-targeted ATP probes with arginine (R)-to-lysine (K) mutations in positions 122 and 126 of the ε-subunit, yielding respective R122K and R126K mutants, which are ATP insensitive, were generated and tested (Supplemental Figure S4, F and J). Of note, in contrast to the CFP/YFP-based probes, the red-shifted, ER-targeted ATP sensors contain TagRFP, the FRET acceptor FP, on the N-terminus and EGFP, the FRET donor protein, on the C-terminus. With those ER-targeted ATP probes containing the wild-type ε-subunit or the N7Q mutation a significant increase of the FRET ratio signal was observed in response to ER Ca2+ depletion independent of the FPs, and hence the position of cysteines, within the different sensors (Supplemental Figure S4, F and J). In line with these findings, the CFP/YFP and the red-shifted ER ATP sensors that contain the ATP-insensitive R122K, R126K mutated ε-subunit did not respond with an increase of the FRET ratio signal upon ER Ca2+ depletion (Supplemental Figure S4, F and J). These observations indicate that exclusively changes in the level of ATP and not a reductive shift of the ER redox account for the Ca2+-coupled signal and point to the redox insensitivity of the ER-targeted ATP probes. Addition of the reducing agent dithiothreitol in permeabilized (Supplemental Figure S4K) and intact cells (Supplemental Figure S4L) minimally affected fluorescence signals of ERAT4.01 in the absence and presence of MgATP, which further confirms the redox stability of the genetically encoded ATP probe and excludes the possibility that a Ca2+-coupled reductive shift of the ER redox affects the ERAT4.01 FRET signal in intact cells in response to Ca2+ depletion.
[ATP]ER is determined by ER Ca2+ content in an inverse manner independent of [Ca2+]cyto and [Ca2+]mito
Ca2+ mobilization from the ER induces an increase of the cytosolic ([Ca2+]cyto) and mitochondrial Ca2+ concentrations ([Ca2+]mito), which facilitate ATP biosynthesis primarily by stimulating mitochondrial enzymes (Denton, 2009; Nakano et al., 2011). So far our data are not conclusive on whether ER ATP levels are elevated by Ca2+-induced activation of ATP biosynthesis or [Ca2+]ER depletion causes the process. To find out whether elevation of [Ca2+]cyto is sufficient to trigger an increase of [ATP]ER, we measured ER ATP signals in INS-1 832/13 cells treated with high K+. Under these conditions, Ca2+ entry via voltage-dependent, L-type Ca2+ channels elevates [Ca2+]cyto without [Ca2+]ER depletion (Alam et al., 2012). The K+-induced cytosolic Ca2+ elevation (Figure 5A, left) did not increase the ERAT4.01 FRET signal in INS-1 832/13 cells (Figure 5A, middle), whereas ER ATP levels dropped during treatment with high K+. The use of D1ER revealed that under these conditions ER Ca2+ levels increased (Figure 5A, right), confirming an inverse correlation between [Ca2+]ER and [ATP]ER (Figure 3E). Moreover, these experiments indicate that cytosolic Ca2+ elevation alone is not sufficient to trigger the ER ATP signal. In an analogous experiment, HeLa cells were treated with an IP3-generating agonist in the presence of extracellular Ca2+, which resulted in a strong cytosolic Ca2+ elevation. However, the ER Ca2+ content was only partially affected due to activation of store-operated Ca2+ entry (SOCE; Smyth et al., 2010) under this condition (Figure 5B). In correlation with the ER Ca2+ content, there was a partial elevation in [ATP]ER, which was further increased by removal of Ca2+ from the medium (Figure 5B). Simultaneous drop of both [Ca2+]cyto and [Ca2+]ER under this condition confirms that the ER ATP increase is determined by the ER Ca2+ content in an inverse manner. Similar results were obtained in INS-1 832/13 cells, in which the SOCE-mediated increase of [Ca2+]cyto and [Ca2+]mito (Supplemental Figure S5) triggered elevation of ATP within mitochondria, whereas under these conditions [ATP]ER was reduced (Figure 5C) during ER Ca2+ refilling (Figure 5D). These findings further demonstrate that, despite Ca2+-induced augmentation of the mitochondrial ATP biosynthesis rate, [Ca2+]ER inversely determines [ATP]ER.
FIGURE 5:
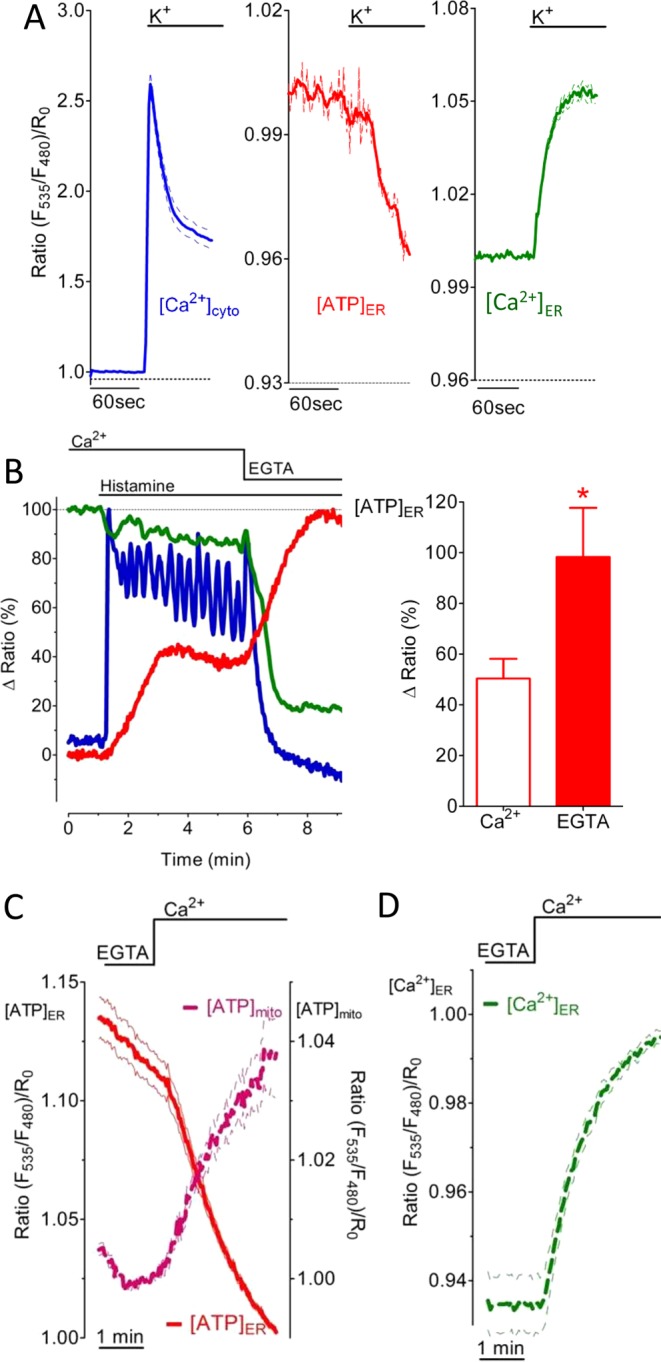
ER Ca2+ content controls [ATP]ER in an inverse manner independent of [Ca2+]cyto and [Ca2+]mito. (A) Average normalized mean ratio signal of [Ca2+]cyto ± SEM over time (left), mean ERAT4.01 signal (middle), and [Ca2+]ER (right) in INS-1 832/13 cells that were treated with 130 mM K+ (n = 21). (B) Ca2+ was mobilized in HeLa cells by 100 μM histamine in the presence of 2 mM extracellular Ca2+ (blue curve) and propagated in the absence of Ca2+ in the medium as indicated. Curves show representative responses in [Ca2+]ER (green), [ATP]ER (red), and [Ca2+]cyto (blue) of HeLa cells expressing D1ER, ERAT4.01, or the cytosolic Cameleon, respectively. Right, relative [ATP]ER increase in presence of Ca2+ (white column, n = 13) or its absence (red column, n = 13). *p < 0.05 vs. ERAT4.01 signal measured in the presence of extracellular Ca2+. (C) Average signals ± SEM over time of [ATP]mito (magenta dotted curve, n = 25) and [ATP]ER (red curve n = 21) in INS-1 832/13 cells upon SOCE. Cells expressed either the mitochondria-targeted ATP probe mtAT1.03 or the ER-targeted ER ATP sensor ERAT4.01. (D) ER Ca2+ refilling upon SOCE measured in INS-1 832/13 cells expressing D1ER (n = 6).
Ca2+-coupled ER ATP signal requires ATP synthesis
So far our data do not answer whether [Ca2+]ER depletion reduces the consumption of ATP within the organelle or evokes ATP transfer across the ER membrane. To clarify this point, we blocked the supply of ATP by inhibition of ATP-generating processes. Inhibition of glycolysis with 2-DG in HeLa cells abolished the [ATP]ER increase (Figure 6, A, top, and B, left), although ER Ca2+ release was unaffected (Figure 6, A, bottom, and B, right). Similar results were obtained when [Ca2+]ER was passively emptied by ionomycin (Supplemental Figure S6A), indicating that the inhibitory effect of 2-DG on the ER ATP signal in HeLa cells was independent of the mode of Ca2+ mobilization. However, inhibiting the ATP synthase with oligomycin A even facilitated the increase in [ATP]ER upon ER Ca2+ depletion in HeLa cells (Figure 6, A and B). These data indicate that the ATP elevation observed in the ER is not due to reduced ATP consumption but due to increased ATP transfer or bioavailability in the organelle upon ER Ca2+ release. Moreover, these data further confirm that in HeLa cells the [Ca2+]ER -controlled ATP increase is mainly dependent on anaerobic glycolysis. However, long-term inhibition of the ATP synthase in HeLa cells with oligomycin A for 30 min was effective in reducing Ca2+-coupled ER ATP increase by 49% (Supplemental Figure S6B). On the other hand, acute inhibition of the ATP synthase in OXPHOS-dependent INS-1 832/13 cells with oligomycin A for 3 min strongly reduced ATP in the ER upon [Ca2+]ER depletion independent of the mode of Ca2+ mobilization (Figure 6, C and D, and Supplemental Figure S6C). These data further indicate that the Ca2+-coupled ER ATP signal requires continuous synthesis and transfer of ATP.
FIGURE 6:

The Ca2+-coupled ER ATP signal requires transfer of ATP across the ER membrane. (A) Individual responses of HeLa cells expressing the ER-targeted ATP sensor ERAT4.01 (top) or the ER Ca2+ probe D1ER (bottom) in response to treatment with 100 μM histamine in the absence of Ca2+. Cells were pretreated with either 2 μM oligomycin A (red dotted curve; n = 24 for [ATP]ER; n = 30 for [Ca2+]ER) or 10 mM 2-DG (blue dotted curve; n = 22 for [ATP]ER; n = 26 for [Ca2+]ER), and control cells were untreated (black curve; n = 18 for [ATP]ER; n = 30 for [Ca2+]ER). As indicated, 2 mM Ca2+ was readded upon washout of the IP3-generating agonists. Curves represent normalized mean ratio ± SEM over time. (B) Increase in [ATP]ER in control HeLa cells (white) and HeLa cells treated with either 2 μM oligomycin A (red) or 2-DG (blue) before Ca2+ mobilization by 100 μM histamine (as shown in A). *p < 0.05 vs. controls. (C) Individual responses of INS-1 832/13 cells expressing the ER-targeted ATP sensor ERAT4.01 (top) or the ER Ca2+ probe D1ER (bottom) in response to cell treatment with 100 μM carbachol in the absence of Ca2+. Cells were pretreated with 2 μM oligomycin A (red dotted curves; n = 63 for [ATP]ER; n = 13 for [Ca2+]ER), and control cells were untreated (black curves; n = 64 for [ATP]ER; n = 19 for [Ca2+]ER). As indicated, 2 mM Ca2+ was readded upon washout of the IP3-generating agonists. Curves represent normalized mean ratio ± SEM over time. (D) Increase in [ATP]ER in cells treated with 2 μM oligomycin A (red) as compared with untreated controls (white). Ca2+ was mobilized by 100 μM carbachol (see C). *p < 0.05 vs. respective controls.
Ca2+-regulated ER ATP increase is highly sensitive to cellular growth and energy status and requires AMPK activity
To correlate the Ca2+-coupled ER ATP elevation with the metabolic status of the cell, we investigated ER ATP signals under different rates of cell growth and substrate availability. For this purpose, we compared ER Ca2+ and ATP signals (Figure 7A) of freshly split HeLa cells (20 h before experiments [20hSC]) with those of HeLa cells that had been split 72 h before experiments (72hSC). The cell growth rate was clearly higher in 20hSC than in 72hSC (Supplemental Figure S7, A–C). The higher growth rate of the 20hSC correlates with a higher metabolic rate in these cells (Supplemental Figure S7D). The ER ATP increase in response to ionomycin was considerably larger in 20hSC than in 72hSC (Figure 7A, top), whereas the respective [Ca2+]ER depletion, which was identified as the main cause of ER ATP increases, did not differ between the two groups (Figure 7A, bottom). These observations show that in response to ER Ca2+ mobilization more ATP is available within the lumen of this organelle in constantly dividing cancerous cells with a high metabolic rate.
FIGURE 7:

The Ca2+-coupled ER ATP transport machinery is highly sensitive to the cellular growth and energy status and requires AMPK activity. (A) Normalized average ratio signals ± SEM of [ATP]ER (top) and [Ca2+]ER (bottom) over time in response to 2 μM ionomycin in Ca2+-free medium of HeLa cells that were split 20 h (20hSC; black curves; n = 26 for [ATP]ER; n = 18 for [Ca2+]ER) or 72 h before measurement (72hSC; green dotted curve; n = 32 for [ATP]ER; n = 21 for [Ca2+]ER). *p < 0.05 vs. controls. Right, relative changes of [ATP]ER (top) and [Ca2+]ER (bottom) deduced from the signals presented. *p < 0.05 vs. 72hSC. (B) Normalized ERAT4.01 FRET signals in HeLa cells that were constantly perfused with medium containing 10 mM glucose (black curve and corresponding white column, n = 13) or when glucose was removed for 4 min (orange curve, orange column, n = 9) before cell treatment with 2 μM ionomycin. *p < 0.05 vs. without glucose. (C) ERAT4.01 mean normalized ratios ± SEM of 72hSC upon treatment with 2 μM ionomycin in Ca2+-free medium. Cells were preincubated in loading buffer with 10 mM glucose (black curve, n = 32) or without glucose (blue dotted curve, n = 32) for 2–8 h before experiments. *p < 0.05 vs. without glucose. (D) ERAT4.01 mean normalized ratio signals ± SEM of 20hSC upon treatment with 2 μM ionomycin in Ca2+-free bathing medium. Cells were preincubated in loading buffer with 10 mM (green dotted curve, n = 26) or without glucose (purple dotted curve, n = 38) for 2–8 h before experiments. *p < 0.05 vs. without glucose. (E) Time course demonstrating the effect of glucose starvation on the ER Ca2+-coupled ER ATP signal in response to 2 μM ionomycin using HeLa cells (n = 3–38). Respective signals were normalized to the average maximal delta ERAT4.01 ratio signal of cells that were kept in 10 mM glucose. (F) Normalized mean ERAT4.01 ratios ± SEM over time of HeLa cells that were kept in glucose-free medium for 5 h. Control cells (black curve, n = 14) and cells that were pretreated with 2 μM oligomycin A for 20 min (red curve, n = 26) were stimulated with 2 μM ionomycin in Ca2+- and glucose-free buffer. *p < 0.05 vs. control. (G) Average normalized ERAT4.01 signals ± SEM over time in control (black curve, n = 30) and HeLa cells treated with siRNA against AMPKα/β (red curve, n = 36) in response to 2 μM ionomycin in Ca2+-free medium. Cells were kept in buffer containing 10 mM glucose. *p < 0.05 vs. control. (H) Experiments as in G, for cells kept in glucose-free medium for 2–8 h. Control cells (black curves, n = 42); siAMPKα/β-treated cells (red curve, n = 27). *p < 0.05 vs. control.
To further investigate correlations between the metabolic rate and Ca2+-controlled ER ATP fluxes, we examined [ATP]ER under conditions of energy stress, which was induced by glucose removal from the cell storage medium. When glucose was removed, the ATP elevation in the ER upon [Ca2+]ER depletion was immediately abolished (within 4 min; Figure 7B), whereas readdition of glucose for 4 min to the medium almost completely restored the signal (Supplemental Figure S7E). This indicates a strong dependence of the Ca2+-controlled ER ATP increase on glucose availability. Of interest, under long-term glucose starvation (2–8 h), the [ATP]ER increase in response to [Ca2+]ER depletion was again observed in both 72hSC (Figure 7C) and 20hSC (Figure 7D), whereas there was still a significant difference in the Ca2+-controlled ER ATP signals of glucose-starved and control cells. The respective [Ca2+]ER depletion was not altered by glucose starvation in the two cell groups (Supplemental Figure S7, F and G). Plotting the Ca2+-coupled ER ATP increase over time showed that the ER ATP signal was initially strongly reduced but recovered after 4 h (Figure 7E). However, the global cellular ATP content was not significantly affected by glucose starvation during this time (Supplemental Figure S7H). These findings point to compensatory mechanisms that specifically maintain Ca2+-coupled ER ATP fluxes under conditions of energy stress. Our data demonstrate that glucose-deprived HeLa cells have a much higher rate of oxygen consumption (Supplemental Figure S7I), reflecting enhanced OXPHOS to compensate for a halt in anaerobic glycolysis. In line with these findings, the restored Ca2+-coupled ER ATP signal in glucose-starved HeLa cells was abolished by inhibition of mitochondrial ATP synthase (Figure 7F), whereas the respective signal in the presence of glucose was only partially reduced by OXPHOS inhibition (Supplemental Figure S6B).
Cells—particularly cancer cells—have sophisticated mechanisms to cope with energy stress (Mathupala et al., 2010; Liang and Mills, 2013). AMPK is a central stress sensor that regulates cellular ATP homeostasis (Hardie et al., 2012). To determine whether this kinase controls the Ca2+-coupled ER ATP increase, we measured ER ATP signals in cells treated with small interfering RNA (siRNA) targeting AMPK. The siRNA-based approach effectively lowered AMPK expression levels in HeLa cells (Supplemental Figure S7J). AMPK-knockdown cells showed reduced ER ATP signals in response to ionomycin (Figure 7, G and H), whereas the respective ER Ca2+ release was not affected (Supplemental Figure S7, K and L). Of note, the reducing effect of AMPK knockdown on the ionomycin-triggered [ATP]ER elevation was observed in both cells maintained in high glucose–containing medium (Figure 7G) and glucose-starved cells (Figure 7H). These findings suggest that AMPK activity is generally fundamental for ER ATP transfer upon [Ca2+]ER mobilization in HeLa cells.
DISCUSSION
The genetically encoded fluorescent ER ATP sensor presented here is a novel tool for monitoring [ATP]ER in living cells with high temporal and spatial resolution. Based on this exceptional advantage compared with existing methods to assess ATP, application of this tool proved for the first time the existence of tightly regulated ATP dynamics within the ER. It unveiled a Ca2+-controlled ER ATP signal, which raises several stimulating questions and novel hypotheses regarding the role of ATP in this organelle.
Many studies proved that the ER needs to be supplied with energy in order to perform a number of vital functions. Although many reports suggest a transfer of energy in the form of ATP into the ER (Clairmont et al., 1992; Hirschberg et al., 1998; Szabadkai et al., 2006; Kornmann and Walter, 2010; Elbaz and Schuldiner, 2011), the experimental proof is limited, mainly due to the lack of sophisticated methods to measure ATP within organelles. Attempts have been made to estimate changes of ATP levels within the ER in living cells by using ER-targeted firefly luciferase (Dorner and Kaufman, 1994). However, this approach does not allow visualization of organelle ATP dynamics in a reversible manner on the single-cell level. Although Willemse et al. (2007) published a cautionary note regarding the interference of ATP with fluorescent proteins, Imamura et al. (2009) developed an efficient CFP-YFP FRET-based ATP sensor capable of detecting ATP in living cells with high spatiotemporal resolution. We modified and targeted this kind of genetically encoded ATP sensor to measure ATP dynamics within the ER. By using pharmacological tools that deplete the cellular ATP content, we showed that the ER-targeted ATP probe senses ATP within the lumen of the ER in a ratiometric (Supplemental Figure S2A) and reversible manner (Figure 2, A and B) in real time (Figure 2). The correlation between ATP dynamics in the ER with that of mitochondria shows that [ATP]ER changes lag only slightly behind changes of mitochondrial ATP levels (Figure 2, E and G), indicating regulated coupling of the two organelles in terms of ATP transfer. Because cytosolic ATP changes did not correlate with that of the ER (Supplemental Figure S2, B–E), this points to a spatial association between the organelles’ ATP pools and confirms the concept of a bioenergetic coupling of these organelles (Bravo et al., 2011; Calì et al., 2013). An analogous functional and spatial coupling was reported for the transfer of lipids (Kornmann and Walter, 2010), chaperones (Sun et al., 2006), and Ca2+ signaling between the ER and mitochondria (Szabadkai et al., 2006; Graier et al., 2007), which is accomplished by physical tethering of the organelles (de Brito and Scorrano, 2008; Merkwirth and Langer, 2008).
The central finding of this work is that the ER Ca2+ concentration is a major regulator of ATP elevation in the ER, once Ca2+ falls below a certain threshold in the organelle (Figure 3E). ATP forms stable complexes with both Ca2+ and Mg2+ ions, whereas in living cells MgATP is the predominant form. Of interest, our experiments demonstrate that the genetically encoded ATP probe detects MgATP exclusively (Figure 4H). Because both Ca2+ and Mg2+ concentrations within the ER are in the high micromolar to millimolar range (Miyawaki et al., 1997; Mooren et al., 2001), CaATP and MgATP complexes might coexist within the organelle. Considering a rather constant Mg2+ concentration within the ER and the higher affinity of Mg2+ to form the MgATP complex, a transformation of CaATP into MgATP can only partially account for the increase of the ERAT4.01 FRET signal we observed in intact cells upon ER Ca2+ depletion. However, in pancreatic acinar cells, ER-dependent Mg2+ and Ca2+ movements were shown in response to cholecystokinin (Mooren et al., 2001), indicating that the formation of MgATP within the ER upon Ca2+ mobilization might occur. Nevertheless, we found several conditions in which the ER Ca2+ depletion was identical despite a significant effect on the ER ATP increase (Figures 6 and 7, C, D, G, and H, and Supplemental Figure S7, F, G, and K). This argues against the possibility that MgATP increases within the ER by Ca2+-dependent Mg2+ entry into the organelle in the cell types used. Because we proved that the fluorescence properties of the ATP probe are Ca2+ and redox insensitive (Figure 4 and Supplemental Figure S4), neither ER Ca2+ fluctuations nor changes in the ER thiol redox directly account for the FRET signal observed. Although the actual physiological meaning of the Ca2+-coupled ER ATP elevation awaits to be explored in detail, the inverse correlation between [Ca2+]ER and [ATP]ER suggests an increased demand of energy in the lumen of the organelle in order to cope with Ca2+-related stress under conditions of cell stimulation. Considering that both [Ca2+]ER and [ATP]ER are major regulators of protein folding within the ER (Braakman and Bulleid, 2011), the inverse correlation of these factors might be essential to sustain the vital functions of the organelle. Moreover, our data further show that [ATP]ER is highly dependent on glycolysis or OXPHOS, as inhibition of these metabolic processes reduces both basal ER ATP levels (Figure 2) and Ca2+-coupled increase of ATP within the lumen of the ER (Figure 6). These findings are in line with several reports demonstrating that inhibition of ATP-generating processes severely impairs ER function (Harding et al., 2002; Schröder and Kaufman, 2005; Yu and Kim, 2010).
Although Ca2+ is known to enhance OXPHOS and, hence, mitochondrial ATP generation (Jouaville et al., 1999; Denton, 2009; Nakano et al., 2011), our data reveal that the increase in [ATP]ER upon ER Ca2+ release is not evoked by Ca2+-stimulated ATP synthesis, but instead [Ca2+]ER is the main determinant of the process (Figure 5 and Supplemental Figure S5). Accordingly, it is tempting to speculate about the existence of a putative ER ATP translocase, which is activated once Ca2+ falls below a certain threshold within the ER. Bioinformatics search tools such as National Center for Biotechnology Information BLAST (Altschul et al., 1990), Target P (Emanuelsson et al., 2007), PROSITE (Sigrist et al., 2002), and TMHMM (Krogh et al., 2001) indeed predict the existence of several ANT-like proteins with Ca2+-binding domains (e.g., EF hands; unpublished data). Although it still needs to be verified experimentally whether such proteins catalyze the transfer of ATP into the ER in a Ca2+-dependent manner, an in silico approach was successfully used to identify the ER-ANT1 in A. thaliana (Leroch et al., 2008). Our data show that the genetically encoded ER ATP sensor is a suitable novel tool to characterize ER ATP dynamics in vivo, which also offers the possibility to identify the elusive ER ATP transporter(s) in future.
Our data show that a change of the metabolic rate by whatever means (e.g., glucose deprivation; Figure 7, B–H) and differential rate of proliferation (Figure 7A) affects the Ca2+-coupled ER ATP increase, indicating that the process is tightly regulated and linked to ATP-generating processes. Of interest, glucose deprivation initially strongly inhibited the ER ATP signal in response to ER Ca2+ depletion, but after ∼4 h the signal was almost completely restored (Figure 7E) by enhanced activity of mitochondrial OXPHOS (Supplemental Figure S7I). These findings suggest compensatory mechanisms that regulate and reestablish the process during energy stress and point to adaptability of the Ca2+-coupled ER ATP regulation. Our data show that AMPK regulates the Ca2+-coupled ATP increase under different conditions, suggesting that this energy stress sensor is a strong regulator of ER ATP homeostasis. AMPK serves as an energy stress sensor that is able to restore and maintain ATP levels by stimulating both glycolysis and OXPHOS while inhibiting ATP-consuming processes (Shaw, 2006; Hardie et al., 2012; Kottakis and Bardeesy, 2012). Hence the clear dependence of Ca2+-coupled ER ATP regulation on AMPK activity reported here (Figure 7, G and H) might indicate that this process is particularly important to balance stress responses. Although the role of [ATP]ER in modulating ER stress responses is not clear, AMPK activation might counteract ER stress–induced cell damage by controlling ER ATP levels.
In summary, with the design and use of an ER-targeted, genetically encoded ATP sensor, we revealed the existence of an abundant [Ca2+]ER-regulated ER ATP increase. Furthermore, the Ca2+-coupled ER ATP signal was tightly linked to ATP generation, and AMPK was found to be an important regulator of this process. Understanding such mechanistic specifics of organelle ATP dynamics might have multiple implications in cell physiology and disease.
MATERIALS AND METHODS
Construction of ER-targeted ATP probes
To engineer ERAT4.01, the ATP-binding box (i.e., ε-subunit) of the F0F1-ATP synthase of B. subtilis was amplified from AT1.03 (Imamura et al., 2009), including restriction sites for SphI and SacI by PCR. Subsequently, the D1 domain (design1 of calmodulin and M13 sequence) of the ER-targeted Ca2+ probe D1ER (Palmer et al., 2004) was exchanged for the ATP-binding box using the restriction enzymes SphI and SacI in the pUC19(+) cloning vector and the complete ER-targeted ATP sensor transferred into the pcDNA3.1(+) expression vector via the restriction sites of HindIII and EcoRI. In analogy, the ER-targeted ATeams ERAT3.01N7Q and ERAT3.01N7Q, R122K, R126K and the respective red-shifted ATP probes ERGRAT, ERGRATN7Q, and ERGRATN7Q, R122K, R126K containing TagRFP on the N-terminus and EGFP on the C-terminus were constructed. For details see Supplemental Figure S4.
Cell culture and transfection
Human umbilical vein endothelial cells (EA.hy926), HeLa cells, and HEK-293 cells were grown in DMEM supplemented with 10% fetal calf serum (FCS). The rat pancreatic insulinoma cell line (INS-1 832/13) was cultured in RPMI 1640 medium containing 10% FCS. All cells were kept at 37°C in 5% CO2. Cells were transfected with respective constructs after reaching 50% confluence using the TransFast transfection reagent from Promega (Madison, WI) as described previously (Waldeck-Weiermair et al., 2012). Cells were transiently transfected with the FRET-based mitochondrial or cytosolic ATP sensor mitAT1.03 or AT1.03 (Imamura et al., 2009) or the ER Ca2+ sensor D1ER (Palmer et al., 2004). Standard AMPKα/β siRNA was obtained from Santa Cruz Biotechnology (Dallas, TX).
ATP and Ca2+ measurements using genetically encoded sensors
Cells transfected with the respective FRET-based sensors were grown on 30-mm glass coverslips. Before experiments, cells were kept in a loading buffer containing (in mM) 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 20 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2.6 NaHCO3, 0.44 KH2PO4, 0.34 Na2HPO4, and 10 d-glucose with 0.1% vitamins, 0.2% essential amino acids, and 1% penicillin/streptomycin for 2–8 h at room temperature. Glucose starvation was induced by incubating cells with loading buffer without glucose. Coverslips were subsequently put into a perfusion chamber and imaged using an AxioVert inverted microscope (Zeiss, Vienna, Austria) with a 40× oil immersion objective. The Ca2+ sensors (Cameleons) and ERAT4.01 were excited at 440 ± 10 nm, and emission was recorded at 480 and 530 nm using a beam splitter or a motorized filter wheel as described previously (Waldeck-Weiermair et al., 2012). Red-shifted, ER-targeted ATP probes (Supplemental Figure S4) were excited at 477 nm, and emission was collected at 510 and 590 nm using a motorized filter wheel. Cell permeabilization was obtained using a mixture of 10 μM digitonin and 2 μM ionomycin in a buffer containing 130 mM KCl, 10 mM HEPES, pH 7.2 (KOH), with or without 2 mM Ca2+ (CaCl2) and with or without 1–10 mM MgATP or 10 mM CaATP.
Characterization of the ATP probe in vitro
The fluorescence spectra of purified AT1.03 in the absence and presence of 100, 200, 300, 400, and 500 μM Ca2+ were measured using a buffer containing 50 mM 3-(N-morpholino)propanesulfonic acid–KOH (pH 7.3), 50 mM KCl, 0.5 mM, MgCl2, and 0.05% Triton X-100 at 37°C with a FP-6500 spectrofluorometer (Jasco, Tokyo, Japan) as previously described (Imamura et al., 2009). Equimolar amounts of MgCl2 were added to obtain MgATP complex.
Confocal imaging
High-resolution images for localizing ERAT 4.01 were acquired by array confocal laser scanning microscopy. The array confocal laser scanning microscope was built on an inverse, fully automatic microscope (Axio Observer.Z1; Zeiss, Göttingen, Germany) equipped with a 100× oil immersion objective (Plan-Fluor ×100/1.45 oil; Zeiss), a Nipkow-based confocal scanner unit (CSU-X1; Yokogawa, Tokyo, Japan), a motorized filter wheel (CSUX1FW; Yokogawa) on the emission side, and an acousto-optical tunable filter–based laser merge module for laser lines 405, 445, 473, 488, 515, and 561 nm (Visitron Systems, Puchheim, Germany). ERAT4.01 was excited at 445 nm; ER RFP was excited at 561 nm. Emission was acquired with a charge-coupled device camera (CoolSNAP-HQ; Photometrics, Tucson, AZ). All devices were controlled by VisiView Premier acquisition software (Visitron Systems).
Measurement of cellular oxygen consumption and extracellular acidification rate
HeLa cells were plated in XF96 polystyrene cell culture microplates (Seahorse Bioscience, North Billerica, MA) at a density of 40,000 cells/well. After overnight incubation, cells were preincubated for 3 h at 37°C in physiological buffer containing glucose and compounds as specified. Before starting the experiment, cells were changed to unbuffered XF assay medium (Seahorse Bioscience) supplemented with 1 mM sodium pyruvate and either 10 mM or no glucose, as indicated.
Measurement of cellular ATP content using high-performance liquid chromatography
Separation of adenine nucleotides was performed on a Hypersil ODS column (5 μm, 250 × 4 mm inner diameter), using a L2200 autosampler, two l-2130 HTA pumps, and a L2450 diode array detector as described recently (Khan et al., 2012).
Quantification of cell proliferation
Cell proliferation rate was determined by real-time phase contrast microscopy, using a Cell-IQ device (Chipman Technology, Tampere, Finland) as described (Toimela et al., 2008). Images were acquired at randomly selected areas of the wells. Confluency and cell number was quantified using Cell-IQ Analyser software.
Statistical analysis
Data shown represent mean ± SEM, where n is the number of single cells of three or more independent experiments or just the number of individual experiments. Statistical analyses were performed with unpaired Student's t test, and p < 0.05 was considered to be significant.
Supplementary Material
Acknowledgments
We thank Sandra Blass for excellent technical assistance and C.J.S. Edgell (University of North Carolina, Chapel Hill, NC) for the EA.hy926 cells. We thank Roger Tsien (University of California, San Diego, La Jolla, CA) for sending us Cameleons and C. B. Newgard (Duke University, Durham, NC) for the INS-1 832/13 cells. We thank Karin Osibow (Medical University of Graz, Graz, Austria), Maud Frieden (University of Geneva, Geneva, Switzerland), and Franck Polleux (Dorris Neuroscience Center, La Jolla, CA) for critically reviewing the manuscript. This work was supported by the Austrian Science Fund (P21857-B18 and P22553-B18). N.V. is supported by the Austrian Science Fund within the DKplus Metabolic and Cardiovascular Disease program (W1226-B18) of the Medical University of Graz.
Abbreviations used:
- AMPK
AMP-activated protein kinase
- ATP
adenosine triphosphate
- BHQ
2,5-di-tert-butylhydroquinone
- CCh
carbachol
- CFP
cyan fluorescent protein
- Cs
cysteines
- 2-DG
2-deoxy-d-glucose
- EGFP
enhanced GFP
- ER
endoplasmic reticulum
- FCS
10% fetal calf serum
- FPs
fluorescent proteins
- FRET
Förster resonance energy transfer
- IP3
inositol 1,4,5-triphosphate
- RFP
red fluorescent protein
- SERCA
sarco/endoplasmic reticulum Ca2+-ATPase
- SOCE
store-operated Ca2+ entry
- UPR
unfolded protein response
- YFP
yellow fluorescent protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-07-0433) December 4, 2013.
*These authors contributed equally.
REFERENCES
- Alam MR, Groschner LN, Parichatikanond W, Kuo L, Bondarenko AI, Rost R, Waldeck-Weiermair M, Malli R, Graier WF. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic β-cells. J Biol Chem. 2012;287:34445–34454. doi: 10.1074/jbc.M112.392084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Avezov E, Cross BCS, Kaminski Schierle GS, Winters M, Harding HP, Melo EP, Kaminski CF, Ron D. Lifetime imaging of a fluorescent protein sensor reveals surprising stability of ER thiol redox. J Cell Biol. 2013;201:337–349. doi: 10.1083/jcb.201211155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee DK. N-glycans in cell survival and death: cross-talk between glycosyltransferases. Biochim Biophys Acta. 2012;1820:1338–1346. doi: 10.1016/j.bbagen.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–249. doi: 10.1016/s0143416002001823. [DOI] [PubMed] [Google Scholar]
- Blom T, Somerharju P, Ikonen E. Synthesis and biosynthetic trafficking of membrane lipids. Cold Spring Harbor Perspect Biol. 2011;3:a004713. doi: 10.1101/cshperspect.a004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- Bravo R, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci. 2011;124:2143–2152. doi: 10.1242/jcs.080762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Calì T, Ottolini D, Negro A, Brini M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+ transfer to sustain cell bioenergetics. Biochim Biophys Acta. 2013;1832:495–508. doi: 10.1016/j.bbadis.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Chia WS, Chia DX, Rao F, Nun SB, Shochat SG. ATP binding to p97/VCP D1 domain regulates selective recruitment of adaptor s to its proximal N-domain. PloS One. 2012;7:e50490. doi: 10.1371/journal.pone.0050490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairmont CA, Maio AD, Hirschberg CB. Translocation of ATP into the lumen of rough endoplasmic reticulum-derived vesicles and its binding to luminal proteins including BiP GRP78 and GRP94. J Biol Chem. 1992;267:3983–3990. [PubMed] [Google Scholar]
- de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Dorner AJ, Kaufman RJ. The levels of endoplasmic reticulum proteins and ATP affect folding and secretion of selective proteins. Biologicals. 1994;22:103–112. doi: 10.1006/biol.1994.1016. [DOI] [PubMed] [Google Scholar]
- Elbaz Y, Schuldiner M. Staying in touch the molecular era of organelle contact sites. Trends Biochem Sci. 2011;36:616–623. doi: 10.1016/j.tibs.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, Heijne GV, Nielsen H. Locating proteins in the cell using TargetP SignalP and related tools. Nat Protoc. 2007;2:953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- Fang M, Shen Z, Huang S, Zhao L, Chen S, Mak TW, Wang X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell. 2010;143:711–724. doi: 10.1016/j.cell.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Graier WF, Frieden M, Malli R. Mitochondria and Ca2+ signaling: old guests, new functions. Pflugers Arch. 2007;455:375–396. doi: 10.1007/s00424-007-0296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenendyk J, Sreenivasaiah PK, Kim DH, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circ Res. 2010;107:1185–1197. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- Han J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15:481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- Hirschberg CB, Robbins PW, Abeijon C. Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu Rev Biochem. 1998;67:49–69. doi: 10.1146/annurev.biochem.67.1.49. [DOI] [PubMed] [Google Scholar]
- Imamura H, Nhat KPH, Togawa H, Saito K, Iino R, Kato-Yamada Y, Noji H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci USA. 2009;106:15651–15656. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R. Regulation of mitochondrial ATP synthesis by calcium: evidence for a long-term metabolic priming. Proc Natl Acad Sci USA. 1999;96:13807–13812. doi: 10.1073/pnas.96.24.13807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Sanyal AJ. Endoplasmic reticulum stress and the unfolded protein response. Clin Liver Dis. 2009;13:581–590. doi: 10.1016/j.cld.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Khan MJ, et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem. 2012;287:21110–21120. doi: 10.1074/jbc.M111.319129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendörfer KU, Then AR, Kearns BG, Bankaitis VA, Mayinger P. Sac1p plays a crucial role in microsomal ATP transport which is distinct from its function in Golgi phospholipid metabolism. EMBO J. 1999;18:1506–1515. doi: 10.1093/emboj/18.6.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh A, Walter P. Structural basis of the unfolded protein response. Annu Rev Cell Dev Biol. 2012;28:251–277. doi: 10.1146/annurev-cellbio-101011-155826. [DOI] [PubMed] [Google Scholar]
- Kornmann B, Walter P. ERMES-mediated ER-mitochondria contacts molecular hubs for the regulation of mitochondrial biology. J Cell Sci. 2010;123:1389–1393. doi: 10.1242/jcs.058636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottakis F, Bardeesy N. LKB1-AMPK axis revisited. Cell Res. 2012;22:1617–1620. doi: 10.1038/cr.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, Heijne GV, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Le G, Neuhof A, Rapoport T. The endoplasmic reticulum membrane is permeable to small molecules. Mol Biol Cell. 2004;15:447–455. doi: 10.1091/mbc.E03-05-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroch M, Neuhaus HE, Kirchberger S, Zimmermann S, Melzer M, Gerhold J, Tjaden J. Identification of a novel adenine nucleotide transporter in the endoplasmic reticulum of Arabidopsis. Plant Cell. 2008;20:438–451. doi: 10.1105/tpc.107.057554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor. Cancer Res. 2013;73:2929–2935. doi: 10.1158/0008-5472.CAN-12-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg effect in carcinogenesis. J Biol Chem. 2002;277:23111–23115. doi: 10.1074/jbc.M202487200. [DOI] [PubMed] [Google Scholar]
- Mathupala SP, Ko YH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta. 2010;1797:1225–1230. doi: 10.1016/j.bbabio.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkwirth C, Langer T. Mitofusin 2 builds a bridge between ER and mitochondria. Cell. 2008;135:1165–1167. doi: 10.1016/j.cell.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Merulla J, Fasana E, Soldà T, Molinari M. Specificity and regulation of the endoplasmic reticulum-associated degradation machinery. Traffic. 2013;14:767–777. doi: 10.1111/tra.12068. [DOI] [PubMed] [Google Scholar]
- Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M, Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997;388:882–887. doi: 10.1038/42264. [DOI] [PubMed] [Google Scholar]
- Miyazaki S. IP3 receptor-mediated spatial and temporal Ca2+ signaling of the cell. Jpn J Physiol. 1993;43:409–434. doi: 10.2170/jjphysiol.43.409. [DOI] [PubMed] [Google Scholar]
- Mohorko E, Glockshuber R, Aebi M. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J Inherit Metab Dis. 2011;34:869–878. doi: 10.1007/s10545-011-9337-1. [DOI] [PubMed] [Google Scholar]
- Mooren FC, Turi S, Gunzel D, Schlue WR, Domschke W, Singh J, Lerch MM. Calcium-magnesium interactions in pancreatic acinar cells. FASEB J. 2001;15:659–672. doi: 10.1096/fj.00-0172com. [DOI] [PubMed] [Google Scholar]
- Naidoo N. ER and aging—protein folding and the ER stress response. Ageing Res Rev. 2009;8:150–159. doi: 10.1016/j.arr.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Nakano M, Imamura H, Nagai T, Noji H. Ca2 regulation of mitochondrial ATP synthesis visualized at the single cell level. ACS Chem Biol. 2011;6:709–715. doi: 10.1021/cb100313n. [DOI] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. 2009;315:2496–2504. doi: 10.1016/j.yexcr.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham HR. The retention signal for soluble proteins of the endoplasmic reticulum. Trends Biochem Sci. 1990;15:483–486. doi: 10.1016/0968-0004(90)90303-s. [DOI] [PubMed] [Google Scholar]
- Rasheva VI, Domingos PM. Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis. 2009;14:996–1007. doi: 10.1007/s10495-009-0341-y. [DOI] [PubMed] [Google Scholar]
- Ron D, Harding HP. Protein-folding homeostasis in the endoplasmic reticulum and nutritional regulation. Cold Spring Harbor Perspect Biol. 2012;4:a013177. doi: 10.1101/cshperspect.a013177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutat Res. 2005;569:29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006;18:598–608. doi: 10.1016/j.ceb.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Shin SJ, Lee WK, Lim HW, Park J. Characterization of the ATP transporter in the reconstituted rough endoplasmic reticulum proteoliposomes. Biochim Biophys Acta. 2000;1468:55–62. doi: 10.1016/s0005-2736(00)00241-8. [DOI] [PubMed] [Google Scholar]
- Sigrist CJA, Cerutti L, Hulo N, Gattiker A, Falquet L, Pagni M, Bairoch A, Bucher P. PROSITE a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3:265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- Smyth JT, Hwang SY, Tomita T, DeHaven WI, Mercer JC, Putney JW. Activation and regulation of store-operated calcium entry. J Cell Mol Med. 2010;14:2337–2349. doi: 10.1111/j.1582-4934.2010.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Wei S, Li C, Chang Y, Chao C, Lai Y. Localization of GRP78 to mitochondria under the unfolded protein response. Biochem J. 2006;396:31–39. doi: 10.1042/BJ20051916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175:901–911. doi: 10.1083/jcb.200608073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toimela T, Tähti H, Ylikomi T. Comparison of an automated pattern analysis machine vision time-lapse system with traditional endpoint measurements in the analysis of cell growth and cytotoxicity. Altern Lab Anim. 2008;36:313–325. doi: 10.1177/026119290803600309. [DOI] [PubMed] [Google Scholar]
- Waldeck-Weiermair M, Alam MR, Khan MJ, Deak AT, Vishnu N, Karsten F, Imamura H, Graier WF, Malli R. Spatiotemporal correlations between cytosolic and mitochondrial Ca2+ signals using a novel red-shifted mitochondrial targeted Cameleon. PloS One. 2012;7:e45917. doi: 10.1371/journal.pone.0045917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemse M, Janssen E, de Lange F, Wieringa B, Fransen J. ATP and FRET—a cautionary note. Nat Biotechnol. 2007;25:170–172. doi: 10.1038/nbt0207-170. [DOI] [PubMed] [Google Scholar]
- Yu SM, Kim SJ. Endoplasmic reticulum stress (ER-stress) by 2-deoxy-D-glucose (2DG) reduces cyclooxygenase-2 (COX-2) expression and N-glycosylation and induces a loss of COX-2 activity via a Src kinase-dependent pathway in rabbit articular chondrocytes. Exp Mol Med. 2010;42:777–786. doi: 10.3858/emm.2010.42.11.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2012;14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.