

A 13-year-old boy presented with a five-month history of nausea and morning vomiting not associated with headaches or abdominal pains. His bowel movements were normal.
He had demonstrated weight loss and reported fatigue, drooling, choking and bilateral calf muscle cramps. There were no visual changes, ataxia, vertigo, rigidity, seizures or psychiatric disturbances.
His neurological examination was significant for tongue fasiculations, dysarthria, and mild weakness and tremor in the right hand, with poor coordination. The remainder of his neurological examination was normal, with no papilloedema.
The differential diagnosis included central nervous system neoplasia, a demyelinating disease, a neurodegenerative disease, a metabolic disorder and a bradykinetic movement disorder such as juvenile Parkinsonism. Other considerations included drug effects or toxicity, and endocrine disorders such as hyperthyroidism. An urgent brain magnetic resonance imaging was scheduled. His thyroid profile, drug screen, electrolyte levels and renal function were all normal.
Magnetic resonance imaging demonstrated bilateral symmetrical striatal and tegmental lesions suggestive of necrosis and/or calcifications (Figure 1). There was diffuse brain atrophy with prominent ventricles, wide occipital horns and prominent pericerebral spaces.
Figure 1).
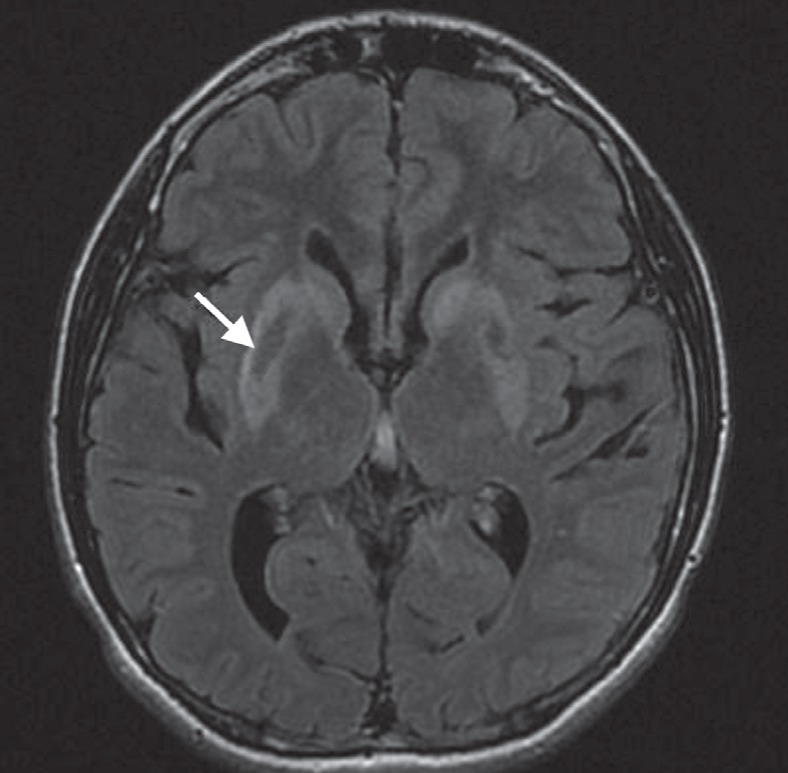
Axial fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging through the basal ganglia. Markedly increased FLAIR signal in the lentiform nuclei and caudate heads bilaterally, as well as the superimposed low signal, likely indicate mineralization centrally within the bilateral lentiforms (arrow)
CASE 2 DIAGNOSIS: WILSON DISEASE AND HEPATOLENTICULAR DEGENERATION
The differential diagnosis was narrowed to a metabolic disease such as a mitochondrial cytopathy or mineralization (copper or iron accumulation). Additional tests confirmed the diagnosis. Slit-lamp examination revealed Kayser-Fleischer rings. His serum ceruloplasmin level was low (93 mg/L; normal >200 mg/L) and his 24 h urinary copper excretion was elevated (6.7 μmol/day; normal <0.6 μmol/day). Serum aminotransferase, alkaline phosphatase and albumin levels were normal, as was a complete blood count. An abdominal ultrasound revealed a coarse liver with splenomegaly.
The above findings were consistent with Wilson disease (WD). Given that normal transaminase levels are unusual in WD in the absence of hepatic synthetic dysfunction, and given the coarse appearance of the liver, a liver biopsy was performed. The biopsy demonstrated moderate portal fibrosis with mild nonspecific hepatitis (Figure 2A). The stain for copper showed positive granular staining in hepatocyte cytoplasm in focal and zonal distributions (Figure 2A, insert). Electron microscopy revealed microvesicular steatosis and mitochondrial changes consistent with WD (Figure 2B). His echocardiogram was normal. The diagnosis of WD was confirmed by the finding of a homozygous mutation in the ATP7B gene (c.1708 G→C) and treatment with chelation therapy was commenced. The patient’s condition stabilized, with some residual evidence of Parkinsonism.
Figure 2).
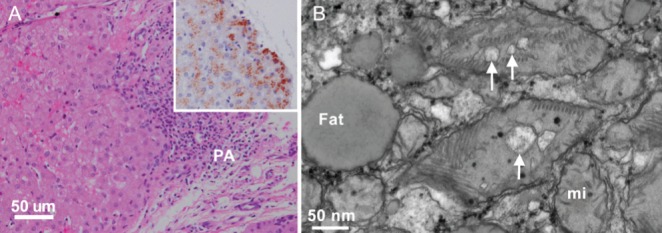
Light and electron microscopy of liver biopsy. A Periportal area with relatively well-preserved hepatocytes (left) and a portal area (PA) with mild to moderate lymphocytic infiltrate and fibrosis (hematoxylin and eosin stain, bar 50 μm). Insert: granular copper deposits in cytoplasm of hepatocytes (rubeanic acid stain for copper). B High-magnification electron micrograph of hepatocyte cytoplasm containing fat droplets (Fat) and mixture of normal mitochondria (mi) and ‘mega’ mitochondria with dilated mitochondrial cristae (arrows, bar 50 nm)
WD is a disorder of copper metabolism that presents with hepatic, neurological, hematological or psychiatric disturbances, and can occur from childhood to among individuals >50 years of age. WD more commonly presents with liver disease in children and younger adults. The presentation can vary and includes asymptomatic elevated aminotransferase levels, recurrent jaundice, fulminant hepatic failure with coagulopathy, encephalopathy and acute Coombs-negative intra-vascular hemolysis; or chronic liver disease with portal hypertension, hepatosplenomegaly, ascites and low albumin levels (1).
Forty per cent of individuals with WD present with neurological involvement including tremors, poor coordination, loss of fine motor control, chorea, rigidity and/or gait disturbances (2). Pseudobulbar involvement, such as dysarthria, drooling and dysphagia, are more common in older individuals but can occur in children and adolescents, as was the case in our patient. Psychiatric manifestations include mood disturbances, phobias, compulsive behaviours and aggression. Cognitive decline may occur.
Kayser-Fleischer rings result from copper deposition in Descemet’s membrane of the cornea and reflect a high degree of copper storage. It is estimated that Kayser-Fleischer rings occur in approximately 90% of WD patients with neurological disease and approximately 50% of those with hepatic manifestations (3). Other findings include renal involvement, arthritis, cardiomyopathy, pancreatitis and rhabdomyolysis.
The prevalence of WD is estimated to be one in 30,000. It is inherited in an autosomal recessive manner. ATP7B is the only gene known to be associated with WD. Given that copper studies are frequently equivocal, molecular genetic testing is becoming increasingly more important in diagnosis and for determining the genetic status of at-risk siblings. Biochemical testing relies on demonstration of abnormal copper parameters, such as low serum ceruloplasmin, high urinary copper excretion and hepatic deposits of copper, all of which were present in our patient. Interestingly, he had no overt physical signs or laboratory evidence of liver disease.
Copper-chelating agents are the first-line therapy for WD. Penicillamine has previously been used as a first-line treatment. Trientine is the second-line treatment for individuals who cannot tolerate penicillamine. Our patient was started on trientine because it is gaining acceptance as a first-line drug due to excellent tolerance (4). He was also treated with oral zinc, which inhibits the absorption of copper from the gastrointestinal tract. Restriction of foods high in copper (eg, chocolate, mushrooms, nuts, shellfish, liver) is important, especially at the beginning of treatment. Liver transplantation is reserved for individuals who fail to respond to medical therapy or present with fulminant hepatic failure. It remains unclear whether liver transplantation should be a primary treatment for severe neurological involvement in WD because there are limited data regarding neurological recovery after transplant.
CLINICAL PEARLS
Although frank neurological presentation of WD occurs more commonly in older individuals, it can occur in children and adolescents in the absence of overt evidence of liver disease.
Molecular testing of the ATP7B gene is clinically available and is becoming increasingly more prudent because biochemical testing does not always support the diagnosis of WD.
REFERENCES
- 1.Wilson DC, Phillips MJ, Cox DW, Roberts EA. Severe hepatic Wilson’s disease in preschool-aged children. J Pediatr. 2000;137:719–22. doi: 10.1067/mpd.2000.108569. [DOI] [PubMed] [Google Scholar]
- 2.Svetel M, Kozic D, Stefanova E, Semnic R, Dragasevic N, Kostic VS. Dystonia in Wilson’s disease. Mov Disord. 2001;16:719–23. doi: 10.1002/mds.1118. [DOI] [PubMed] [Google Scholar]
- 3.Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson’s disease: An update. Hepatology. 2008;47:2089–111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]
- 4.Askari FK, Greenson J, Dick RD, Johnson VD, Brewer GJ. Treatment of Wilson’s disease with zinc. XVIII. Initial treatment of the hepatic decompensation presentation with trientine and zinc. J Lab Clin Med. 2003;142:385–90. doi: 10.1016/S0022-2143(03)00157-4. [DOI] [PubMed] [Google Scholar]