

Abstract
Demand for palm oil has been increasing by an average of ∼8% the past decade and currently accounts for about 59% of the world's vegetable oil market. This drives the need to increase palm oil production. Nevertheless, due to the increasing need for sustainable production, it is imperative to increase productivity rather than the area cultivated. Studies on the oil palm genome are essential to help identify genes or markers that are associated with important processes or traits, such as flowering, yield and disease resistance. To achieve this, 294,115 and 150,744 sequences from the hypomethylated or gene-rich regions of Elaeis guineensis and E. oleifera genome were sequenced and assembled into contigs. An additional 16,427 shot-gun sequences and 176 bacterial artificial chromosomes (BAC) were also generated to check the quality of libraries constructed. Comparison of these sequences revealed that although the methylation-filtered libraries were sequenced at low coverage, they still tagged at least 66% of the RefSeq supported genes in the BAC and had a filtration power of at least 2.0. A total 33,752 microsatellites and 40,820 high-quality single nucleotide polymorphism (SNP) markers were identified. These represent the most comprehensive collection of microsatellites and SNPs to date and would be an important resource for genetic mapping and association studies. The gene models predicted from the assembled contigs were mined for genes of interest, and 242, 65 and 14 oil palm transcription factors, resistance genes and miRNAs were identified respectively. Examples of the transcriptional factors tagged include those associated with floral development and tissue culture, such as homeodomain proteins, MADS, Squamosa and Apetala2. The E. guineensis and E. oleifera hypomethylated sequences provide an important resource to understand the molecular mechanisms associated with important agronomic traits in oil palm.
Introduction
The oil palm is a perennial crop that belongs to the family Arecaceae and the genus Elaeis [1]. There are two species in the genus - Elaeis guineensis (EG), the African oil palm and Elaeis oleifera (EO), of American origin [2]. EG is widely grown in the humid tropics (South-East Asia, Equatorial America, Africa and South Pacific) [3], and has become one of the most important crop in Malaysia and Indonesia. In order to remain competitive with other vegetable oil crops, there is a need to boost its yield and improve oil quality, for both of which deciphering its genome is key – to better understand the complexity of gene expression and interactions. One of the methods used is to identify genes expressed in a tissue of interest. The expressed sequence tag (EST) approach coupled with conventional sanger sequencing [4] was initially used to obtain information on gene diversity and mRNA expression patterns from various oil palm tissues [5]–[7]. However, the method is limited in utility, mostly identifying the abundantly expressed genes [8]. Although alternatives, such as constructing normalized cDNA libraries [9] have been tried, the method was deemed technically demanding.
The development of next generation sequencing (NGS) resolved these issues and identification of low abundance genes was thus made possible [10]. In oil palm, NGS sequencing was able to provide an in depth view of the genes expressed in flowers and fruit development. Comparing flowers of normal and abnormal clonal palms, Shearman and colleagues [11] identified a large number of differentially expressed genes, including those involved in chromatin remodelling and histone methylation. The abnormal palms in the study produced mantled fruits, a form of abnormality observed in palms produced via somatic embryogenesis. Shearman and colleagues [11] results are encouraging as previous studies have linked the occurrence of mantled fruits to changes in methylation [12]–[14]. In fruit development, Bourgis et al. [15] and Tranbarger et al. [16] were able to determine the expression of a new oil palm WRINKLED1 (WRI1) homolog, known to be involved in fatty acid biosynthesis in other plants. The expression of the gene correlated with those of several fatty acid biosynthetic genes in the mesocarp of oil palm. Nevertheless, the master regulator of WRI1 remains elusive [15], [16]. In both cases, access to the whole complement of genes – which can only be achieved by whole genome sequencing would enable hypothesis-driven experiments to be carried out and allow further investigations.
However, whole genome sequencing for complex organisms is costly and requires specialized expertise to navigate the data. This is more so for oil palm, with a genome of ∼1,800 Mb [17] is much larger than most oil seed crops [18]–[21] and model crops, such as rice (420 to 466 Mb) [22], [23] and Arabidopsis thaliana (∼135 Mb) [24]. However, the availability of NGS technology has recently allowed the sequencing of the oil palm genome [25]. Nevertheless, generating genomic sequence information through methylation filtration, a technique that allows for the preferential selection of hypomethylated regions of the genome [26], [27] and Sanger technology provides a comprehensive view of the genic regions of the genome. The method is based on chemical discrimination of repeated DNA from genes by certain strains of bacteria [27] resulting in the generation of a comprehensive gene coverage without the need for whole genome sequencing. The GeneThresher® (GT) methylation filtration technique has been validated in over a dozen plant genomes spanning all the major branches of the plant kingdom. It has been employed to generate comprehensive gene sets in ryegrass, clover, corn [28] and sorghum [29], where in sorghum, up to 96% of the genes were successfully tagged. The GT sequences were also an important source of microsatellite markers for application in genetic diversity [30] and genetic mapping [31] research programmes. This study reports on the sequencing and characterization of the hypomethylated regions of the oil palm genome, which is an important resource that focuses on the active regions of the genome.
Materials and Methods
Genomic library construction and methylation filtering
Nuclear genomic DNA was purified from the spear leaf of 7 EG and 2 EO palms, randomly sheared and size selected (0.6 to 1.4 kb). The fragments were ligated to a plasmid vector and transformed into DH5a (methylation filtering strain) or DH10b (non-methylation filtering strain) to generate GT (filtered) or whole genome (UF, unfiltered) libraries, respectively. UF libraries were used as negative control to determine the efficacy of filtered GT libraries. Nine filtered and nine unfiltered libraries were constructed (Table S1). The transformed strains were plated, DNA isolated from colonies, and end-sequenced with 3730 sequencing technology (Life Technologies Corp). For the oil palm bacterial artificial chromosomes (BAC), high molecular weight nuclear DNA was purified from an EG palm, embedded in agarose plugs, partially digested with HindIII, size selected and cloned into the CopyControl™ pCC1BAC™ (HindIII Cloning-Ready) Vector. DNA from individual BAC was prepared and four equimolar pools were constructed (∼44 BAC/pool) representing ∼10 megabases of the oil palm genome. Paired-end libraries were constructed from a 3–4 kb fraction of randomly sheared pooled BAC DNA using Roche 454 titanium kits, and sequenced to ∼30 fold coverage using Roche 454 XL sequencing technology.
Sequence assembly
A graph-based clustering algorithm (MCL) and CAP3 [32] assembler were used for assembly. Graph based sequence clustering uses a data structure in which each sequence is a node of a graph, and each edge is a weighted connection between sequences. When clustering paired-end sequence data, edges are entered both to indicate sequence similarity, and mate-pairs, with the weighting based on alignment score for similar sequences, and a nominal weighting for mate-pairs. An initial own-versus-own BLAST [33] of sequences was performed, using an e-value cut-off of 1e−10 to ensure that the sequences that overlap by ≥40 base pairs (the minimum to join a contig), should report a BLAST hit. In the initial own-versus-own run, only the best hit of each sequence (apart from itself) was reported, so that each sequence only formed a link to at most one other sequence in the graph. The MCL algorithm was then executed to form clusters, which were then assembled into first-pass contigs. In addition, most singletons were identified in this phase, and excluded from further processing. The first pass analysis was designed to remove most of the redundancy in the data. A second-pass analysis was then initiated, in which an own-versus-own BLAST of the first-pass contigs against themselves was executed to report all hits. This forms the complete graph with each first-pass contig having outgoing graph links to all other first-pass contigs it hits, and clusters were formed again. These clusters-of-first-pass-contigs were then used to partition the original sequences into final bins of sequences. The first-pass contigs were discarded once the second pass clustering was completed. The final bins of original sequences were assembled using CAP3.
Filter Power
Filter power (FP), which is the ratio of the probability that a filtered read sampled a gene coding sequence over the probability that an unfiltered read sampled a gene coding sequence was calculated according to Bedell et al. [29] (Materials S1). The estimation of FP was validated using a second method based on the statistics of the sequence assembly, following Whitelaw et al. [28], where the number of islands observed in the filtered and unfiltered assemblies were used to infer the effective sizes of the genomes sampled, using the formula of Lander and Waterman [34]. Details of the adaptation of the Lander Waterman formula and of related calculations are described in Materials S2. The size of the genome sampled was estimated by dividing the oil palm genome size (∼1,800 Mb for both species) by FP.
Gene models and protein translations
Ab initio gene models were predicted using Augustus [35], SNAP [36] and GeneMark [37] implemented in MAKER [38]. Models trained on maize and rice data were used in Augustus and SNAP respectively, as they were the only monocot models in those programs. GeneMark, on the other hand, was trained on data from the oil palm BAC contigs. As MAKER carries out evidence based gene model predictions, sequences from the Swiss-Prot protein database were used as evidence of expressed genes to improve the gene predictions. High quality gene models ≥300 bp long that had MAKER's AED (annotation edit distance) scores of <0.1 were selected for further analysis. Sequences with AED scores ≥0.1 were also selected if longer than 300 bp and had a BLAST hit with e-value of ≤1e−20 to sequences in at least one of the public databases (RefSeq plant RNA, RefSeq plant protein and Swiss-Prot protein). MAKER also provided protein translations of the predicted gene models. These were compared to NCBI's RefSeq plant protein database to detect any frame shift error. The gene models were also compared to the oil palm genome [25] using BLASTN with an e-value cutoff of 1e−20. The top hit was used as the putative location of the gene in the genome.
Gene tagging and coverage
Oil palm BAC was used to estimate the percentage of genes tagged by the methylation filtration (MF) method. This involved, firstly, repeat masking of the EG gene transcripts and then performing iterative subtractive hybridization, in which the transcripts were searched against the BAC genes with a stringent e-value of 1e−20. Each iteration used the top hit to mask the BAC gene sequence. The BLAST-and-mask-top-hit iteration was carried out until no further hits were obtained. The BAC gene models predicted by MAKER and sampled by the transcripts were then indicated and quantified by the masked sequences that had been subtracted. To further bracket the estimation of gene space sampling and minimize the number of false positive BAC gene predictions, a smaller BAC gene space represented by alignments of the BAC to known plant RefSeq proteins was determined. The locations where the plant RefSeq proteins aligned were used to calculate the “RefSeq Gene Estimates”.
Comparison of GT sequences to EST and transcriptome sequences
Sanger EST reads from MPOB and Genbank, and 454 EG transcriptome contigs from two recent oil palm publications [15], [16] were compared to the EG and EO genomic sequence assemblies using BLASTN. The EST and transcriptome sequences were also assembled using CD-HIT-EST [39] at a sequence identity threshold of 0.95 and maximum unmatched percentage of 0.05 to form non-redundant EST clusters. The EST clusters were compared to the GT assemblies using BLASTN. An EST was considered tagged if the match had an e-value of not more than 1e−20.
Global comparison and identification of conserved genes
Arabidopsis and date palm genes were downloaded from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/) and WCMC-Q (http://qatar-weill.cornell.edu/research/datepalmGenome/download.html) websites, respectively. The genes were compared to the oil palm unique sequences (contigs and singletons) using TBLASTN. Comparisons of the Arabidopsis to date palm genes and vice versa were carried out using BLASTP. Reciprocal best BLAST hit method was used to determine potential orthologs. The date palm and oil palm potential orthologs were annotated via BLASTP and BLASTX analysis respectively to Genbank's non-redundant (nr) protein database. Ortholog pairs were annotated as known genes if at least one of the pair had significant similarity to a known gene. All analysis was performed at an e-value cutoff of 1e−20.
Gene ontology (GO)
The GO terms which arose from the BLAST results (searches against UniProt and NCBI databases) and InterProScan [40] were mapped onto the Plant GO Slim annotations using CateGOrizer [41] (formerly known as GO Terms Classifications Counter) and the occurrences counted using the single occurrence count option. Predicted genes were functionally annotated via BLAST searches against NCBI RefSeq plant, A. thaliana mRNA, Oryza sativa mRNA and Swiss-Prot protein databases. All contigs, as well as their corresponding predicted transcripts were also annotated with protein domain and other related information using InterProScan.
Microsatellite
Microsatellite analysis was carried out using Sputnik (http://espressosoftware.com/sputnik/index.html). A microsatellite had to be at least six di-, five tri- or four tetranucleotide repeat-units long. One imperfection every 10 repeats was allowed.
Single nucleotide polymorphism (SNP)
Overlapping reads were identified from ACE files by using a Python script. Each nucleotide position in the alignment was interrogated and putative SNP identified only when each allele was supported by at least 2 separate reads. SNP density was calculated by dividing the total number of SNPs by the length of regions with sequence coverage between 4 and 30.
Transcription factors (TF)
TF of A. thaliana, O. sativa subspecies indica and japonica, Triticum aestivum as well as Vitis vinifera in PlantTFDB [42] were downloaded. Oil palm gene models were compared to the PlantTFDB sequences using BLASTP (cutoff: 1e−20) and validated using HMMPfam. They were characterized as TF if they had a significant hit to the PlantTFDB sequences and contained at least one of the key domains of their respective TF family.
Resistance genes
Plant resistance (R) genes were downloaded from PRGdb (http://prgdb.crg.eu/) [43] and converted to a BLAST database. The downloaded R genes were also classified into six classes, as per Yun [44] and Song et al. [45]. The first class, R gene Pto was classified based on the presence of the kinase protein domains [46], [47] while class 2 R genes contain CC (coil-coiled) –NBS (nucleotide binding site) –LRR (leusine rich repeat) protein domains [48]. R genes that contain the TIR (toll interleukin receptor)-NBS-LRR domains was classified as class 3 [49]. In class 4, the genes contain LRR–TM (transmembrane) [50] while class 5 R genes contain the LRR-TM-kinase domains [51]. The last category, uncategorized R genes, are those with domains that cannot be grouped into any of the above mentioned classes [51]. For each class, the R genes were aligned and a HMM model was generated using HMMER [52]. The HMM models were used to identify oil palm R gene homologs, which were validated via BLASTP comparison to RefSeq and the downloaded R genes, and domain search via InterProScan. An e-value cutoff of 1e−20 was used for the BLASTP analysis. Information on the protein domains and their locations were used to define the domain signature of each class. For classes 4 and 5, TMHMM (Transmembrane HMM) [53] analysis was carried out to identify transmembrane regions. R genes were clustered using ClusterW [54] prior to phylogenetic analysis using MEGA5 [55].
microRNAs
The EG and EO contigs were searched against the whole hairpin sequences of miRBase [56] using BLAST. Regions of the contigs with full-length match and few mismatches - typically with 95% identity to microRNAs (miRNAs), were considered as perfect matches. However, regions with very similar but imperfect matches (≥85% similarity; score ≥100), had their secondary structures predicted using the Vienna package [57]. The secondary structure of both the stem-loop and the sequence around the hit region were predicted using RNAfold [58]. The predicted structures were aligned with the RNAdistance program. If the structure around the match showed similarity to the loop, it was considered as a partial match. Mature miRNAs were predicted using MatureBayes [59].
Results and Discussion
Assembly of E. guineensis and E. oleifera sequences
A total 461,286 methylation-filtered and UF sequences were generated from 246,801 plasmids from the respective EG and EO libraries (Table 1). The sequences were analysed and filtered prior to sequence assembly. Methylation-filtered and UF sequence data were combined to improve the quality of both E. guineensis (306,558 EGs) and E. oleifera (154,728 EOs) assemblies. An additional 559 DNA sequences (434 EGs and 125 EOs) from Genbank were also included in their respective assemblies, mainly to increase the number of SNPs detected.
Table 1. Assembly statistics of EG and EO genomic sequences.
| Assembly | EG01 | EO01 |
| Description | EG genomic sequence | EO genomic sequence |
| Input: | ||
| Reads(clones) | 306,558(164,224) | 154,728(82,577) |
| Public | 434 | 125 |
| Result: | ||
| No. Contigs | 45,370 | 18,836 |
| No. Singletons (≥50 bp) | 155,442 | 92,446 |
| No. Singletons (<50 bp) | 17,405 | 8,556 |
| Total Unique Sequences | 200,812 | 111,282 |
| Total Length of Unique Sequences (nt) | 137,247,669 | 66,077,552 |
| % Unique are Contigs* | 23% | 17% |
| % Reads in Contigs | 44% | 35% |
| N50 Length | 1,166 | 1,053 |
| Max Length | 8,319 | 7,186 |
| Mean Length | 1,063 | 909 |
Percentage of unique sequences that are represented by contigs.
After quality assessment and a size cutoff of 50 bp, 94.6% (289,587) and 94.7% (146,297) of the EG and EO reads respectively were included in the assembly. For EG, the assembly (EG01) produced 45,370 contigs while 155,442 remained as singletons. The N50 of the assembly was 1,166 bp. The EO sequence assembly (EO01) revealed 18,836 contigs and 92,446 singletons with N50 of 1,053 bp. Table 1 summarizes the statistics of EG01 and EO01. These sequences represent an important resource for the research community, especially since there are only limited numbers of oil palm genomic sequences targeting coding regions in the public domain. The collections of sequences are available for download at http://genomsawit.mpob.gov.my and have been registered at NCBI under the E. guineensis and E. oleifera BioProject accessions PRJNA217845 and PRJNA217846 respectively.
Size of genome space sampled by methylation filtration
Previous studies in maize [28], [60], [61], land plants [62], sorghum [29], cowpea [63] and Oryza glaberrima [64] have shown that MF enriches for sequences from the gene space. Using the method of Bedell et al. [29] (Materials S1), the filter power (FP) of EG was estimated as 2.0 to 2.8, and 2.5 to 2.6 for EO. Based on the estimated FP, the sampled genome size was 643 Mb to 900 Mb for EG, and 692 Mb to 720 Mb for EO. Taking the average FP of EG as 2.4 (the range was 2.0 to 2.8), the estimated hypomethylated space of EG is 705 Mb, similar to the size of the Sorghum bicolor genome (Figure 1). Similarly, EO had an average FP of 2.6 and a genome space of 692 Mb. These represent a 2.5 (EG) and 2.6 (EO) fold reduction from the original palm genomes.
Figure 1. Hypomethylated regions of the oil palm genome sampled by GT Technology.
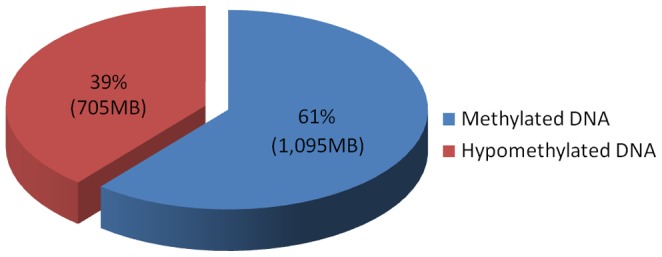
MF reduced the oil palm genome by 61%, thereby allowing sampling of 705 Mb of the hypomethylated region while filtering out 1,095 Mb of the 1,800 Mb genome.
The estimation of FP on the EG sequences was supported by a second method, a modification of the technique described by Whitelaw et al. [28] using the formula of Lander and Waterman [34]. Modifications were made for the GT assemblies used, where unlike previously, the MF and UF reads were assembled together to obtain contigs. The Lander Waterman formula was adapted to obtain the number of islands expected in a mixed assembly of sequences obtained by sampling from two genomes of different effective sizes. This was used to infer the size of the filtered genome, given the known size of the unfiltered genome and the number of islands observed in the mixed assembly. Using this approach, the genome space estimated for EG was 563 Mb with a FP of 3.2. In EO, the sequence coverage was insufficient to perform the analysis. The genome sampling method requires at least 0.1x coverage of the genome to be reliable. Nevertheless, both methods showed that the MF libraries had FP of at least 2.0. Gene enrichment in the MF sequences of oil palm was similar to the 2.47 to 2.83-fold enrichment reported in soybean, potato and oilseed rape, and higher than the 1.89-fold enrichment in rice [62].
Gene models
The oil palm gene models were predicted using MAKER, an evidence-based gene prediction tool. MAKER uses a combination of gene prediction software along with alignments to known transcripts and proteins in producing high quality gene predictions. A total 80,297 gene models were predicted for the EG01, EO01 and BAC contigs. Considering only the high quality transcripts, the number was reduced to 5,504 (166 in BAC, 3,954 in EG01 and 1,385 in EO01 contigs) (File S1). The predicted transcripts were searched against the recently released oil palm genome build [25] to determine whether GT genes were represented in the chromosomes. A total 3,934 EG01 and 1,375 EO01 gene models were successfully placed (Table 2). Of these, 3,048 EG01 and 1,049 EO01 gene models were identified on the 16 oil palm chromosomes [25]. The predicted genes were evenly distributed on all the chromosomes. The location of the GT sequences could help to pinpoint the exonic regions of the oil palm genome.
Table 2. Identification of GT gene models in the oil palm EG5 chromosomes.
| EG5 Chromosomes | Predicted Transcripts | |
| EG01 | EO01 | |
| EG5_Chr1 | 369 | 139 |
| EG5_Chr2 | 297 | 106 |
| EG5_Chr3 | 296 | 106 |
| EG5_Chr4 | 238 | 91 |
| EG5_Chr5 | 248 | 75 |
| EG5_Chr6 | 171 | 50 |
| EG5_Chr7 | 178 | 79 |
| EG5_Chr8 | 175 | 60 |
| EG5_Chr9 | 138 | 51 |
| EG5_Chr10 | 174 | 57 |
| EG5_Chr11 | 131 | 33 |
| EG5_Chr12 | 151 | 45 |
| EG5_Chr13 | 121 | 36 |
| EG5_Chr14 | 145 | 44 |
| EG5_Chr15 | 126 | 42 |
| EG5_Chr16 | 90 | 35 |
| Other scaffolds | 886 | 326 |
| Total hits | 3934 | 1375 |
Gene tagging and coverage
Oil palm BAC were used to estimate the percentage of genes tagged by MF. Full length repeat-masked EG01 contigs and singletons were subtracted in silico from the BAC gene models using the iterative BLAST method. The BAC gene sequences sampled were then indicated and quantified. The analysis showed at least 62% of the gene space on the BAC was sampled. The analysis is indicated in Table 3 as the “Predicted Gene Estimates”.
Table 3. Estimates of percentage BAC gene space sampled.
| Estimated % Gene Space Sampled | Pool A* | Pool B* | Pool C* | Pool D* |
| Predicted Gene Estimates | 77% | 79% | 62% | 71% |
| RefSeq Gene Estimates | 71% | 77% | 68% | 66% |
| Masked Sanger EST contigs and singletons | 33% | 36% | 34% | 31% |
| (25,781 sequences, 15 Mb) | ||||
| Masked 454 transcriptome | 36% | 35% | 40% | 47% |
| (70,729 sequences, 69 Mb) |
Pool A, B, C and D (∼44 BAC/pool) are equimolar pools representing ∼10 megabases of the oil palm genome.
Steps were taken to minimize false BAC gene predictions as these can inflate the gene space. For EG01, this was done by sampling smaller BAC gene spaces, represented by alignments of BAC to known plant reference sequence proteins. BAC were positionally annotated with known plant RefSeq proteins and 157 BAC sequence transcripts corresponding to these gene positions extracted (Table 4). The same stringent in-silico subtractive hybridization of full length repeat-masked EG01 contigs and singletons subtracted from this reduced set of BAC gene transcripts resulted in the “RefSeq Gene Estimates” in Table 3.
Table 4. Reduced BAC gene space annotated by plant RefSeq orthologs.
| Pool A | Pool B | Pool C | Pool D | |
| No. transcripts | 49 | 46 | 27 | 35 |
| Mean transcript length | 1,145 | 1,059 | 1,150 | 992 |
| Maximum transcript length | 3,423 | 2,934 | 3,642 | 2,100 |
To evaluate gene space coverage, EG01 sampling of this reduced space was compared with that achieved by two other gene sampling methods, EST and 454 transcriptome sequencing. EST contigs assembled from Sanger EST reads (MPOB and Genbank), and 454 EG transcriptome contigs [15], [16] were repeat masked and compared to the reduced set of BAC gene transcripts using the same iterative method described above. The results showed that the gene space sampled in EG01 was higher than that obtained using EST sequencing and from the 454 sequenced transcriptome. This shows the efficiency of the method for gene discovery. Nevertheless, it is important to note that the number of ESTs and transcriptome data were low and specific only to the tissues sampled. Most of the ESTs [6], [7], [9], [65] were sampled from tissue culture materials while the transcriptome data was mainly sampled from mesocarp tissues [15], [16]. The BACs were randomly sampled and genes expressed in these tissues might not be represented in these BACs. This could have resulted in the low level of BAC gene space sampled by the transcripts.
Comparison of GT sequences to EST and transcriptome sequences
In order to determine whether the available oil palm ESTs were tagged by the MF sequences, the GT assemblies were compared to the EST and transcriptome contig sequences. EG01 was able to tag between 64% and 78% of the EST sequences. Nevertheless, to obtain a better estimate of the number of ESTs tagged, the GT assemblies were compared to a non-redundant set of ESTs. The analysis showed that EG01 sequences were able to tag a high percentage of the EST clusters (72%). The results obtained were comparable to those from cowpea, where 73.7% of the EST dataset matched the GT sequences [63]. In EO01, the percentage was lower since the EST and transcriptome sequences were mostly obtained from EG. Differences between the two oil palm species most likely accounted for the reduced similarity. Figure 2 show the percentages of EST and transcriptome sequences that have hits to EG01 and EO01 sequences. Interestingly, comparison of the EG01 and EO01 gene models showed that 23.3% and 21.4% of the gene models respectively, were absent from the EST and transcriptome data (Table 5). The GT sequences not only had a high coverage of the available EST sequences, it was also able to identify additional genes that would be an important resource for research. The list of genes not tagged by the EST data is in File S2.
Figure 2. BLASTN analysis of oil palm EST and transcriptome sequences to EG01 and EO01.

The percentage of EST, transcriptome and Cluster sequences that have significant similarity (≤1e−20) to EG01 and EO01 sequences are shown in green and yellow respectively. Cluster is a set of non-redundant sequences generated from the assembly of the EST and transcriptome data by CD-HIT-EST.
Table 5. Comparison of predicted oil palm gene models against EST and transcriptome data.
e-value cutoff: 1e−20.
Global comparison of EG01 sequences to Arabidopsis and date palm genes
The oil palm genomic sequences were compared to Arabidopsis and date palm genes to determine the coverage of the genes tagged by the EG01 data. The Arabidopsis and date palm dataset contains 35,386 [66] and 28,890 [67] genes respectively. In the first analysis, the Arabidopsis sequences were used as TBLASTN queries against EG and as BLASTP queries for the date palm protein sequences. There were 15,431 (44%) and 24,604 (70%) Arabidopsis genes with matches to oil palm and date palm genes, respectively. The fewer hits to oil palm were not surprising considering the low coverage of its GT sequences in this study. As date palm is the closest related plant genome to oil palm to be sequenced, the analysis was repeated using date palm genes as query in searches against oil palm EG and Arabidopsis protein sequences. This provided a better representation for cross species gene annotation.
In the analysis, 17,838 (62%) date palm sequences had hits to oil palm while 19,489 (68%) had matches to Arabidopsis. A total 371 date palm proteins had matches to EG but not to Arabidopsis. Reciprocal best BLAST hits of these sequences identified 192 potential orthologs of date palm and oil palm that did not have any similarity to Arabidopsis genes. Comparison to Genbank's nr protein database showed that five of these genes had similarity to repeat elements and were removed from further analysis. About 50% of the putative orthologs did not have any similarity to sequences in Genbank. The remaining sequences had hits to known [58] and uncharacterized [34] genes, such as hypothetical genes or putative proteins, of which 19 had significant similarity only to monocotyledon genes in the nr database. The list of these orthologs is available in File S3. These genes may be useful to study conserved functions between oil palm and date palm. Overall, even at low coverage, the GT method was able to tag a high percentage of genes in the date palm genome and identify genes that are conserved between date palm and oil palm.
Gene ontology (GO)
BLAST results from the functional annotations analysis (specifically, searches against the UniProtKB and RefSeq plant mRNA databases) of the good quality gene models were searched against the UniProt and NCBI databases for GO terms using a set of custom scripts. Final results of the non-redundant GO analysis were merged based on results from the BLAST and InterProScan searches. Table 6 shows the domain annotation and summary of the GO search results. The GO analysis results indicated that the predicted genes were distributed in different functional classes (Figure S1). More importantly, similar trends were observed in both EG and EO functional classification, although, as expected there were differences in the number of genes in each functional class. The three top level categories were Molecular Function (ML), Biological Process (BP) and Cellular Component (CC). It is worthwhile to note that 75% of the predicted genes in EG were assigned to ML, while 43% and 41% were categorized with BP and CC terms, respectively. Notably, a similar trend was observed in EO. The results also showed that 18% of the predicted genes could not be annotated with GO terms. Analysis of their annotation showed that more than 50% of them had similarity to hypothetical proteins.
Table 6. Summary of domain, sub-cellular localisation and GO annotation.
| Dataset | EG01 Contigs | EO01 Contigs | BAC Contigs |
| Predicted Genes with Domain annotations | 2,861 | 1,013 | 86 |
| Predicted Genes with SignalP predictions | 581 | 183 | n/a |
| Predicted Genes with TargetP predictions | 148 | 48 | n/a |
| Predicted Genes with GO Molecular Function terms | 2,960 | 1,068 | 129 |
| Predicted Genes with GO Biological Process terms | 1704 | 636 | 96 |
| Predicted Genes with GO Cellular Component terms | 1623 | 622 | 59 |
A more comprehensive insight into ML revealed that the top subcategories for EG and EO were molecular function [GO:0003674], catalytic activity [GO:0003824] and transferase activity [GO:0016740]. The majority of the predicted genes annotated under the GO term BP category were regulating transcription, DNA-dependence [GO:0006355] and proteolysis [GO:0006508]. Interestingly, the GT data did not have over-representation of highly expressed genes, such as ribosomal genes, as seen in the ribosomal and cytoskeletal peaks in the BAC data in Figure S1c. This suggests that the GT sequences were randomly distributed in the hypomethylated regions of the genome. The gene ontology results are given in File S1.
The gene models (3,954 EG and 1,385 EO) were also mapped onto the Kyoto Encyclopedia of Genes and Genomes (KEGG) orthology database [68], which enable reconstruction of the KEGG pathways. A total 488 EG01 genes were assigned to KO (KEGG Ortholog), of which 317 were mapped onto 71 pathways. As for EO, out of 223 KO annotated genes, 161 were successfully assigned and mapped onto 56 pathways. In EG, oxidative phosphorylation [25], ribosome [23] and glycolysis/gluconeogenesis [16] are the most abundant. A similar result was observed in EO, where oxidative phosphorylation [28] represented the most common pathway, followed by photosynthesis [15]. Table S2 shows the number of genes categorized into KEGG pathways.
Microsatellites
Microsatellites, also known as Simple Sequence Repeats (SSR), are important sources of molecular markers for genetic studies. The earliest exploitation of a sizeable number of EG genomic SSR for genetic mapping was by Billotte and colleagues [69] with 369 SSR of dinucleotide (GA and GT) and trinucleotide (CCG) repeats used. Although the researchers were able to show the effectiveness of SSR as molecular markers, the number of SSR used was small. Hence the GeneThresher sequences were mined for di-, tri- and tetranucleotide repeats, where 23,621 and 10,131 SSR were identified from EG01 and EO01, respectively (Table 7). The dinucleotide repeats in the assembled sequences of both oil palm species exceeded those of tri- and tetranucleotide repeats. This was consistent with what has been reported earlier by Tranbarger et al. [70] who found that dinucleotide repeats were the most abundant EST-SSR (36%) in oil palm followed by tri- (24%) and tetra- (29%) motifs. The most frequent dinucleotides in their transcriptome data were those with 6 repeat motifs, compared to 17–18 repeat motifs in the genomic SSR reported by Billotte et al. [69], [71]. The authors concluded that there was a higher frequency of lower number of repeat motifs in the coding region. The current MF data reflected this pattern as it also mainly covered the genic regions. Nevertheless, the MF data also revealed a high percentage of AT dinucleotide with 40 repeat motifs (Figure 3). This suggests that the MF sequences also contained sequences from non-genic regions, most likely the flanking regions of genes.
Table 7. Summary of di-, tri- and tetranucleotide repeat motifs in EG01, EO01 and BAC.
| Data Set | Dinucleotides | Trinucleotides | Tetranucleotides | Total |
| EG01 | 14, 910 | 5,152 | 3,559 | 23,621 |
| EO01 | 6,366 | 2,247 | 1,518 | 10,131 |
| BAC | 594 | 328 | 247 | 1,169 |
Figure 3. Distribution of dinucleotide repeats observed in EG01 SSR.

The AC, AG, AT and CG repeats are represented in blue, red, green and purple respectively. The total number of observations for each repeat are represented by the height of the respective column.
Among dimerics in EG01, the AG motif was the most abundant repeat with 28.62%, followed by AT (26.06%) and AC (8.18%) respectively (Table S3). A similar trend was also observed in EO01 (Table S4). Low et al. [7] identified similar trends in dinucleotide repeat motifs and repeat numbers in oil palm ESTs with AG/CT (67%) and AT (21%) the most abundant, followed by AC/GT (11%) and CG (0.3%). The AG/CT dinucleotide repeat motif was consistent with the high frequencies in the genic regions of A. thaliana [72] and rice [73]. Similar patterns were also noted for EST-SSR in peanut [74] and cacao [75]. The CG motif was generally in low abundance in both Elaeis species.
Although the trinucleotide repeats are not the most prevalent SSR in the hypomethylated regions of oil palm, they are of interest as they are found predominantly in the exonic regions. Low et al. [7] compared the distribution of a small number of oil palm full-length EST-SSR and found the mono- and di-nucleotide repeats in the untranslated regions (UTR), whereas trinucleotides were in both UTR and open reading frames (ORF), with a preference for ORF. Zhang and colleagues [72] observed that the trinucleotides, followed by the hexanucleotides accounted for 92.6% of the SSR in the coding regions of A. thaliana. A similar observation was also made by Toth et al. [76] that trimers and hexamers were rampant in the exon regions of eukaryotic genomes. In EG01 and EO01, the most abundant trimers were AAG (5.94%, 6.11%), AAT (5.86%, 5.89%), AGG (3.29%, 3.35%) and CCG (1.99%, 2.06%), respectively (Tables S3 and S4). The trend is similar to the patterns observed by Low et al. [7] in oil palm ESTs, where the most prevalent trinucleotides were AAG/CTT (23%), AGG/CCT (13%), CCG/CGG (11%) and AAT/ATT (11%). Although the most prevalent trimer in both the genomic and EST-SSR was AAG, the genomic sequences had a higher representation of AAT repeats compared to EST-SSR. The high abundance of the tri-repeat motif of AAG in EG and EO was similar to that reported in the EST sequences of cotton (G. hirsutum, G. arboretum and G. raimondii) [77] and cucumber [78], respectively. A slightly different frequency of repeat motifs of AAG and AAC was observed in the exonic regions of embryophyta [76]. Zhang et al. [72] also observed that the AAG motif was the most prominent repeat in the 5′UTR region of A. thaliana.
In the hypomethylated region of Elaeis, the trimer motif CCG was also observed albeit at low percentage of 1.99–2.06%. This is different from other monocots (maize and wheat), where the CCG motif alone accounted for half of the trinucleotide repeats in rice and is also moderately rich in other plants [79]. Yonemaru et al. [80] also found that the most frequent trimers in S. bicolor were CGC/GCG. This shows that diverse taxonomic groups exhibit different tendencies for SSR types which are also influenced by their genomic locations [76]. In the 33 different tetranucleotide repeat motifs found in the Elaeis sequences, AAAT motif was the most frequent, followed by AAAG and ACAT. Similar motifs were also found in the A. thaliana genome, which is AT-rich [81], and in the genome of cotton G. raimondii [77].
The MF sequences proved to be an important source of SSR markers. In fact, the utility of SSR from methylation filtered sequences for oil palm genetic diversity analysis and genetic mapping was demonstrated by Zaki et al. [30] and Ting et al. [31], respectively. The SSR markers from MF sequences have several advantages. One is that they show increased polymorphism associated with genomic based SSR markers compared to EST-SSR. At the same time, since they represent mostly genic regions, they can be used to target candidate genes, similar to EST-SSR. The locations of SSR in EG01 and EO01 are given in File S4.
Single nucleotide polymorphism
Evolution of the techniques for identification of molecular markers in recent years has led to the discovery of SNPs, which is a single base difference in a DNA sequence with an alternative of two possible nucleotides at a specific location on the chromosome [82]. Initially, the number of putative SNPs identified was 36,138 for EG01 and 14,640 for EO01 contigs. However, to avoid false positives, SNPs with extraordinarily high coverage depth (>30; 2 standard deviation from mean) were excluded from further analysis. This removed SNPs in repetitive or duplicated regions of the genome, leaving 28,842 and 12,578 for EG and EO, respectively. The list of SNPs in EG01 and EO01 are available in File S4. The SNP densities were 2.30 and 2.83 per 100 bp for EG and EO, respectively. However, a previous study on oil palm ESTs showed a density of 1.36 SNPs per 100 bp [83]. As the SNP density in the genic region is expected to be lower than the non-genic region, it is likely that the GT SNP densities are not reflective of the SNP densities of the oil palm genome. The sequence coverage needs to be increased to have a better estimate of the SNP density. Sequence depth values across all contigs for EG and EO are provided in Figure S2. The depths varied widely as most positions within most contigs are only supported by a single read.
The SNPs were grouped into either transition (C/T or G/A) or transversion (C/G, A/T, C/A or T/G) nucleotide substitutions. The frequency of transition exceeded transversion (Table 8), similar to that reported for 1,317 SNPs mined from 5,452 oil palm sequences from seven tissues [83]. Similar trends were observed in maize [84], S. bicolor [85] and ginger [86]. In Table 8, the number of SNPs observed in the EG and EO contigs for both transition type SNPs (G/A and C/T) showed no significant difference. However, in transversions, the A/T type SNPs were more frequent than other transversions, and collectively accounted for 44.2% (EG) and 41.3% (EO) of all transversions. The overall transition vs transversion ratio in EO was 7.52, which indicates higher transitions over transversions. In EG, the ratio was slightly lower (7.17), consistent with Riju and Arunachalam [87] who identified an overall EO transition vs transversion ratio of 1.40 for EO and 1.02 for EG. They opined that the transition vs transversion rate is important to understand DNA evolution, with a low value indicative of high genetic divergence and vice versa. Interestingly, the lower divergence of EO vis-à-vis EG had been demonstrated experimentally using various marker systems, such as SSR [31], [88] and even a small number of SNPs [89].
Table 8. Summary of SNPs.
| EG01 Contigs | EO01 Contigs | |
| Transitions | ||
| C/T | 12,391 | 5,638 |
| G/A | 12,397 | 5,464 |
| Transversions | ||
| A/T | 1,928 | 866 |
| C/G | 180 | 97 |
| G/T | 696 | 226 |
| A/C | 650 | 287 |
| Total | 28,242 | 12,578 |
In barley [90], higher polymorphism rate was observed for transition SNPs (71%) vs transversions (29%), information of possible importance for identification of informative markers. Categorizing the SNPs into transition and transversions could potentially improve efficiency and reduce the number of non-polymorphic SNP markers. Furthermore, as the GT data represent exonic regions of the genome that encode for genes and their regulatory regions, identification of non-synonymous SNPs in genes associated with traits could potentially provide insight into the modulation of the trait. This was recently reported when an important monogenic trait (SHELL) in oil palm was shown to be caused by two independent SNPs in a single gene. The mutations disrupt the DNA-binding domain of a MADS-box gene homologue of SEEDSTICK, resulting in three different EG fruit forms - dura, pisifera and the hybrid tenera. This single gene is responsible for the hybrid vigour or heterosis observed in the tenera fruit form of oil palm [91]. The polymorphisms in the gene will prove to be an important diagnostic assay for commercial seed production and to enhance breeding activities in oil palm.
Transcription factors
Transcription factors (TF) help regulate gene expression and are an integral part in the development of an organism. The number of TF in plant genomes is large - 6 to 9% of the coding regions that code for TF [24], [42], [92], [93]. Analysis of Musa acuminata genome recently showed that 8.6% or 3,155 of its protein-coding gene models coded for TF. This represents one of the highest numbers of TF identified in a sequenced plant genome [93]. The evolution of a large number of TF could explain the diversity and complexity observed in plants. In oil palm, comparison of the EG01 and EO01 gene models to TF from A. thaliana, O. sativa, T. aestivum and V. vinifera from the PlantTFDB database [42] showed that both libraries contained 37 TF gene families, while the BAC sequences were able to identify two additional gene families. A total of 178 and 61 transcriptional factors were identified from the EG01 and EO01 gene models, respectively. The numbers of GT sequences for each gene family are listed in Table 9.
Table 9. Oil palm TF in EG01, EO01 and BAC sequences.
| Transcription Factor | EG | EO | BAC | Transcription Factor | EG | EO | BAC |
| AP2 | 4 | 1 | GRF | 2 | |||
| ARF | 7 | 2 | HB-other | 1 | |||
| ARR-B | 2 | 1 | HD-ZIP | 12 | 5 | ||
| BBR/BPC | 2 | HSF | 1 | ||||
| BES1 | 1 | LBD | 4 | ||||
| bHLH | 12 | 6 | M-type | 1 | |||
| bZIP | 8 | 6 | MYB | 12 | 5 | ||
| C2H2 | 16 | 3 | MYB_related | 2 | |||
| C3H | 5 | 2 | NAC | 10 | 4 | ||
| CAMTA | 1 | NF-X1 | 1 | ||||
| CO-like | 1 | NF-YB | 3 | 1 | |||
| CPP | 1 | 1 | Nin-like | 1 | |||
| Dof | 7 | RAV | 1 | ||||
| E2F/DP | 2 | SBP | 3 | 1 | |||
| EIL | 1 | SRS | 1 | ||||
| ERF | 13 | 8 | 1 | TALE | 10 | 1 | |
| FAR1 | 1 | TCP | 4 | 1 | |||
| G2-like | 6 | 2 | WOX | 1 | |||
| GATA | 2 | 1 | WRKY | 7 | 2 | ||
| GRAS | 15 | 4 | Total | 178 | 61 | 3 |
Ethylene Response Factor (ERF) is the one of the biggest group of TF identified. This is not surprising as the ERF family is the most abundant TF in PlantTFDB and second most abundant in DRTF (rice transcriptional factor database) [94]. The analysis also revealed five Apetala2 (AP2) and one RAV genes. These three gene families belong to the AP2/ERF TF superfamily involved in responding to plant biotic and abiotic stress [95], [96]. The ERF sub-family is also known for its involvement in regulating the expression of pathogenesis-related (PR) genes and could play a role in the transduction of various signals to a suite of downstream defence genes [97]. These genes are important for studies related to how oil palm defends itself against pathogens, especially the fungus Ganoderma, which is the cause of a major oil palm disease in Malaysia and Indonesia.
Analysis of the GT sequences also revealed TF associated with floral development and tissue culture, such as homeodomain proteins, MADS, Squamosa (SBP) and Apetala2 (AP2). These genes are involved in floral organ patterning and are expressed in different stages of floral development [98]. In Arabidopsis, AP2 is required for the specification of the first and second whorl organ identities [99]. MADS box genes are also hypothesized to be involved in clonal abnormality, namely mantled flowers in ramets [100]. Although clonal abnormality in oil palm has been associated with changes in methylation, the role of MADS box genes in this phenomenon is being investigated. In fact, methylation changes in this group of genes that determine the ABC model for floral development could be pivotal in the clonal abnormality phenomenon observed [101]. In this study, a MADS box gene was identified in the GT dataset. An additional putative MADS box gene with significant similarity (5e−27) to a MIKC gene but only containing the k domain was also identified. The sequence did not contain the MADS domain and was thus not included in Table 9.
The TF dataset represents an important resource not only to study floral development and stress responses but also other important mechanisms in oil palm. Another important application of the TF data is that 58 of these sequences contained SSR or SNPs, which can help localize the genes on the oil palm genetic maps. As some genes of similar function tend to cluster in the genome, identification of the genetic loci would allow researchers to test other genes/markers flanking the TF for association with specific traits. The list of TF is available in File S1.
Resistance gene homolog
Fungi cause three major diseases in oil palm - Fusarium wilt, bud rot and basal stem rot. Identification of the genes involved in pathogenicity and resistance is an important step towards identifying disease tolerant/resistant palms. As such, efforts have been made to identify oil palm pathogenesis-related genes (R genes homolog) in the EG01 and EO01 gene models. R genes play an important role in the early stages of plant defense mechanism [102], [103]. They have distinct interactions with specific molecules secreted by the pathogen into plant cells during invasion [104]–[106]. Comparison of the predicted amino acid sequences from EG01 and EO01 to known R genes revealed 52 EG and 13 EO R gene homologs (File S1).
Of these, only two class 1 R genes from EG and one from EO containing both the kinase and Pto domains were identified. The analysis also revealed that class 2 was the largest group of R genes identified. Homology search and InterProScan confirmed the presence of 29 EG class 2 R genes. They represent ∼44.6% of the oil palm R genes identified, in line with previous reports that the NBS-LRR group is the largest class of R genes [107]. Barbosa-da-silva and colleagues [50] also reported that R genes with NBS-LRR domain properties are the largest group and contained the most functionally defined R genes. We suspect that this class is an important component of the plant immune response system. The NBS domain is involved in ATP binding and hydrolysis, while the LRR domain is the determinant of response specificity [108]. However, no class 2 R gene was identified in the EO01 data, probably due to the lower coverage of the EO libraries. The R genes in EO01 were probably partial length and categorized as class 6 (uncategorized).
The analysis also did not reveal any class 3 R genes in the EG01 and EO01 data. This was not surprising as class 3 R genes are predominantly found in dicotyledons. The only monocotyledon TIR-NBS-LRR R gene identified was reported in the Triticum-Thinopyrus line [109]. Oil palm, being a monocotyledon, is not expected to have homologues of class 3 R genes. Homologues to class 4, 5 and 6 were also identified. Eight EG01 gene models were classified as class 4 R genes. An additional seven EG01 and three EO01 gene models that contained the LRR-TM-kinase domains were classified as class 5 R gene. The final class of R genes, the ‘uncategorised’, contained 15 gene models.
Classification of the oil palm R genes homologs were further verified using phylogenetic analysis (Figure 4). The analysis showed three distinct clades, representing class 1, 2, and a combination of class 4 and 5. Class 4 and 5 share the same clade because both classes contained the LRR and TM domains. Class 5 can be differentiated by an additional kinase domain. Class 6 R genes (uncategorised) were not included in the phylogenetic analysis. The phylogenetic analysis generally concurred with the classification of the genes. The R genes identified in this study would facilitate the understanding of how oil palm defends itself against diseases such as bud rot and basal stem rot, which have devastated large tracts of oil palm plantations. Combining knowledge of R genes and associating it with quantitative trait loci analysis of germplasm/breeding populations for disease resistance [110] would help with future development of elite oil palm varieties.
Figure 4. Phylogenetic analysis of EG and EO R genes.

Class 1, 2, 4 and 5 are represented by blue, red, black and green circles respectively.
microRNAs
miRNAs are short sequences from a class of RNAs ∼18 to 24 nt in length. They are produced by dicer-catalyzed excision from stem-loop precursors and play an important role in diverse organisms [111]. The functional role of miRNAs can be elucidated by the identification of their mRNA targets [112]. At present, most of the plant miRNAs identified belong to the model plant A. thaliana. Nevertheless, there is a growing resource of miRNAs from other plants, such as rice, tomato and sorghum [112]–[114]. In this study, a homology approach was used to identify oil palm miRNAs. The EG01 and EO01 contigs were searched against the stem-loop precursors of miRBase [56]. As mature miRNAs are short, using the stem-loop precursors provide a longer sequence for comparison to identify conserved regions.
Forty miRNAs were identified from the contigs. Of them, 28 were predicted in EG01 contigs where 10 contigs gave perfect hits and 18 partial matches. In EO01 contigs, nine gave perfect hits and three partial matches to known miRNAs in the registry. Stringent parameters with 85% similarity cutoff and a score of ≥100 were used to avoid false positives. The list of predicted miRNAs for EG01 and EO01 is shown in Table S5. However, the predictions are dependent on the miRNAs deposited in miRBase. The small number of predicted miRNAs obtained was most likely due to the lack of closely related species in miRBase. The quality of the predicted putative miRNAs was further verified by looking at mismatches in the hit regions. This was to ensure that the mismatches did not break the secondary structure and only fell on the open-loop regions. As a result, 14 predicted mature sequences were retrieved from MatureBayes program as potential oil palm miRNAs (Table 10).
Table 10. List of predicted mature miRNAs from EG01 and EO01 contigs.
| Contigs | Best Hits with miRNAs in miRBase | Match Status | Predicted Mature miRNA* |
| EGC01043189 | peu-MIR2911 | Perfect | gcggcgacccgcucucgccgcg |
| EGC01002494 | peu-MIR2911 | Perfect | gcggcgacccgcucucgccgcg |
| EGC01007640 | peu-MIR2911 | Perfect | gcggcgacccgcucucgccgcg |
| EGC01002621 | peu-MIR2916 | Perfect | ccugaaagcaacauccgccgau |
| EGC01009851 | peu-MIR2916 | Perfect | gaagacgaucagauaccguccu |
| EGC01006056 | peu-MIR2911 | Perfect | gcggcgacccgcucucgccgcg |
| EGC01005984 | peu-MIR2911 | Perfect | gcggcgacccgcucucgccgcg |
| EGC01029522 | ptc-MIR156j | Perfect | ugaugcagagcuccaugcaucc |
| EOC01000015 | peu-MIR2916 | Perfect | ugggggcucgaagacgaucagau |
| peu-MIR2914 | |||
| peu-MIR2910 | |||
| EOC01008865 | vvi-MIR319f | Perfect | gaugcaaugggucuugcauguc |
| EOC01001645 | sbi-MIR167g | Perfect | ggcaucgggggcgcaacgcccu |
| EOC01006693 | ptc-MIR319e | Perfect | gcuuccuucagcccacucaugg |
| EOC01010601 | vvi-MIR845a | Perfect | cucauccaagaucuagaggaaa |
| EOC01007557 | vvi-MIR845b | Perfect | cccuucaguccaaucggcgggc |
Mature miRNAs were predicted using MatureBayes program.
Target prediction of the 14 potential miRNAs identified one target mRNA transcript that is similar to the Rab21-family small GTPase, which is a small GTP-binding protein of the Ras superfamily [115]. Identification of only a single target gene is not surprising as most of the oil palm mRNA transcripts available are not full-length and probably lack the UTR regions. Nevertheless, identification of the Rab protein is interesting as it plays an important role in regulating intracellular vesicle trafficking. In plants, Rab proteins have been implicated in transport between the endoplasmic reticulum and Golgi apparatus, trafficking of soluble cargo, fusion of endocytic vesicles and vesicular transport along microtubules. Studies in Arabidopsis have also identified that certain Rab proteins are influenced by hormones, such as ethylene and auxin [116]. It would be interesting to determine the expression of the oil palm miRNAs, and its interaction with the Rab21 transcript. Looking forward, the recently released oil palm genome data will provide valuable information for further characterization of the oil palm miRNAs.
Supporting Information
Gene ontology classification of EG01, EO01 and BAC sequences. Three GO categories, [A] Molecular function (ML) [B] Biological process (BP), and [C] Cellular component (CC) terms were mapped to Plant Slim GO annotations using CateGOrizer.
(DOCX)
Depth at SNP positions for [A] EG01 and [B] EO01 contigs. The red line indicates the cut-off of two standard deviation from mean, where the SNPs on the left of the line were defined as unreliable.
(DOCX)
E. guineensis and E. oleifera filtered and unfiltered genomic library information.
(DOCX)
Categorization of EG and EO genes into KEGG pathways.
(DOCX)
Di-, tri- and tetranucleotide repeats identified in EG01.
(DOCX)
Di-, tri- and tetranucleotide repeats identified in EO01.
(DOCX)
List of perfect and partial match miRNAs from EG01 and EO01 contigs.
(DOCX)
Gene information and annotations.
(XLSX)
List of genes not tagged by oil palm ESTs.
(XLSX)
List of orthologs.
(XLSX)
List of SSRs and SNPs.
(XLS)
Formula to calculate gene enrichment.
(DOCX)
Formula to calculate genome sampling.
(DOCX)
Acknowledgments
A special appreciation is extended to Dr. Cheah Suan Choo, who conceptualized and embarked on the project. We also wish to thank Nathan Lakey and Mohd A. Budiman from Orion Genomics LLC for their assistance in the project, especially for construction of the GT and UT libraries and valuable discussions on the project. Special thanks to the Breeding and Genetics Group for the supply of palms for sequencing and Dr. Ravigadevi for her help and support. The authors would also like to thank Anar Khan for her valuable thoughts and assistance with data analysis. Last but not least, we wish to extend our appreciation to the Datuk Dr. Choo Yuen May for her support and encouragement for the project.
Funding Statement
This work was supported by special MPOB grant (R001702000-RB01-J). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Dransfield J, Uhl NW, Asmussen CB, Baker WJ, Harley MM, et al.. (2008) Genera Palmarum. Evolution and classification of the palms. 2nd ed. Royal Botanic Gardens: Kew Publishing. [Google Scholar]
- 2.Corley RHV, Tinker PB (2003) The oil palm. 4th ed. Oxford: Blackwell Science. [Google Scholar]
- 3.Latiff A (2000) The biology of the genus Elaeis. In: Basiron, Y, Jalani, B S, Chan KW, editor. Advances in oil palm research, Volume 1. Malaysian Palm Oil Board. pp. 19–38. [Google Scholar]
- 4. Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, et al. (1991) Complementary DNA sequencing: expressed sequence tags and human genome project. Science 252: 1651–1656. [DOI] [PubMed] [Google Scholar]
- 5. Jouannic S, Argout X, Lechauve F, Fizames C, Borgel A, et al. (2005) Analysis of expressed sequence tags from oil palm (Elaeis guineensis). FEBS Letters 579: 2709–2714 Available: http://www.ncbi.nlm.nih.gov/pubmed/15862313. Accessed 2013 May 30 [DOI] [PubMed] [Google Scholar]
- 6. Ho CL, Kwan YY, Choi MC, Tee SS, Ng WH, et al. (2007) Analysis and functional annotation of expressed sequence tags (ESTs) from multiple tissues of oil palm (Elaeis guineensis Jacq.). BMC Genomics 8: 381 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2222642&tool=pmcentrez&rendertype=abstract. Accessed 32013 May 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Low ETL, Alias H, Boon SH, Shariff E, Tan CYA, et al. (2008) Oil palm (Elaeis guineensis Jacq.) tissue culture ESTs: identifying genes associated with callogenesis and embryogenesis. BMC Plant Biology 8: 62 Available: http://www.biomedcentral.com/1471-2229/8/62/. Accessed 2013 June 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Feng S, Wang X, Zhang X, Dang PM, Holbrook CC, et al. (2012) Peanut (Arachis hypogaea) Expressed Sequence Tag Project: Progress and Application. Comparative and Functional Genomics 2012: 373768 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3382957&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chan PL, Ma LS, Low ETL, Shariff EM, Ooi LCL, et al. (2010) Normalized embryoid cDNA library of oil palm (Elaeis guineensis). Electronic Journal of Biotechnology 13: 14 Available: http://www.ejbiotechnology.info/content/vol13/issue1/full/14/index.html. Accessed 2013 June 5 [Google Scholar]
- 10. Wang W, Wang Y, Zhang Q, Qi Y, Guo D (2009) Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC genomics 10: 465 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2763888&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shearman JR, Jantasuriyarat C, Sangsrakru D, Yoocha T, Vannavichit A, et al. (2013) Transcriptome analysis of normal and mantled developing oil palm flower and fruit. Genomics 101: 306–312 Available: http://www.ncbi.nlm.nih.gov/pubmed/23474141. Accessed 2013 June 3 [DOI] [PubMed] [Google Scholar]
- 12. Jaligot E, Rival A, Beulé T, Dussert S, Verdeil JL (2000) Somaclonal variation in oil palm (Elaeis guineensis Jacq.): the DNA methylation hypothesis. Plant Cell Reports 19: 684–690 Available: http://link.springer.com/10.1007/s002999900177 [DOI] [PubMed] [Google Scholar]
- 13. Matthes M, Singh R, Cheah SC, Karp A (2001) Variation in oil palm (Elaeis guineensis Jacq.) tissue culture-derived regenerants revealed by AFLPs with methylation-sensitive enzymes. Theoretical and Applied Genetics 102: 971–979 Available: http://link.springer.com/10.1007/s001220000491 [Google Scholar]
- 14. Lucia G, Castiglione MR, Martini G, Geri C, Ronchi VN (2007) Methylated DNA sequence extrusion during plant early meiotic prophase. Caryologia 60: 279–289. [Google Scholar]
- 15. Bourgis F, Kilaru A, Cao X, Ngando-Ebongue GF, Drira N, et al. (2011) Comparative transcriptome and metabolite analysis of oil palm and date palm mesocarp that differ dramatically in carbon partitioning. Proceedings of the National Academy of Sciences 108: 12527–12532 Available: http://www.ncbi.nlm.nih.gov/pubmed/21709233. Accessed 2013 May 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tranbarger TJ, Dussert S, Joët T, Argout X, Summo M, et al. (2011) Regulatory mechanisms underlying oil palm fruit mesocarp maturation, ripening, and functional specialization in lipid and carotenoid metabolism. Plant Physiology 156: 564–584 Available: http://www.ncbi.nlm.nih.gov/pubmed/21487046. Accessed 2013 May 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rival A, Beule T, Barre P, Hamon S, Duval Y, et al. (1997) Comparative flow cytometric estimation of nuclear DNA content in oil palm (Elaeis guineensis Jacq) tissue cultures and seed-derived plants. Plant Cell Reports 16: 884–887 10.1007/s002990050339 [DOI] [PubMed] [Google Scholar]
- 18. Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183. [DOI] [PubMed] [Google Scholar]
- 19. Ragupathy R, Rathinavelu R, Cloutier S (2011) Physical mapping and BAC-end sequence analysis provide initial insights into the flax (Linum usitatissimum L.) genome. BMC Genomics 12: 217 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3113786&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. The Brassica rapa Genome Sequencing Consortium (2011) The genome of the mesopolyploid crop species Brassica rapa. Nature Genetics 43: 1035–1039. [DOI] [PubMed] [Google Scholar]
- 21. Zhang H, Miao H, Wang L, Qu L, Liu H, et al. (2013) Genome sequencing of the important oilseed crop Sesamum indicum L. Genome Biology 14: 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goff SA, Ricke D, Lan TH, Presting G, Wang R, et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92–100 Available: http://www.ncbi.nlm.nih.gov/pubmed/11935018. Accessed 2013 May 24 [DOI] [PubMed] [Google Scholar]
- 23. Yu J, Hu S, Wang J, Wong GKS, Li S, et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79–92 Available: http://www.ncbi.nlm.nih.gov/pubmed/11935017. Accessed 2013 June 6 [DOI] [PubMed] [Google Scholar]
- 24. The Arabidopsis Genome Initiative (2000) Analysis of the genome sequence of the Flowering plant Arabidopsis thaliana. Nature 408: 796–815. [DOI] [PubMed] [Google Scholar]
- 25. Singh R, Ong-Abdullah M, Low ETL, Manaf MAA, Rosli R, et al. (2013) Oil palm genome sequence reveals divergence of interfertile species in Old and New worlds. Nature 500: 335–339 Available: http://www.ncbi.nlm.nih.gov/pubmed/23883927. Accessed 2013 Aug 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rabinowicz PD, Schutz K, Dedhia N, Yordan C, Parnell LD, et al. (1999) Differential methylation of genes and retrotransposons facilitates shotgun sequencing of the maize genome. Nature Genetics 23: 305–308. [DOI] [PubMed] [Google Scholar]
- 27.Budiman MA, Singh R, Low ETL, Nunberg A, Citek R, et al.. (2005) Sequencing of the oil palm genespace. Proceedings of PIPOC2005 International Palm Oil Congress (Agriculture, Biotechnology and Sustainability Conference). Kuala Lumpur. pp. 628–639.
- 28. Whitelaw CA, Barbazuk WB, Pertea G, Chan AP, Cheung F, et al. (2003) Enrichment of gene-coding sequences in maize by genome filtration. Science 302: 2118–2120 Available: http://www.ncbi.nlm.nih.gov/pubmed/14684821. Accessed 2013 June 5 [DOI] [PubMed] [Google Scholar]
- 29. Bedell JA, Budiman MA, Nunberg A, Citek RW, Robbins D, et al. (2005) Sorghum genome sequencing by methylation filtration. PLoS Biology 3: 103–115 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=539327&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zaki NM, Singh R, Rosli R, Ismail I (2012) Elaeis oleifera Genomic-SSR Markers: Exploitation in Oil Palm Germplasm Diversity and Cross-Amplification in Arecaceae. International Journal of Molecular Sciences 13: 4069–4088 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3344202&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ting NC, Zaki NM, Rosli R, Low ETL, Ithnin M, et al. (2010) SSR mining in oil palm EST database: application in oil palm germplasm diversity studies. Journal of Genetics 89: 135–145 Available: http://www.springerlink.com/index/141254V246XV1064.pdf [DOI] [PubMed] [Google Scholar]
- 32. Huang X (1999) CAP3: A DNA sequence assembly program. Genome Research 9: 868–877 Available: http://www.genome.org/cgi/doi/10.1101/gr.9.9.868. Accessed 2013 May 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lander ES, Waterman MS (1988) Genomic mapping by fingerprinting random clones: a mathematical analysis. Genomics 2 Available: http://www.sciencedirect.com/science/article/pii/0888754388900079. Accessed 2013 June 5 [DOI] [PubMed] [Google Scholar]
- 35. Stanke M, Steinkamp R, Waack S, Morgenstern B (2004) AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Research 32: W309–W312 Available: http://www.ncbi.nlm.nih.gov/pubmed/15215400. Accessed 2013 May 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Korf I (2004) Gene finding in novel genomes. BMC Bioinformatics 5: 59 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=421630&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Besemer J, Lomsadze A, Borodovsky M (2001) GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Research 29: 2607–2618 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=55746&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cantarel BL, Korf I, Robb SMC, Parra G, Ross E, et al. (2008) MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Research 18: 188–196 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2134774&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics (Oxford, England) 22: 1658–1659 Available: http://www.ncbi.nlm.nih.gov/pubmed/16731699. Accessed 2013 Aug 6 [DOI] [PubMed] [Google Scholar]
- 40. Zdobnov EM, Apweiler R (2001) InterProScan-an intergration platform fo the signature-recognition methods in InterPro. Bioinformatics 17: 847–848 Available: http://www.ncbi.nlm.nih.gov/pubmed/11590104 [DOI] [PubMed] [Google Scholar]
- 41. Joslyn CA, Mniszewski SM, Fulmer A, Heaton G (2004) The gene ontology categorizer. Bioinformatics 20: i169–i177 Available: http://www.ncbi.nlm.nih.gov/pubmed/15262796. Accessed 2013 June 5 [DOI] [PubMed] [Google Scholar]
- 42. Zhang H, Jin J, Tang L, Zhao Y, Gu X, et al. (2011) PlantTFDB 2.0: update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Research 39: D1114–D1117 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3013715&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanseverino W, Hermoso A, D'Alessandro R, Vlasova A, Andolfo G, et al. (2013) PRGdb 2.0: towards a community-based database model for the analysis of R-genes in plants. Nucleic Acids Research 41: : D1167–71. Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3531111&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 5. [DOI] [PMC free article] [PubMed]
- 44. Yun C (1999) Classification and function of plant disease resistance genes. Plant Pathology Journal 15: 105–111 Available: http://www.ppj-online.org/folder.php?a=down&id=42922 [Google Scholar]
- 45. Song WY, Pi LY, Wang GL, Gardner J, Holsten T, et al. (1997) Evolution of the rice Xa21 disease resistance gene family. The Plant Cell 9: 1279–1287 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=156997&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martin GB, Brommonschenkel SH, Chunwongse J, Frary A, Ganal MW, et al. (1993) Map-based cloning of a protein kinase gene conferring disease resistance in tomato. Science 262: 1432–1436 Available: http://www.ncbi.nlm.nih.gov/pubmed/7902614 [DOI] [PubMed] [Google Scholar]
- 47. Peraza-Echeverria S, James-Kay A, Canto-Canché B, Castillo-Castro E (2007) Structural and phylogenetic analysis of Pto-type disease resistance gene candidates in banana. Molecular Genetics and Genomics 278: 443–453 Available: http://www.ncbi.nlm.nih.gov/pubmed/17587056. Accessed 2013 June 5 [DOI] [PubMed] [Google Scholar]
- 48. Van der Linden CG, Wouters DCAE, Mihalka V, Kochieva EZ, Smulders MJM, et al. (2004) Efficient targeting of plant disease resistance loci using NBS profiling. Theoretical And Applied Genetics 109: 384–393 Available: http://www.ncbi.nlm.nih.gov/pubmed/15057419. Accessed 2013 May 31 [DOI] [PubMed] [Google Scholar]
- 49. Meyers BC, Dickerman AW, Michelmore RW, Sivaramakrishnan S, Sobral BW, et al. (1999) Plant disease resistance genes encode members of an ancient and diverse protein family within the nucleotide-binding superfamily. The Plant Journal 20: 317–332 Available: http://www.ncbi.nlm.nih.gov/pubmed/10571892 [DOI] [PubMed] [Google Scholar]
- 50. Barbosa-da-silva A, Wanderley-nogueira AC, Silva RRM, Berlarmino LC, Soares-cavalcanti NM, et al. (2005) In silico survey of resistance (R) genes in Eucalyptus transcriptome. Genetics and Molecular Biology 28: 562–574. [Google Scholar]
- 51. Wanderley-Nogueira AC, Soares-Cavalcanti NM, Morais DA, Belarmino LC, Barbosa-Silva A, et al. (2007) Abundance and diversity of resistance genes in the sugarcane transcriptome revealed by in silico analysis. Genetics and Molecular Research 6: 866–889. [PubMed] [Google Scholar]
- 52. Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Research 39: W29–37 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3125773&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology 305: 567–580 Available: http://www.ncbi.nlm.nih.gov/pubmed/11152613. Accessed 2013 May 31 [DOI] [PubMed] [Google Scholar]
- 54. Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research 22: 4673–4680 Available: http://nar.oxfordjournals.org/cgi/doi/10.1093/nar/22.22.4673. Accessed 2013 Aug 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular biology and evolution 28: 2731–2739 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3203626&tool=pmcentrez&rendertype=abstract. Accessed 2013 Aug 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Griffiths-Jones S, Saini HK, Van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Research 36: D154–D158 Available: http://www.ncbi.nlm.nih.gov/pubmed/17991681. Accessed 2013 May 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL (2008) The Vienna RNA websuite. Nucleic Acids Research 36: W70–W74 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2447809&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hofacker IL, Stadler PF (2006) Memory efficient folding algorithms for circular RNA secondary structures. Bioinformatics 22: 1172–1176 Available: http://www.ncbi.nlm.nih.gov/pubmed/16452114. Accessed 2013 May 22 [DOI] [PubMed] [Google Scholar]
- 59. Gkirtzou K, Tsamardinos I, Tsakalides P, Poirazi P (2010) MatureBayes: a probabilistic algorithm for identifying the mature miRNA within novel precursors. PloS ONE 5: e11843 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2917354&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Palmer LE, Rabinowicz PD, O'Shaughnessy AL, Balija VS, Nascimento LU, et al. (2003) Maize genome sequencing by methylation filtration. Science 302: 2115–2117. [DOI] [PubMed] [Google Scholar]
- 61. Martienssen RA, Rabinowicz PD, O'Shaughnessy AL, McCombie WR (2004) Sequencing the maize genome. Current Opinion in Plant Biology 7: 102–107 Available: http://www.ncbi.nlm.nih.gov/pubmed/15003207. Accessed 2013 June 6 [DOI] [PubMed] [Google Scholar]
- 62. Rabinowicz PD, Citek R, Budiman MA, Numberg A, Bedell JA, et al. (2005) Differential methylation of genes and repeats in land plants. Genome Research 15: 1431–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Timko MP, Rushton PJ, Laudeman TW, Bokowiec MT, Chipumuro E, et al. (2008) Sequencing and analysis of the gene-rich space of cowpea. BMC Genomics 9: 103 Available: http://www.ncbi.nlm.nih.gov/pubmed/18304330. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sakai H, Ikawa H, Tanaka T, Numa H, Minami H, et al. (2011) Distinct evolutionary patterns of Oryza glaberrima deciphered by genome sequencing and comparative analysis. The Plant Journal 66: 796–805 Available: http://www.ncbi.nlm.nih.gov/pubmed/21323774. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lin HC, Morcillo F, Dussert S, Tranchant-Dubreuil C, Tregear JW, et al. (2009) Transcriptome analysis during somatic embryogenesis of the tropical monocot Elaeis guineensis: evidence for conserved gene functions in early development. Plant Molecular Biology 70: 173–192 Available: http://www.ncbi.nlm.nih.gov/pubmed/19199047. Accessed 2013 June 5 [DOI] [PubMed] [Google Scholar]
- 66. Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, et al. (2008) The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Research 36: D1009–D1014 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2238962&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Al-Dous EK, George B, Al-Mahmoud ME, Al-Jaber MY, Wang H, et al. (2011) De novo genome sequencing and comparative genomics of date palm (Phoenix dactylifera). Nature biotechnology 29: 521–527 Available: http://www.ncbi.nlm.nih.gov/pubmed/21623354. Accessed 2013 Aug 6 [DOI] [PubMed] [Google Scholar]
- 68. Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, et al. (2006) From genomics to chemical genomics: new developments in KEGG. Nucleic acids research 34: D354–7 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1347464&tool=pmcentrez&rendertype=abstract. Accessed 2013 Aug 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Billotte N, Marseillac N, Risterucci AM, Adon B, Brottier P, et al. (2005) Microsatellite-based high density linkage map in oil palm (Elaeis guineensis Jacq.). Theoretical and Applied Genetics 110: 754–765 Available: http://www.ncbi.nlm.nih.gov/pubmed/15723275. Accessed 2013 June 5 [DOI] [PubMed] [Google Scholar]
- 70. Tranbarger TJ, Kluabmongkol W, Sangsrakru D, Morcillo F, Tregear JW, et al. (2012) SSR markers in transcripts of genes linked to post-transcriptional and transcriptional regulatory functions during vegetative and reproductive development of Elaeis guineensis. BMC Plant Biology 12: 1 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3282652&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Billotte N, Risterucci AM, Barcelos E, Noyer JL, Amblard P, et al. (2001) Development, characteristics and across-taxa utility of oil palm (Elaeis guineensis Jacq.) microsatellite markers. Genome 44: 413–425. [PubMed] [Google Scholar]
- 72. Zhang L, Yuan D, Yu S, Li Z, Cao Y, et al. (2004) Preference of simple sequence repeats in coding and non-coding regions of Arabidopsis thaliana. Bioinformatics 20: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 73. Lawson MJ, Zhang L (2006) Distinct patterns of SSR distribution in the Arabidopsis thaliana and rice genomes. Genome Biology 7: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liang X, Chen X, Hong Y, Liu H, Zhou G, et al. (2009) Utility of EST-derived SSR in cultivated peanut (Arachis hypogaea L.) and Arachis wild species. BMC Plant Biology 9: 35 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2678122&tool=pmcentrez&rendertype=abstract. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Riju A, Rajesh MK, Sherin PTPF, Chandrasekar A, Apshara SE, et al. (2009) Mining of expressed sequence tag libraries of cacao for microsatellite markers using five computational tools. Journal of Genetics 88: 217–225 Available: http://www.ncbi.nlm.nih.gov/pubmed/19700860 [DOI] [PubMed] [Google Scholar]
- 76. Tóth G, Gáspári Z, Jurka J (2000) Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Research 10: 967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yuan D, Liang S, Lin Z, Zhang X (2012) In silico comparative analysis of EST-SSRs in three cotton genomes. African Journal of Biotechnology 11: 13269–13371 10.5897/AJB11.655 [DOI] [Google Scholar]
- 78. Cavagnaro PF, Senalik DA, Yang L, Simon PW, Harkins TT, et al. (2010) Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genomics 11: 569 Available: http://www.biomedcentral.com/1471-2164/11/569. Accessed 2013 June 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with non-repetitive DNA in plant genomes. Nature Genetics 30: 194–200 10.1038/ng822 [DOI] [PubMed] [Google Scholar]
- 80. Yonemaru J, Ando T, Mizubayashi T, Kasuga S, Matsumoto T, et al. (2009) Development of genome-wide simple sequence repeat markers using whole-genome shotgun sequences of sorghum (Sorghum bicolor (L.) Moench). DNA Research 16: 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, et al. (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156: 847–854 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1461288&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vignal A, Milan D, SanCristobal M, Eggen A (2002) A review on SNP and other types of molecular markers and their use in animal genetics. Genetics Selection Evolution 34: 275–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Riju A, Chandrasekar A, Arunachalam V (2007) Mining for single nucleotide polymorphisms and insertions/deletions in expressed sequence tag libraries of oil palm. Bioinformation 2: 128–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Batley J, Barker G, O'Sullivan H, Edwards KJ, Edwards D (2003) Mining for single nucleotide polymorphisms and insertions/deletions in maize expressed sequence tag data. Plant Physiology 132: 84–91 Available: http://www.plantphysiol.org/content/132/1/84.short [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Singhal D, Gupta P, Sharma P, Kashyap N, Anand S, et al. (2011) In-silico single nucleotide polymorphisms (SNP) mining of Sorghum bicolor genome. African Journal of Biotechnology 10: 580–583 Available: http://www.academicjournals.org/ajb/PDF/pdf2011/24Jan/Singhal et al.pdf. Accessed 2013 June 6 [Google Scholar]
- 86. Chandrasekar A, Riju A, Sithara K, Anoop S, Eapen SJ (2009) Identification of single nucleotide polymorphism in ginger using expressed sequence tags. Bioinformation 4: 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Riju A, Arunachalam V (2009) Interspecific differences in single nucleotide polymorphisms (SNPs) and indels in expressed sequence tag libraries of oil palm Elaeis guineensis and E. oleifera. Nature Precedings [Google Scholar]
- 88. Singh R, Zaki NM, Ting NC, Rosli R, Tan SG, et al. (2008) Exploiting an oil palm EST database for the development of gene-derived SSR markers and their exploitation for assessment of genetic diversity. Biologia 63: 227–235 Available: http://www.springerlink.com/index/10.2478/s11756-008-0041-z. Accessed 2013 June 5 [Google Scholar]
- 89.Ooi LCL, Maizura I, Rajinder S (2007) SNP markers in oil palm (Elaeis spp.): Discovery and applications. International Plant and Animal Genome XV Conference. San Diego. p. 177.
- 90. Soleimani VD, Baum BR, Johnson DA (2003) Efficient validation of single nucleotide polymorphisms in plants by allele-specific PCR, with an example from barley. Plant Molecular Biology Reporter 21: 281–288. [Google Scholar]
- 91. Singh R, Low ETL, Ooi LCL, Ong-Abdullah M, Ting NC, et al. (2013) The oil palm SHELL gene controls oil yield and encodes a homologue of SEEDSTICK. Nature 500: 340–344 Available: http://www.ncbi.nlm.nih.gov/pubmed/23883930. Accessed 2013 Aug 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mitsuda N, Ohme-Takagi M (2009) Functional analysis of transcription factors in Arabidopsis. Plant Cell Physiology 50: 1232–1248 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2709548&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. D'Hont A, Denoeud F, Aury JM, Baurens FC, Carreel F, et al. (2012) The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature 488: 213–217 Available: http://www.nature.com/doifinder/10.1038/nature11241. Accessed 2013 May 22 [DOI] [PubMed] [Google Scholar]
- 94. Gao G, Zhong Y, Guo A, Zhu Q, Tang W, et al. (2006) DRTF: a database of rice transcription factors. Bioinformatics 22: 1286–1287 Available: http://www.ncbi.nlm.nih.gov/pubmed/16551659. Accessed 2013 June 5 [DOI] [PubMed] [Google Scholar]
- 95. Zhang G, Chen M, Li L, Xu Z, Chen X, et al. (2009) Overexpression of the soybean GmERF3 gene, an AP2/ERF type transcription factor for increased tolerances to salt, drought, and diseases in transgenic tobacco. Journal of Experimental Botany 60: 3781–3796 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2736888&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mizoi J, Shinozaki K, Yamaguchi-Shinozaki K (2012) AP2/ERF family transcription factors in plant abiotic stress responses. Biochimica et Biophysica Acta 1819: 86–96 Available: http://www.ncbi.nlm.nih.gov/pubmed/21867785. Accessed 2013 May 30 [DOI] [PubMed] [Google Scholar]
- 97. Xu ZS, Chen M, Li LC, Ma YZ (2011) Functions and application of the AP2/ERF transcription factor family in crop improvement. Journal of Integrative Plant Biology 53: 570–585 Available: http://www.ncbi.nlm.nih.gov/pubmed/21676172. Accessed 2013 May 23 [DOI] [PubMed] [Google Scholar]
- 98. Vijayraghavan U (2001) How plants pattern flowers: Lessons from molecular genetic studies of flowering in Arabidopsis thaliana a model plant. Current Science 80: 233–243. [Google Scholar]
- 99. Irish VF (1999) Patterning the flower. Developmental Biology 209: 211–220. [DOI] [PubMed] [Google Scholar]
- 100. Alwee SS, Van der Linden CG, Van der Schoot J, de Folter S, Angenent GC, et al. (2006) Characterization of oil palm MADS box genes in relation to the mantled flower abnormality. Plant Cell, Tissue and Organ Culture 85: 331–344 Available: http://www.springerlink.com/index/10.1007/s11240-006-9084-4. Accessed 2013 June 5 [Google Scholar]
- 101. Jaligot E, Adler S, Debladis E, Beulé T, Richaud F, et al. (2011) Epigenetic imbalance and the floral developmental abnormality of the in vitro-regenerated oil palm Elaeis guineensis. Annals of Botany 108: 1453–1462 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3219487&tool=pmcentrez&rendertype=abstract. Accessed 30 May 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lorang JM, Sweat TA, Wolpert TJ (2007) Plant disease susceptibility conferred by a “resistance” gene. Proceedings of the National Academy of Sciences 104: 14861–14866 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1976202&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Tameling WIL, Takken FLW (2007) Resistance proteins: scouts of the plant innate immune system. European Journal of Plant Pathology 121: 243–255 Available: http://www.springerlink.com/index/10.1007/s10658-007-9187-8. Accessed 2013 May 24 [Google Scholar]
- 104. Bent AF (1996) Plant disease resistance genes: function meets structure. The Plant Cell 8: 1757–1771 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=161313&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dangl JL, Jones JDG (2001) Plant pathogens and integrated defence responses to infection. Nature 411: 826–833 Available: http://www.nature.com/nature/journal/v411/n6839/abs/411826a0.html [DOI] [PubMed] [Google Scholar]
- 106. Deslandes L, Olivier J, Peeters N, Feng DX, Khounlotham M, et al. (2003) Physical interaction between RRS1-R, a protein conferring resistance to bacterial wilt, and PopP2, a type III effector targeted to the plant nucleus. Proceedings of the National Academy of Sciences 100: 8024–8029 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=164706&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. McDowell JM, Woffenden BJ (2003) Plant disease resistance genes: recent insights and potential applications. Trends in Biotechnology 21: 178–183 Available: http://linkinghub.elsevier.com/retrieve/pii/S0167779903000532. Accessed 2013 May 29 [DOI] [PubMed] [Google Scholar]
- 108. Belkhadir Y, Subramaniam R, Dangl JL (2004) Plant disease resistance protein signaling: NBS-LRR proteins and their partners. Current opinion in plant biology 7: 391–399 Available: http://www.ncbi.nlm.nih.gov/pubmed/15231261. Accessed 2013 Aug 8 [DOI] [PubMed] [Google Scholar]
- 109. Tarr DEK, Alexander HM (2009) TIR-NBS-LRR genes are rare in monocots: evidence from diverse monocot orders. BMC Research Notes 2: 197 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2763876&tool=pmcentrez&rendertype=abstract. Accessed 2013 May 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Boyd LA, Ridout C, O'Sullivan DM, Leach JE, Leung H (2013) Plant-pathogen interactions: disease resistance in modern agriculture. Trends in genetics: TIG 29: 233–240 Available: http://www.ncbi.nlm.nih.gov/pubmed/23153595. Accessed 2013 Aug 9 [DOI] [PubMed] [Google Scholar]
- 111. Bartel DP (2004) MicroRNAs: Genomics, biogenesis, mechanism, and function genomics: The miRNA genes. Cell 116: 281–297 Available: http://www.ncbi.nlm.nih.gov/pubmed/14744438 [DOI] [PubMed] [Google Scholar]
- 112. Molesini B, Pii Y, Pandolfini T (2012) Fruit improvement using intragenesis and artificial microRNA. Trends in biotechnology 30: 80–88 Available: http://www.ncbi.nlm.nih.gov/pubmed/21871680. Accessed 2013 Sept 4 [DOI] [PubMed] [Google Scholar]
- 113. Itaya A, Bundschuh R, Archual AJ, Joung JG, Fei Z, et al. (2008) Small RNAs in tomato fruit and leaf development. Biochimica et biophysica acta 1779: 99–107 Available: http://www.ncbi.nlm.nih.gov/pubmed/18078843. Accessed 2013 Aug 22 [DOI] [PubMed] [Google Scholar]
- 114. Zhang X, Zou Z, Zhang J, Zhang Y, Han Q, et al. (2011) Over-expression of sly-miR156a in tomato results in multiple vegetative and reproductive trait alterations and partial phenocopy of the sft mutant. FEBS letters 585: 435–439 Available: http://www.ncbi.nlm.nih.gov/pubmed/21187095. Accessed 2013 Aug 9 [DOI] [PubMed] [Google Scholar]
- 115. Opdam FJ, Kamps G, Croes H, van Bokhoven H, Ginsel La, et al. (2000) Expression of Rab small GTPases in epithelial Caco-2 cells: Rab21 is an apically located GTP-binding protein in polarised intestinal epithelial cells. European journal of cell biology 79: 308–316 Available: http://www.ncbi.nlm.nih.gov/pubmed/10887961 [DOI] [PubMed] [Google Scholar]
- 116. Ma QH (2007) Small GTP-binding Proteins and their Functions in Plants. Journal of Plant Growth Regulation 26: 369–388 Available: http://link.springer.com/10.1007/s00344-007-9022-7. Accessed 2013 Sept 3 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gene ontology classification of EG01, EO01 and BAC sequences. Three GO categories, [A] Molecular function (ML) [B] Biological process (BP), and [C] Cellular component (CC) terms were mapped to Plant Slim GO annotations using CateGOrizer.
(DOCX)
Depth at SNP positions for [A] EG01 and [B] EO01 contigs. The red line indicates the cut-off of two standard deviation from mean, where the SNPs on the left of the line were defined as unreliable.
(DOCX)
E. guineensis and E. oleifera filtered and unfiltered genomic library information.
(DOCX)
Categorization of EG and EO genes into KEGG pathways.
(DOCX)
Di-, tri- and tetranucleotide repeats identified in EG01.
(DOCX)
Di-, tri- and tetranucleotide repeats identified in EO01.
(DOCX)
List of perfect and partial match miRNAs from EG01 and EO01 contigs.
(DOCX)
Gene information and annotations.
(XLSX)
List of genes not tagged by oil palm ESTs.
(XLSX)
List of orthologs.
(XLSX)
List of SSRs and SNPs.
(XLS)
Formula to calculate gene enrichment.
(DOCX)
Formula to calculate genome sampling.
(DOCX)