

Abstract
Primary cardiac neoplasms are extremely rare. Angiosarcoma is the most commonly seen histological subtype and is characterized by its permeating and destructive nature. Unfortunately, primary cardiac angiosarcoma is often overlooked as an initial diagnosis because of its rarity. Since the time it was first identified in 1934, little progress has been made in improving survival outcome. Complete or partial surgical resection is still the best option for palliation, with little hope for cure. Improvements have been made in the ability to view and distinguish tumors. Echocardiography is one of the most useful diagnostic tools because of its high sensitivity; therefore, CT and MR images are often used to detect sites of metastatic disease. Immunohistochemistry staining can also be employed as an adjunctive diagnostic tool. CD31, CD34, FLI-1, and von Willebrand factor are the most commonly used markers in detecting tumors of endothelial origin. However, due to the vast heterogeneity within a tumor, immunohistochemistry staining can be quite variable. Surgical resection remains the standard modality of treatment. Primary cardiac angiosarcoma is largely resistant to chemotherapy and/or radiation. However, the exact benefit and its place in a multimodality treatment regimen are still under investigation.
Keywords: primary cardiac angiosarcoma, immunohistochemistry, angiosarcoma, endothelial, CD31, FLI-1, CD34, cytokeratin, vimentin
Background
Realdo Columbus first identified the presence of a heart tumor in 1559. However, it was not until 1934 that the first clinical diagnosis of a primary heart sarcoma was reported [1]. Primary cardiac neoplasms are extremely rare. The autopsy incidence is 0.0001–0.030% [2], or around 1 in every 500 cardiovascular surgical cases [3]. The majority of tumors are benign (75%). Of the remaining 25% of tumors that are identified as being malignant, cardiac sarcomas comprise 95% of cases. Primary cardiac angiosarcoma is the most common histological subtype and constitutes 30% of those cases [4].
Angiosarcomas are uncommon malignant neoplasms that most commonly occur in the skin, breast, liver, spleen, and deep tissue. Almost 50% of angiosarcomas occur in the head and neck [41]. Being a very aggressive neoplasm, they have a high rate of local recurrence and systemic metastases. Various parameters, including the primary site, metastases at diagnosis, and grade of the tumor, can indicate a poor prognosis. While surgery is the primary method of treatment, combination with chemotherapy and radiation is usually preferred. In primary cardiac angiosarcoma, males are usually affected more often than females, in a 2–3/1 ratio [5]. Although cases have been identified among a wide range of ages, most patients that are affected are younger than 65 years of age [6]. Casha [7] reported a case of primary cardiac angiosarcoma with familial incidence. The report described a 24-year-old woman with primary cardiac angiosarcoma who had a father that died at the age of 31 with the same tumor; histological and immunohistochemical features were identical. There have been 4 known reports of familial cardiac angiosarcomas since then. The patients had a mean survival time of 4 months compared to a 5-year survival rate of 14% in patients with sporadic angiosarcoma [8]. Angiosarcoma is most commonly found in the right atrium and frequently interferes with neighboring structures, resulting in congestive heart failure, pericardial effusion, and cardiac tamponade. The prognosis is poor and most patients succumb to disease within months of diagnosis [9]. The rarity of this diagnosis has made it difficult to standardize therapy, but surgical treatment is usually preferred. The exact benefits of chemotherapy and radiation are still being investigated [10].
Previous studies and reviews have focused primarily on cardiac neoplasms as a whole. This paper aimed to consolidate information from each of these disparate reports in order to provide the most comprehensive understanding of primary cardiac angiosarcoma to date, with emphasis on diagnostic modalities and the preferred management strategies based on recent literature findings.
Anatomy and Histology
Primary cardiac angiosarcoma is an endothelial cell tumor. Nearly 90% of tumors occur in the right atrium as a multicentric mass. It is characterized by an aggressive and permeating growth within the surrounding myocardial wall, but can project into or fill the atrial chamber and invade the vena cava and tricuspid valve [11]. Less than 5% occur in the left atrium or ventricles [4]. Though the pericardium is commonly involved in right-sided angiosarcoma, as represented by a rate of 61% in a study by Butany and Yu [12], it can also serve as the primary and only site of tumor growth [4]. Butany and Yu [12] also reported 2 cases of angiosarcoma originating in the right coronary artery. On gross examination, the tumors are usually hemorrhagic and necrotic, with a dark red or brown appearance (Figure 1) [4,11]. If the angiosarcoma spreads into the pericardium, it produces a thickened, gray-black layer [11].
Figure 1.
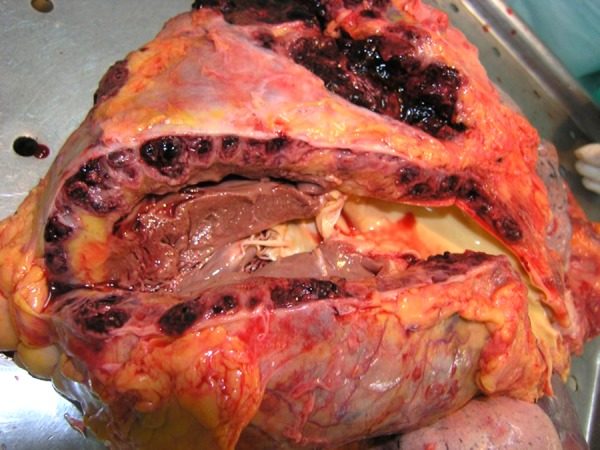
Gross autopsy specimen from a 75-year-old male patient showing hemorrhagic angiosarcoma infiltrating the wall of the right ventricle.
Histologically, angiosarcomas consist of highly variable endothelium-lined channels [9,11]. Common characteristics include anastomosing vascular channels, which vary in size based on frequency of mitoses, solid spindle cell areas with minimal or no apparent vascular spaces, foci of endothelial tufting, and lack of calcification [4,9,13,14]. Cardiac angiosarcomas have well differentiated vascular channels mixed with poorly differentiated solid areas of epithelioid cells and spindle cells. Tumors have been reported to have spindle cell proliferation that resembles Kaposi’s sarcoma. The tumor cells have hyperchromatic, pleomorphic nuclei with frequent mitoses. There are 3 patterns seen histologically: a vascular area with anatomizing channels, a solid high-grade epithelioid area, and a spindle cell Kaposi-like area [15].
Immunohistochemistry
Immunohistochemistry is used to further differentiate angiosarcomas from other soft tissue neoplasms, but it should only be used as an adjunctive diagnostic tool. Staining is quite variable due to vast cellular heterogeneity within an angiosarcoma, as well as differences based on tumor location. In general, a combination of endothelial markers is needed for diagnosis.
Table 1 describes commonly used vascular markers for identifying endothelial angiosarcomas. CD31 and FLI-1 (Friendly leukemia virus integration-1) are very sensitive for indicating both benign and malignant vascular tumors (Figure 2) [16–20]. CD34 is also useful, but is less sensitive [19,20]. Positive staining for CD31 and CD34 demonstrate the presence of vascular lumens throughout the lesion [21]. von Willebrand factor is expressed, but is considered the least sensitive marker for angiosarcoma, especially when the tumor is inadequately differentiated [17,19,20]. Meis-Kindblom’s [22] study found factor VIII monoclonal antibody to be the most sensitive endothelial marker, but Suvarna [23] states that factor VIII lacks sensitivity and does not stain positive in most cases. Cytokeratin is positive in approximately one-third of angiosarcomas, although most of these cases are epithelial cell type-dominant tumors [19,22]. Cytokeratin shows weak focal staining in angiosarcomas; strongly diffuse staining suggests an alternative diagnosis of mesothelioma or carcinoma [23]. Vimentin is positive in most endothelial angiosarcomas, but is also positive in other cellular lineages [17,22]. Due to this non-specificity in staining, the presence of either cytokeratin or vimentin is not a definitive discriminant between angiosarcoma and other soft tissue neoplasms. The markers BNH9 and p53 are also of interest. BNH9 is a monoclonal antibody against blood group-related H and Y antigen. Studies by Meis-Kindblom [22] show angiosarcomas with a high positivity for BNH9, while Delsol et al. [24] demonstrate that BNH9 is negative for all soft tissue sarcomas except angiosarcomas. A study of p53 mutations in angiosarcomas showed that abnormalities of the tumor suppressor gene contributed to tumor formation and that frequency of mutation differed by the site of the tumor origin [25]. Another marker, Ki67, is often positive and is used as a prognostic factor. High values (≥10%) have been statistically correlated with poor survival outcome [22]. Ki-67 staining determined the proliferation rate of cardiac angiosarcomas to be 20–40% [15]. Fernandes [21] found alpha-smooth muscle actin staining positive in the vessels of the cardiac angiosarcoma lesion that was being studied. These results were corroborated by Meis-Kindblom [22], who found that alpha-smooth muscle actin was in 24% of the evaluated tumors. Alpha-smooth muscle actin is frequently found in the pericytes that encircle the neoplastic endothelial cells [22]. Cardiac angiosarcomas have also shown strong, diffuse reactivity to Wilms Tumor-1. This can be very helpful in distinguishing the other types of cardiac sarcomas (synovial sarcoma, leiomyosarcoma, unclassified) from cardiac angiosarcomas [15].
Table 1.
Common vascular markers for endothelial angiosarcoma.
| Immunostain | Antigen Description | Result | Comment | Source |
|---|---|---|---|---|
| BNH9 | Monoclonal antibody against blood group related H and Y antigen | Positive 72% | Strongly positive, diffuse; negative for all other soft tissue sarcomas except angiosarcoma | Delsol et al., 1991; Meis-Kindblom and Kindblom, 1998 |
| CD31 | Transmembrane glycoprotein; seen in inflammation and angiogenesis | Positive 90% | Highly sensitive and specific for vascular neoplasm; also identifies macrophages, megakaryocytes, and plasma cells | De Young et al., 1998; Weiss and Goldblum, 2001; Pusztaszeri et al., 2006; Folpe, 2010 |
| CD34 | Hematopoietic progenitor cell antigen | Positive 50–74% | Less sensitive than CD31; prevalent in areas of enhanced vascular differentiation; also seen in epithelioid sarcoma | Heim-Hall and Yohe, 2008; Folpe, 2010 |
| Cytokeratin | Intermediate filament | Positive 35% | Focally expressed; often associated with epithelial angiosarcoma and carcinomas | Meis-Kindblom and Kindblom, 1998; Folpe, 2010 |
| FLI-1* | ETS family of transcription factors; involved in cell proliferation and tumorigenesis | Positive 100% | Extremely sensitive and specific for diagnosing vascular tumors; can be seen on hematopoietic cells; also used to diagnose Ewing/PNET | Pusztaszeri et al., 2006; Heim-Hall and Yohe, 2008 |
| Ki67 | Proliferation index | High in 83% | Prognostic factor; considered high if Ki67 value ≥10% | Meis-Kindblom and Kindblom, 1998 |
| p53 | Tumor suppressor | Positive 20% | Considered positive if nuclear staining ≥20% | Naka et al., 1997; Meis-Kindblom and Kindblom, 1998 |
| Vimentin | Intermediate filament | Positive | Accents endothelial cells and vessel lumen formation; also expressed in melanoma and lymphoma | Meis-Kindblom and Kindblom, 1998; Weiss and Goldblum, 2001 |
| von Willebrand factor** | Glycoprotein; mediates platelet adhesion | Positive | Focal appearance, highly specific; least sensitive marker, especially if inadequately differentiated; also expressed by megakaryocytes | Weiss and Goldblum, 2001; Heim-Hall and Yohe, 2008; Folpe, 2010 |
Friend leukemia virus integration-1;
Factor VIII related antigen.
Figure 2.
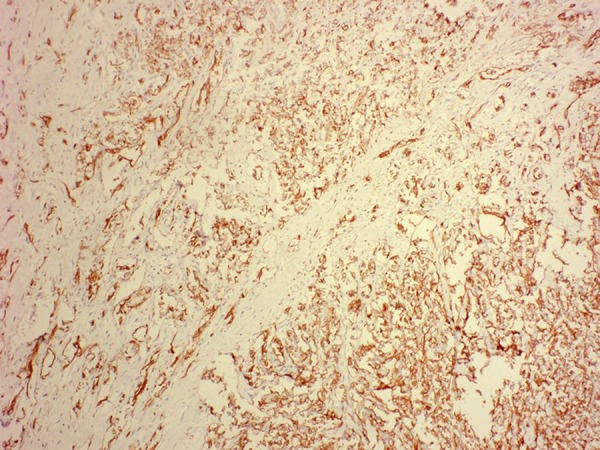
Histological specimen obtained from the tumor in Figure 1. Staining with the vascular marker CD31 reveals endothelial lined channels within the tumor parenchyma.
Symptoms
If symptomatic, primary cardiac angiosarcomas result in nonspecific constitutional symptoms like shortness of breath, weight loss, and anemia-related fatigue and malaise [4,26]. Butany and Yu [12] reported 46% of patients presenting with chest pain as a chief complaint. The most common complaint is dyspnea [15]. This generalized symptomatology contributes to the difficulty in diagnosing such cases. More specific clinical findings usually manifest later in the course of the disease and are dependent on the extent of infiltration within the cardiac wall, infiltration into neighboring structures, and the extent of metastases. Patients are usually asymptomatic until the tumor has increased to a certain size or the patient has developed regional spread or metastases.
In addition, necrosis of the myocardial wall can lead to myocardial rupture [27]. The pericardium is frequently involved with right-sided angiosarcoma; cardiac tamponade and pericardial effusion are common complications. In a study by Hong et al. [2], 56% of the patients presented with pericardial effusion with or without cardiac tamponade. Right-sided congestive heart failure and systemic symptoms secondary to obstruction of blood flow are other frequent clinical manifestations [6]. Other signs of right atrial tumor include valvular and caval obstruction leading to stenosis, systemic or pulmonary emboli, and supraventricular arrhythmias [28].
Diagnosis
Early diagnosis of primary cardiac angiosarcoma remains elusive. Generalized symptoms and rarity of the disease often prevent clinicians from including this disease in an initial differential diagnosis. However, with new imaging technology, early detection of cardiac neoplasms has improved. The rate of ante mortem tissue diagnosis has improved from only 4 of 41 cases in a 1968 review, to 50% in 1986, 70% in 1992, and 82% in 2000 [12]. Physical examination, ECG, and cardiac catheterization are all used in evaluation. Imaging studies like CT, nuclear magnetic resonance, ultrasound, and echocardiography have emerged as preferred methods.
Echocardiography is the mainstay of evaluating cardiac tumors. Transesophageal echocardiography has 97% sensitivity in detecting cardiac masses [29]. Centofanti [30] considers echocardiography the most important diagnostic tool in identifying cardiac tumors. The location, shape, size, attachment, and mobility of a tumor can be found via transthoracic echocardiography, whereas transesophageal echocardiography provides information about the insertion point of the tumor. However, the authors suggested that if the patient’s age is above 50, coronary arteriography should be performed to evaluate the coronary arteries.
Metastases are often widespread at the time of diagnosis [11]. The lungs are the most common site of metastatic disease, but the liver, lymph nodes, bone, adrenal glands, and spleen may also be involved (Figure 3) [12]. Butany and Yu [12] reported a notable increase in brain metastases among surgically treated patients. They postulated that the higher incidence may be due to greater dissemination of malignant cells into the systemic circulation with tumor dissection. In addition to echocardiography, CT should be used to gain a better understanding of the cardiac tumor anatomy and to check for systemic metastasis [31]. Cardiac MRI is better than CT in characterizing the soft tissue and distinguishing between different abnormalities specific to the myocardium. Cardiac MRI can help distinguish between thrombi and tumors in the cardiac cavity [29]. Kim et al. [32] described 2 distinct patterns visible on MR images, one of which is based on the tendency for angiosarcoma to be hemorrhagic and necrotic.
Figure 3.

Gross autopsy specimen from a 75-year-old male patient showing several hemorrhagic liver metastases secondary to a primary cardiac angiosarcoma.
This results in localized nodular areas of increased signal intensity, dispersed among areas of low to intermediate signal intensity, to produce a cauliflower-like appearance. The second pattern is based on diffuse pericardial infiltration, in which linear contrast material enhancement along vascular pools demonstrates a sunray appearance [32,33]. Pericardiocentesis and tissue biopsy are also used for diagnosis [26]. Pericardial fluid cytology is unreliable and should not be used. Malignant cells are very rarely found in the bloody fluid, even when the tumor has invaded the pericardium [29].
In a study by Ge [15], all the patients underwent open cardiac biopsy or surgical resection for tissue diagnosis. Although all of the patients’ tumors were located in the right atrium, endomyocardial biopsies were negative [15]. Rettmar et al. [34] agreed that endomyocardial biopsies are a poor diagnostic tool, reporting that only 50% of their samples were diagnostic [34]. For an accurate diagnosis, the authors suggest surgical exploration and intraoperative frozen sections [15].
Treatment
The prognosis for primary cardiac angiosarcoma is poor, with a mean survival of 3.8±2.5 months without surgical resection [12,35]. There have been no clear prognostic factors. Tumor grade, mitotic rate, age, sex, growth pattern, margin status, and proliferation rate have not shown any consistent pattern regarding prognosis. Ge’s [15] study showed that tumor size and the extent of regional spread of the tumor was associated with time of survival. However, the study was limited by its small sample size [15]. Therapy is not standardized and the exact benefit of adjunctive chemotherapy and/or radiation is still unknown. The challenges lie in the rarity of this disease, the commonly advanced stage at diagnosis, and its aggressive course. However, given the tumor’s high fatality rate, an aggressive approach utilizing a multimodality regimen seems to be favored. Thus, a number of different treatment combinations have been utilized, with varying results. There have been reports of survival times ranging from 12 to 30 months in patients with various combinations of surgery, chemotherapy, radiation, and/or transplantation [4].
Surgery is a commonly chosen therapy, particularly in the setting of localized disease, in that it seems to provide the best option for palliative care and potential cure [12]. Patients treated surgically show a post-operative survival of 2–55 months, with a median survival of 14 months [5]. In studies utilizing medical therapeutics alone, 90% of patients died within 9–12 months [3]. Average survival among patients with heart transplantation, complete resection, or partial resection of tumors appears to be more or less equal [12,39].
Wide resection has been universally accepted as the main treatment modality. Wide resection provides a definitive histological diagnosis and relieves severe mechanical obstruction of cardiac output [30]. Studies have reported difficulty in achieving complete resection of tumors with negative margins. In cases in which complete resection is possible, patients commonly experience local recurrence [29].
Orthotopic heart transplantation has been utilized in patients with high-grade angiosarcomas. Although it was shown that heart transplantation and post-operative chemotherapy did not change long-term outcome, many of these patients are young and have no other options [36]. The authors concluded that a multidisciplinary approach including systemic chemotherapy and local radical resection will give patients with primary cardiac angiosarcoma the best chance of survival [36]. A combination of resection, chemotherapy, and radiation therapy has led to reports of survival up to 3 years [30,40]. However, Hong et al. [2] report poor outcomes with cardiac transplantation and therefore advise against it.
The primary site of the tumor may influence treatment planning. Right-sided tumors appear to be more bulky and infiltrative, with greater likelihood for metastases [3]. Blackmon and Reardon [3] suggested that patients with these tumors may benefit from neoadjuvant chemotherapy, with the intent of shrinking tumor size to maximize the possibility of total surgical resection. They reported a median survival time of 27 months, with 1 exceptional case of a patient still alive 9.5 years after surgery. Blackmon and Reardon [3] also noted that although left-sided tumors are less infiltrative, heart failure is commonly seen early in the disease course, and neoadjuvant chemotherapy is usually contraindicated prior to surgery.
Although it appears that most angiosarcomas are resistant to chemotherapy and radiation, aggressive treatment does seem to contribute to reducing tumor size. Chemotherapy and radiation are important in treatment of angiosarcomas because of the high probability of metastases [37]. Cisplatin, cyclophosphamide, dacarbazine, doxorubicin, ifosfamide, mitomycin-C, paclitaxel, and vincristine are commonly prescribed agents in standard chemotherapy [5,38]. Immunotherapy has also been used, with 1 report showing a 30-month survival following a combination of surgery, chemotherapy, and recombinant IL-2 therapy [5,28]. Most of the literature on cardiac angiosarcomas is based on clinical cases or single-institution studies. Management strategies, prognostic factors, and outcomes are difficult to assess overall; thus, no standardized treatment algorithms exist [2].
Conclusions
Primary cardiac angiosarcomas are an extremely rare tumor with a fatal prognosis. Due to their aggressive and invasive behavior, early diagnosis is imperative for prolonged survival. Unfortunately, this tumor is often overlooked due to its minimal incidence and broad-spectrum of clinical manifestations. If there is any degree of suspicion, clinicians should not discount cardiac angiosarcoma in the initial differential diagnosis. Early diagnosis can prevent tumor progression and metastasis, and can also provide an opportunity to assess different therapeutic combinations and modalities and develop a more standardized treatment regimen for cure or long-term palliative care. For now, surgical resection seems to provide the best chance for prolonging survival. Further investigation of endothelial angiosarcoma markers may provide insight into the etiology of tumor genesis at its site of primary origin and may also provide a more clear and precise profile of angiosarcoma. Until then, a broad-spectrum immunohistochemistry panel should be used and results should be assessed and combined with other diagnostic modalities to aid in the diagnosis and treatment of angiosarcoma.
Footnotes
Source of support: Self financing
References
- 1.Barnes AB, Beaver DC, Snell AM. Primary sarcoma of the heart: report of a case with electrocardiographic and pathological studies. Am Heart J. 2003;9:480–91. [Google Scholar]
- 2.Look-Hong NP, Pandalai PK, Hornick JL, et al. Cardiac Angiosarcoma Management and Outcomes: 20-year single-institution experience. Ann Surg Oncol. 2012;19:2707–15. doi: 10.1245/s10434-012-2334-2. [DOI] [PubMed] [Google Scholar]
- 3.Blackmon SH, Reardon MJ. Surgical treatment of primary cardiac sarcomas. Tex Heart Ins J. 2009;36:451–52. [PMC free article] [PubMed] [Google Scholar]
- 4.Ferguson ER, Walsh GL. Sarcomas of the heart and great vessels. In: Pollock RE, editor. Soft tissue sarcomas. Hamilton: CB Decker Inc; 2002. pp. 155–56.pp. 158–60. [Google Scholar]
- 5.Antonuzzo L, Rotella V, Mazzoni F, et al. Primary cardiac angiosarcoma: a fatal disease. Case Rep Med. 2009:591512. doi: 10.1155/2009/591512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hamidi M, Moody JS, Weigel TL, Kozak KR. Primary cardiac sarcoma. Ann Thorac Surg. 2010;90:176–81. doi: 10.1016/j.athoracsur.2010.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casha A, Davidson L, Roberts P, Unnikrishnan R. Familial angiosarcoma of the heart. Elsevier. 2002;124:392–94. doi: 10.1067/mtc.2002.122314. [DOI] [PubMed] [Google Scholar]
- 8.Keeling IP, Ploner F, Rigler B. Familial Cardiac Angiosarcoma. Ann Thorac Surg. 2006;82:1570–76. doi: 10.1016/j.athoracsur.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Glancy EL, Morales JB, Roberts WC. Angiosarcoma of the heart. Am J Cardiol. 1968;21:413–19. doi: 10.1016/0002-9149(68)90144-6. [DOI] [PubMed] [Google Scholar]
- 10.Bakaeen FG, Reardon MJ, Coselli JS, et al. Surgical outcome in 85 patients with primary cardiac tumors. Am J Surg. 2003;186:641–47. doi: 10.1016/j.amjsurg.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 11.Silver MD, Gotlieb AI, Schoen FJ. Cardiovascular pathology. 3rd ed. Vol. 399. Philadelphia: Churchill Livingstone; 2001. p. 598. [Google Scholar]
- 12.Butany J, Yu W. Cardiac angiosarcoma: two cases and a review of the literature. Can J Cardiol. 2000;16:197–205. [PubMed] [Google Scholar]
- 13.Anderson RH, Berry CL, Hutt MSR, et al. The pathology of the heart. Oxford: Blackwell Scientific Publications; 1975. p. 427. [Google Scholar]
- 14.Burke AP, Cowan D, Virmani R. Primary sarcomas of the heart. Cancer. 1992;69:387–95. doi: 10.1002/1097-0142(19920115)69:2<387::aid-cncr2820690219>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 15.Ge Y, Ro JY, Kim D, et al. Clinicopathologic and immunohistochemical characteristics of adult primary cardiac angiosarcomas: analysis of 10 cases. Ann Diagn Pathol. 2011;15:262–67. doi: 10.1016/j.anndiagpath.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 16.De Young BR, Frierson HF, Jr, Ly MN, et al. CD31 immunoreactivity in carcinomas and mesotheliomas. Am J Clin Pathol. 1998;110:374–77. doi: 10.1093/ajcp/110.3.374. [DOI] [PubMed] [Google Scholar]
- 17.Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 4th ed. Philadelphia: Mosby-Harcourt; 2001. pp. 917–54.pp. 22 [Google Scholar]
- 18.Pusztaszeri MP, Seelentag W, Bosman FT. Immunohistochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J Histochem Cytochem. 2006;54:385–95. doi: 10.1369/jhc.4A6514.2005. [DOI] [PubMed] [Google Scholar]
- 19.Folpe A. Vascular tumors of soft tissue. In: Folpe A, Inwards CY, editors. Bone and soft tissue pathology. Philadelphia: Saunders Elsevier; 2010. pp. 186–88. [Google Scholar]
- 20.Heim-Hall J, Yohe SL. Application of immunohistochemistry to soft tissue neoplasms. Arch Pathol Lab Med. 2008;132:476–89. doi: 10.5858/2008-132-476-AOITST. [DOI] [PubMed] [Google Scholar]
- 21.Fernandes CP, Oliveira FA, Costa FW, et al. Clinical, histological, and immunohistochemical features of mandibular metastasis from primary cardiac angiosarcoma. Oral Surg, Oral Med, Oral Path, Oral Rad. 2013;116:121–27. doi: 10.1016/j.oooo.2012.12.017. [DOI] [PubMed] [Google Scholar]
- 22.Meis-Kindblom JM, Kindblom LG. Angiosarcoma of soft tissue: a study of 80 cases. Am J Surg Pathol. 1998;22:683–97. doi: 10.1097/00000478-199806000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Rassi DM, Davies SJ. Cardiac Tumors. In: Suvarna S, editor. Cardiac Pathology: A Guide to Current Practice. Springer; New York: 2012. p. 215. [Google Scholar]
- 24.Delsol G, Blancher A, al Saati T, et al. Antibody BNH9 detects red blood cell-related antigens on anaplastic large cell (CD30+) lymphomas. Br J Cancer. 1991;64:321–26. doi: 10.1038/bjc.1991.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naka N, Tomita Y, Nakanishi H, et al. Mutations of p53 tumor-suppressor gene in angiosarcoma. Int J Cancer. 1997;71:952–55. doi: 10.1002/(sici)1097-0215(19970611)71:6<952::aid-ijc7>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 26.Brandt RR, Arnold R, Bohle RM, et al. Cardiac angiosarcoma: case report and review of the literature. Z Kardiol. 2005;94:824–28. doi: 10.1007/s00392-005-0296-0. [DOI] [PubMed] [Google Scholar]
- 27.Truong PT, Jones SO, Martens B, et al. Treatment and outcomes in adult patients with primary cardiac sarcoma: the British Columbia Cancer Agency experience. Ann Surg Oncol. 2009;6:3358–65. doi: 10.1245/s10434-009-0734-8. [DOI] [PubMed] [Google Scholar]
- 28.Ananthasubramaniam K, Farha A. Primary right atrial angiosarcoma mimicking acute pericarditis, pulmonary embolism, and tricuspid stenosis. Heart. 1999;81:556–58. doi: 10.1136/hrt.81.5.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riles EG, Gupta S, Wang DD, Tobin K. Primary cardiac angiosarcoma: A diagnostic challenge in a young man with recurrent pericardial effusions. Exp Clin Cardiol. 2012;17:39–42. [PMC free article] [PubMed] [Google Scholar]
- 30.Centofanti PR, Di Rosa E, Deorsola L, et al. Primary Cardiac Tumors: Early and Late Results of Surgical Treatment in 91 Patients. Ann Thorac Surg. 1999;68:1236–41. doi: 10.1016/s0003-4975(99)00700-6. [DOI] [PubMed] [Google Scholar]
- 31.Tang KS, Shang QL, Zhou QC, et al. Primary Cardiac Angiosarcoma with Spontaneous Ruptures of the Right Atrium and Right Coronary Artery. Echocardiography. 2013;30:156–60. doi: 10.1111/echo.12176. [DOI] [PubMed] [Google Scholar]
- 32.Kim DM, Hong JH, Kim SY, et al. Primary cardiac angiosarcoma presenting with cardiac tamponade. Korean Circ J. 2010;40:86–89. doi: 10.4070/kcj.2010.40.2.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Araoz PA, Eklund HE, Welch TJ, Breen JF. CT and MR imaging of primary cardiac malignancies. Radiographics. 1999;19:1421–34. doi: 10.1148/radiographics.19.6.g99no031421. [DOI] [PubMed] [Google Scholar]
- 34.Rettmar KS, Stierle U, Sheikhzadeh A, Diederich KW. Primary angiosarcoma of the heart. Report of a case and review of the literature. Jpn Heart J. 1993;34:667–83. doi: 10.1536/ihj.34.667. [DOI] [PubMed] [Google Scholar]
- 35.Janigan DT, Husain A, Robinson NA. Cardiac angiosarcomas: A review and a case report. Cancer. 1986;57:852–59. doi: 10.1002/1097-0142(19860215)57:4<852::aid-cncr2820570428>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 36.Uberfuhr PM, Meiser B, Fuchs A, et al. Heart Transplantation: An Approach to Treating Primary Cardiac Sarcoma? J Heart Lung Transplant. 2002;21:1135–39. doi: 10.1016/s1053-2498(02)00409-6. [DOI] [PubMed] [Google Scholar]
- 37.Oh SY, Yeom SY, Kim KH. Clinical Implication of Surgical Resection for the Rare Cardiac Tumors Involving Heart and Great Vessels. J Korean Med Sci. 2013;28:717–24. doi: 10.3346/jkms.2013.28.5.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simpson L, Kumar SK, Okuno SH, et al. Malignant primary cardiac tumors: review of a single institution experience. Cancer. 2008;112(11):2440–46. doi: 10.1002/cncr.23459. [DOI] [PubMed] [Google Scholar]
- 39.Piazza N, Chugtai T, Toledano K, et al. Primary cardiac tumors: eighteen years of surgical experience on 21 patients. Can J Cardiol. 2004;20:1443–48. [PubMed] [Google Scholar]
- 40.Sinatra RB, Brancaccio G, di Gioia CR, et al. Integrated approach for cardiac angiosarcoma. Int J Card. 2003;88:301–4. doi: 10.1016/s0167-5273(02)00388-1. [DOI] [PubMed] [Google Scholar]
- 41.Sturgis EM, Potter BO. Sarcomas of the head and neck region. Curr Opin Oncol. 2003;15:239–52. doi: 10.1097/00001622-200305000-00011. [DOI] [PubMed] [Google Scholar]