

Abstract
The Wilms tumor protein 1 (WT1) transcription factor has been associated in malignant melanoma with cell survival and metastasis, thus emerging as a candidate for targeted therapy. A lysine–arginine rich peptide, WT1-pTj, derived from the ZF domain of WT1 was evaluated as an antitumor agent against A2058 human melanoma cells and B16F10-Nex2 syngeneic murine melanoma. Peptide WT1-pTj quickly penetrated human melanoma cells and induced senescence, recognized by increased SA-β-galactosidase activity, enhanced transcriptional activity of p53, and induction of the cell cycle inhibitors p21 and p27. Moreover, the peptide bound to p53 and competed with WT1 protein for binding to p53. WT1-pTj treatment led to sustained cell growth suppression, abrogation of clonogenicity and G2/M cell cycle arrest. Notably, in vivo studies showed that WT1-pTj inhibited both the metastases and subcutaneous growth of murine melanoma in syngeneic mice, and prolonged the survival of nude mice challenged with human melanoma cells. The 27-amino acid cell-penetrating WT1-derived peptide, depends on C3 and H16 for effective antimelanoma activity, inhibits proliferation of WT1-expressing human tumor cell lines, and may have an effective role in the treatment of WT1-expressing malignancies.
Keywords: Malignant melanoma, Wilms tumor 1 (WT1), Senescence, p53
Abbreviations: C PEP, control peptide; CL-ELISA, chemiluminescence ELISA; CPP, cell-penetrating peptide; pTj, Trojan peptide; SA-βGal, senescence-associated β-galactosidase; TMZ, Temozolomide; WT1, Wilms tumor protein 1; ZF, zinc-finger
Highlights
-
•
WT1-pTj is a Lys-Arg-rich peptide derived from the ZF domain of Wilms tumor protein 1.
-
•
WT1-pTj peptide protects syngeneic mice against metastatic melanoma.
-
•
WT1-pTj delays melanoma development after tumor induction and early growth.
-
•
Trojan WT1-pTj delays melanoma cell cycle, binds to and activates p53.
-
•
WT1-pTj activates p21 and p27 and induces senescence features.
1. Introduction
The Wilms tumor protein 1 (WT1) is a transcription factor that plays an important role in cellular development and cell survival. It is abnormally expressed in several human cancers and could be a target of therapeutic agents. Structurally, the WT1 protein contains four C2H2 Krüpple-like zinc-fingers (ZF) in the C-terminal domain, which are important for DNA binding, RNA binding and interaction with other proteins [1]. Originally described as a tumor suppressor gene in pediatric nephroblastoma [2,3], the overexpression, conformational changes and cytoplasmic localization of WT1 in different malignancies such as mesothelioma, ovarian cancer, leukemia, osteosarcomas, glioblastomas and malignant melanomas [4,5] demonstrated oncogenic properties of this protein. In malignant melanoma, the most aggressive and lethal form of skin cancer, WT1 expression has been reported in more than 80% tumor cells but not in epidermal keratinocytes, melanocytes and benign melanocytic nevi in vivo [6]. Notably, RNAi silencing of WT1 induces apoptosis in B16F10 murine melanoma cells [7] and displays antimetastatic activity [8].
The oncogenic role of WT1 in cancer stimulates attempts at neutralizing this tumor-associated antigen. Recently, the anticancer therapy that employs peptides, which can directly target cancer cells, has emerged as an alternate strategy to restrain the progression of tumor growth and metastases [9]. Antitumor peptides may act binding to and inhibiting oncogenes or proteins with aberrant expression in tumor cells. They cause cell cycle arrest and/or induce apoptosis, block signaling mediators and receptors, inhibit angiogenesis, and mediate tumor environment homing of cytotoxic peptide sequences [10–15]. Certain peptides are cell-penetrating (CPPs) or Trojan peptides, with short amphipathic and cationic sequences that permit their penetration across the cell membrane, and thus exert direct anticancer activity [16]. These peptides may be carriers of a variety of antitumor molecules [17,18].
In the present work, we show that a novel WT1-derived peptide (WT1-pTj) is a cell-penetrating antitumor agent that suppresses both proliferation and clonogenicity of B16F10-Nex2 melanoma cells through an irreversible G2/M cell cycle arrest and induction of cellular senescence. In addition to morphological changes and irreversible growth inhibition, senescent cells expressed the senescence-associated β-galactosidase and formed hetero-chromatin foci [19], associated with enhanced transcriptional activation of p53, and accumulation of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1, which have been used as markers of senescence [20].
Most importantly, WT1-pTj displayed a remarkable antimetastatic activity in the syngeneic B16F10-Nex2 melanoma model and prolonged survival of nude mice subcutaneously challenged with human A2058 melanoma cells. Both results emphasize the potential of this novel antitumor peptide to be developed as a therapeutic drug.
The potential use of bioactive peptides as anti-cancer drugs has been investigated in our laboratory and considerable progress has been made using peptides derived from immunoglobulins and from transcription factors [21,22].
2. Materials and methods
2.1. Peptides
A 27-residue synthetic peptide (WT1-pTj) corresponding to amino acids 349–375 of the human WT1 protein (GenBank: CAI95758) and the control peptide (C PEP, with C3A and H16A) were synthesized by Peptide 2.0 Inc. (Chantilly, VA) at 90–99% purity, with amidated C-terminal amino acid, and were completely solubilized in PBS or culture medium. The WT1-pTj peptide is 100% identical to the related sequence of mouse WT1 protein, corresponding to amino acids 426–452 (GenBank: NP659032). Structures and molecular masses of the peptides are depicted on Table 1.
Table 1.
Peptide sequences and molecular mass.
| Name | Sequence | Mass (Da) |
|---|---|---|
| WT1-pTj | KDCERRFSRSDQLKRHQRRHTGVKPFQ-NH2 | 3395.84 |
| b-WT1-pTj | biotin-CGGKDCERRFSRSDQLKRHQRRHTGVKPFQ-NH2 | 3839.38 |
| FITC-WT1-pTj | KDCERRFSRSDQLKRHQRRHTGVKPFQK-FITC | 3914.53 |
| C PEP | KDAERRFSRSDQLKRAQRRHTGVKPFQ-NH2 | 3297.71 |
2.2. Cell lines and culture conditions
Cell lines were originally obtained from Ludwig Institute for Cancer Research, São Paulo, Brazil, or donated by Prof. Luis F. Lima Reis, Hospital Sirio-Libanez, São Paulo, Brazil. These are long established cell lines, acquired from public culture collections or transferred from Ludwig Institute in New York, and maintained in appropriate conditions to serve as standard tumor cell lines for local studies and collaborative research. Animal experiments were carried out using protocols approved by the Ethics Committee for Animal Experimentation of Federal University of Sao Paulo, Brazil (CEP No. 1280/10).
The murine melanoma B16F10-Nex2 subline was established at the Experimental Oncology Unit (UNONEX), Federal University of São Paulo, UNIFESP, as described [23] and used ever since in subcutaneous and metastatic syngeneic models in mice. The human tumor cell lines A2058 and SK-MEL-28 (melanoma), MCF-7 and MDA-MB231 (breast carcinoma), OVCAR-3 (ovarian carcinoma) and HL-60 (acute leukemia) were maintained in complete medium consisting of RPMI-1640 (Gibco, Grand Island, NY) supplemented with 10 mM N-2-hydroxyethylpiperazine-N2 ethanesulphonic acid (HEPES; Sigma–Aldrich, St. Louis, MO), 24 mM sodium bicarbonate, 40 mg/l gentamicin (Hipolabor, Minas Gerais, Brazil), pH 7.2, and 10% fetal bovine serum (FBS; Gibco, Grand Island, NY).
The human fibroblast cell line HFF and mouse embryonic fibroblasts (MEFs) were a gift from Luis F. Lima Reis, Hospital Sirio-Libanez, São Paulo. The HFF cell line was maintained in minimum essential medium Eagle (Gibco, Grand Island, NY) with 2 mM l-glutamine and Earle's balanced saline solution adjusted to contain 1.5 g/l sodium bicarbonate, 0.1 mM non-essential amino acids, and 1.0 mM sodium pyruvate, and 10% FBS. MEF cell line was maintained in DMEM (Gibco, Grand Island, NY) medium supplemented as described above. All cell lines were cultured at 37 °C, in humid atmosphere and 5% CO2.
2.3. Confocal microscopy
A2058 human melanoma cells (4 × 104) were plated in round coverslips and incubated at 37 °C for 24 h. Cells were treated with 0.5 mM fluorescein isothiocyanate (FITC)-labeled-WT1-pTj for 1 h at 37 °C. After incubation, tumor cells were fixed and permeabilized with methanol for 15 min at room temperature, followed by blocking with 0.2% gelatin for 10 min at 37 °C. Nuclear staining was carried out with 10 μg/ml of DAPI (Invitrogen, Eugene, OR). Cells were then washed in PBS and coverslips mounted onto slides with 4 μl Vectashield (Sigma, St. Louis, MO) and imaged using a Carl Zeiss LSM780 confocal microscope (Jena, Germany).
For time dependent WT1-pTj localization, A2058 cells were incubated for 15 min, 1 and 24 h with 0.5 mM biotinylated-WT1-pTj (b-WT1-pTj), washed 3 times in PBS, and fixed with 3.7% paraformaldehyde for 30 min. Cells were then permeabilized in 0.1% Triton X-100 for 30 min followed by blocking for 1 h with 150 mM NaCl, 50 mM Tris and 0.25% BSA, all from Sigma–Aldrich, MO. After washing in PBS, cells were incubated with 10 μg/ml DAPI and streptavidin-FITC (1:200 from stock solution) used as secondary fluorophore for biotinylated peptide, and were both incubated for 15 min at 37 °C. Stained cells on coverslips were examined for peptide intracellular localization using a Confocal Leica SP5 microscope with a 100× oil immersion objective (Leica, Wetzlar, Germany). The Z series was acquired according to sampling criteria built into the software for sequential imaging of DAPI, which stains the nucleus (blue, excitation/emission = 350/470 nm) and FITC, which monitors peptide localization (green, at excitation/emission = 488/525 nm). Images were processed with the ImageJ software (http://rsb.info.nih.gov/ij/).
2.4. Cell viability
The effects of WT1-pTj and C PEP on cell viability were determined by Trypan blue exclusion assay. For EC50 determination, cells (5 × 103/well) were seeded and cultivated in 96-well plates for 12 h at 37 °C. Cells were incubated with increasing concentrations of the peptides (0–1 mM) for 24 h, were detached with Trypsin–EDTA 0.25% solution (Sigma–Aldrich, St. Louis, MO) and the number of viable cells counted using a Neubauer chamber (Electron Microscopy Sciences, Hatfield, PA). The growth kinetics of A2058 cells (103/well) during 96 h in presence of WT1-pTj or C PEP at different concentrations was determined by cell counting every 24 h.
2.5. Colony formation
Anchorage-independent growth of peptide-treated melanoma cells was determined in soft agar. A2058 cells (103) in culture medium with 10% FBS, 0.35% agar and 0.5 mM WT1-pTj or C PEP, were plated on a bottom agar layer containing medium, 10% FBS and 1% agar in a 6-well culture plate. After 6 days of cell plating, more peptide (0.5 mM WT1-pTj or C PEP) and culture medium (300 μl) were added to the corresponding wells. Colonies were stained with 0.5% crystal violet in 70% ethanol and counted 11 days after initial treatment using a vertical microscope.
Alternatively, clonogenicity was tested in peptide-treated A2058 cells (103) seeded in 6-well plates. After 5 days, 500 μl of 0.5 mM WT1-pTj or C PEP or culture medium were added in the corresponding wells. 10 days after plating, colonies were stained with 0.5% crystal violet in 70% ethanol for 10 min and counted in a stereomicroscope.
To assess the colony formation ability after drug removal, A2058 cells (103) were left untreated or treated with 0.5 mM WT1-pTj, 0.5 mM C PEP or 0.5 mM Temozolomide (TMZ; Sigma–Aldrich, St. Louis, MO). After 4 days, cells were harvested and equal numbers of untreated and treated cells were seeded (103) in 6-well plates and cultivated for additional 4 days in fresh media. Colonies were stained and quantified as mentioned above.
2.6. Chemiluminescence ELISA (CL-ELISA)
Binding of p53 to WT1-pTj was examined by CL-ELISA. Recombinant p53 (Novus Biotechnology, Littleton, CO), 100 ng in 50 μl of carbonate buffer (15 mM Na2CO3, 35 mM NaHCO3, 0.2 g/l NaN3, pH 9.6) was left to adhere overnight at 4 °C onto wells of maxisorp opaque ELISA plates (Nunc™, Nalge Nunc International, Dusseldorf, Germany). Plates were blocked for 1 h at 37 °C with 1% BSA in PBS-Tween 0.05%. After 3 washes in PBS-Tween 0.05%, 1 μM biotinylated-WT1-pTj was incubated for 1 h at 37 °C. Streptavidin-peroxidase (1:2000; Sigma–Aldrich, St. Louis, MO) was added and the reaction was evaluated by chemiluminescence using ECL solution (Millipore, Billerica, MA) in a luminometer (SpectraMax, Molecular Devices Software Pro 5.2, Sunnyvale, CA) at 470 nm. Chemiluminescence readings are expressed as Relative Luminescence Units (RLU).
Competitive ELISA was performed to investigate whether WT1-pTj competes with WT1 protein for binding to p53. To that end, 100 ng of recombinant p53 protein in carbonate buffer was dispensed in wells of ELISA microplate (96-well) and left overnight at 4 °C. Blocking and washing preceded the addition of 100 ng of recombinant WT1 (Jena Bioscience, Jena, Germany), followed by 1-h incubation at 37 °C. After washing 3 times in PBS-Tween 0.05%, WT1-pTj or C PEP were incubated at different concentrations for 1 h at 37 °C. Binding of WT1 to p53 was detected using anti-WT1 monoclonal antibody (1:1000, Millipore, Billerica, MA). After incubation for 1 h at 37 °C, the plate was extensively washed in PBS-Tween 0.05% and the secondary anti-mouse IgG coupled to peroxidase (1:2000; Sigma–Aldrich, St. Louis, MO) was added. The plate was washed 3 times in PBS and the reaction was evaluated by chemiluminescence as described above.
2.7. Luciferase reporter assay
Detection of p53 activity was performed using the Cancer 10-pathway Reporter Array (SA Biosciences, Fredrick, MD) following the manufacturer's instructions. After A2058 melanoma cells transfection, they were left untreated or treated with 0.5 mM WT1-pTj or 0.5 mM C PEP, and further incubated for 24 h. Luciferase activity was measured using the Dual Luciferase Assay system (Promega, Madison, WI) on a luminometer (SpectraMax, Molecular Devices Software Pro 5.2, Sunnyvale, CA). Firefly luciferase was the experimental reporter and Renilla luciferase was the internal control for normalizing transfection efficiencies.
2.8. SA-β-galactosidase activity
A2058 cells (103) were cultivated in 6-well plates and then left untreated or treated with 0.5 mM WT1-pTj, 0.5 mM C PEP or 0.5 mM TMZ (Positive control) [24] for 4 days. Cells were washed to remove the peptides and TMZ, following incubation with complete fresh medium for additional 3 days. SA-β-galactosidase activity was detected with the Senescence β-galactosidase Kit (Cell Signaling, Beverly, MA), following the manufacturer's instructions. To quantify SA-β-galactosidase activity, at least 100 cells were counted in three random fields using an inverted light microscope.
2.9. Lysosomal vacuolation observed with acridine orange
Lysosomes from A2058 cells (103/well) plated in 4-well chambers and treated or not with 0.5 mM WT1-pTj or 0.5 mM C PEP for 4 days at 37 °C, were observed following incubation with 1 μg/ml acridine orange (Sigma–Aldrich, St. Louis, MO) for 15 min. A Nikon BioStation IM-Q inverted microscope (red, excitation/emission = 490/650 nm) was used.
2.10. Cell cycle analysis
A2058 cells were seeded in 12-well plates at 2 × 105 cells per well and incubated overnight without serum for cell cycle synchronization. Cells were treated or not with 0.5 mM WT1-pTj or 0.5 mM C PEP for 24 h. After incubation, cells were harvested with trypsin–EDTA 0.25%, washed with PBS and centrifuged. Pellets were stained with propidium iodide (PI) in 0.1% Triton X-100, 0.1% sodium citrate, 50 μg/ml PI (Sigma–Aldrich, St. Louis, MO) and 300 μg/ml RNAse A (Invitrogen, Eugene, OR) for 30 min at 4 °C in the dark. Flow cytometric analysis used a FACSCanto II flow cytometer (Becton Dickinson, San Jose, CA). Post-acquisition analysis used the FlowJo software (Tree Star Inc., Ashland, OR). For evaluation of DNA content, 20,000 events were acquired from at least three independent experiments.
2.11. Western blotting
A2058 cells (106) treated with 0.5 mM WT1-pTj or 0.5 mM C PEP for 24 h after cell cycle synchronization or untreated cells (Control) were lysed for protein extraction with RIPA buffer (50 mM Tris–Cl, pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease and phosphatase inhibitors (Sigma–Aldrich, St. Louis, MO). After 20 min incubation on ice, the cell lysates were collected after centrifugation at 1500 rpm. The protein concentration of lysates was determined by Bradford method (Bio-Rad, Hercules, CA). Proteins (15 μg) were separated by SDS–PAGE and transferred to a nitrocellulose membrane (Millipore, Billerica, MA). Immunoblotting was run with antibodies against p53, phospho-p53 (Ser15, Ser392), p27, p21, cyclin B1, phospho-cdc2 (Tyr15), and β-actin, all purchased from Cell Signaling Technology (Beverly, MA). β-actin was used as the protein loading control. Secondary antibodies conjugated with IgG horseradish peroxidase were purchased from Sigma–Aldrich (St. Louis, MO) and immunoreactivity was detected using the Immobilon solution (Millipore, Billerica, MA). Protein bands were detected using the UVItec Alliance gel documentation system (UVItec, Cambridge, UK).
2.12. Experimental melanoma models in vivo
The Ethics Committee for animal experimentation of Federal University of São Paulo approved all experiments using mice, CEP No. 1280, 2010. All in vivo experiments were performed at least twice.
In the lung metastasis model, male, six-to-eight week-old, C57BL/6 mice were challenged e.v. with 5 × 105 syngeneic B16F10-Nex2 melanoma cells in 100 μl of PBS. Animals (n = 5 per group) were treated the day after tumor challenge, with 5 daily i.p. doses of 300 μg of C PEP, 300 μg of WT1-pTj or vehicle (PBS). After 14 days, lungs were collected from animals of each group, and inspected for lung colonization.
In the syngeneic subcutaneous (s.c.) tumor model, B16F10-Nex2 cells (105/animal) were inoculated into the right flank of C57BL/6 mice. Once nodules reached 200 mm3, mice (n = 5 per group) received 5 i.p. doses of 300 μg of WT1-pTj, 300 μg of C PEP or PBS on alternate days. The human A2058 melanoma cells (4 × 106/animal) were injected subcutaneously in athymic nude mice (n = 5 per group), treated with 300 μg of C PEP, 300 μg of WT1-pTj or vehicle (PBS) for 5 consecutive days. Peritumor therapy started the day after inoculation of melanoma cells. The tumor volume (V), measured daily with a caliper, was calculated by the formula V = 0.52 × d2 × D, where d and D are the short and long diameters of the tumor, respectively. Mice were sacrificed when the tumor size reached 3000 mm3.
2.13. Statistical analysis
Data are expressed as mean values ± standard deviations (SD) of multiple replicates. All data are representative of at least two independent experiments. p-Values were calculated by Student's t-test and considered significant if less than 0.05. Statistical differences among treated groups in the s.c. tumor model were assessed by the Gehan–Breslow–Wilcoxon test. The Kaplan–Meier method was used to calculate survival curves, and log-rank test was used to compare the survival rates of different groups. All statistical analyses were done using Prism Graphpad Software (San Diego, CA).
3. Results
3.1. WT1-pTj displays effective antimelanoma activity in vivo
The WT1-pTj peptide but not the C PEP control peptide, with two amino acids replaced by alanine, significantly reduced the number of lung metastatic nodules in the syngeneic B16F10-Nex2 melanoma model (Fig. 1A). Melanoma cells were injected endovenously in C57Bl/6 mice and treatment consisted in 5 daily i.p. injections of 300 μg of either peptide or vehicle (PBS), starting the day after tumor cell challenge.
Fig. 1.
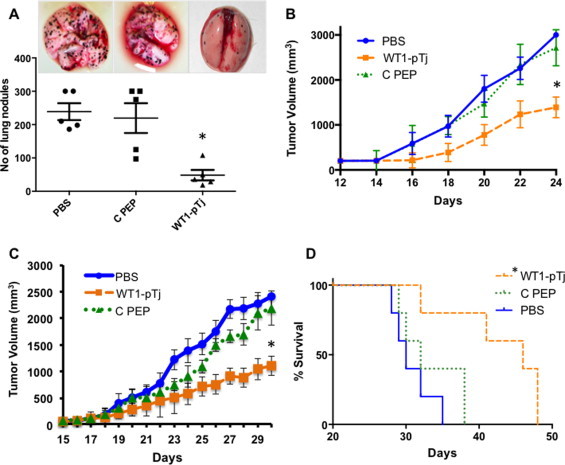
Protective effect of WT1-pTj in vivo. (A) Lung colonization of B16F10-Nex2 endovenously inoculated in C57BL/6 mice. Animals received 5 i.p. doses of 300 μg of C PEP, 300 μg of WT1-pTj or vehicle (PBS) on consecutive days; *p = 0.02 versus C PEP. (B) Effect of WT1-pTj on subcutaneous growth of B16F10-nex2 melanoma tumor. Once nodules reached 200 mm3, mice were randomized and received 5 alternate i.p. injections of 300 μg of WT1-pTj or C PEP; *p < 0.05 versus C PEP. (C) Effect of WT1-pTj on A2058 tumor growth in nude mice. Animals were treated with peritumor injections of 300 μg of WT1-pTj, 300 μg of C PEP or vehicle (PBS) for 5 consecutive days, starting on day 1 post-tumor cell challenge; *p < 0.05 in comparison with C PEP. (D) Survival of nude mice grafted with human melanoma after treatment with WT1-pTj, C PEP or PBS; n = 5 mice per group; *p = 0.0185 in comparison with C PEP.
By using subcutaneous B16F10-Nex2 melanoma graft in syngeneic mice, the tumor was left to grow until it reached 200 mm3. Peptides were then injected i.p., with 5 doses of 300 μg/animal in alternate days. WT1-pTj rather than the C PEP control clearly delayed melanoma growth, even after tumor establishment (Fig. 1B).
The WT1-pTj protective activity was also evaluated using a human melanoma xenograft in nude mice. Animals were challenged with human melanoma cells s.c., and treated daily, for 5 days, with 300 μg WT1-pTj or C PEP. Peritumor therapy started one day after inoculation of melanoma cells. As observed in Fig. 1C, WT1-pTj was effective in delaying the growth of human melanoma-bearing nude mice using in situ injections. Moreover, the survival of mice treated with WT1-pTj was significantly prolonged in comparison to that of the PBS and C PEP control groups (Fig. 1D).
It should be noted that in all in vivo experiments mice maintained healthy physical appearance, normal activity levels and normal weight throughout the study period, showing no toxic effects of peptides. Notwithstanding, an effective antimetastatic activity and a remarkable therapeutic efficacy of WT1-pTj peptide against melanoma were shown.
3.2. WT1-pTj peptide rapidly translocates into human melanoma cells
The direct effects of WT1-pTj on human melanoma cells were investigated. Considering the characteristics of the peptide, Trojan-like properties, and effects related to zinc-finger interactions could be predicted. In fact, WT1-pTj is a 27-mer lysine–arginine rich peptide, from the C-terminal region of WT1, which contains part of the second zinc-finger (ZF) domain of WT1. The analogous peptide, with alanine substitutions at cysteine-3 and histidine-16, both involved in the ZF motif, was used as a suitable control peptide (C PEP). Viable non-permeabilized A2058 human melanoma cells were exposed to 0.5 mM FITC-labeled WT1-pTj for 1 h. Using confocal microscopy, we observed that the peptide entered the cells showing a diffuse distribution in the cytoplasm and was also seen in the nucleus within 1 h, confirming the Trojan nature of the peptide (Fig. 2A).
Fig. 2.
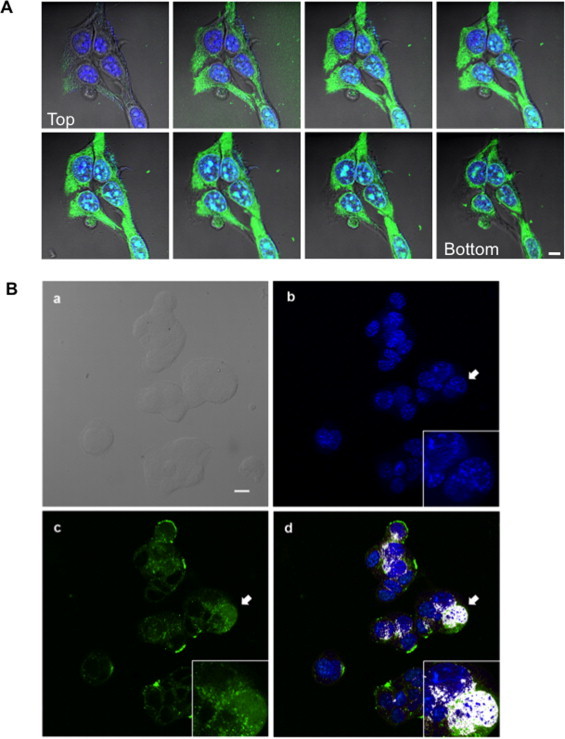
Intracellular distribution of WT1-pTj in A2058 human melanoma cells. (A) Cells were treated with FITC-labeled WT1-pTj for 1 h at 37 °C and visualized by confocal microscopy. Sequence (Z-stacks) showing the distribution of FITC-labeled peptide (green) and the nuclei stained with DAPI (blue). Bar = 10 μm. (B) Co-localization of WT1-pTj and nuclear DNA in human melanoma cells. A2058 cells were treated with biotinylated WT1-pTj for 24 h, fixed with paraformaldehyde, and permeabilized with Triton X-100. (a) Differential interference contrast (DIC) image; (b) Cells stained with DAPI; (c) Cells treated with streptavidin-FITC; (d) Merge, showing nuclear colocalization of WT1-pTj. Inserts: high magnification of cells indicated by arrows. Scale bar = 10 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
A2058 cells were also incubated with biotinylated-WT1-pTj (b-WT1-pTj) for 15 min, 1 and 24 h, then were fixed and permeabilized, followed by incubation with streptavidin-FITC and DAPI. Images taken 15 min after exposure to 0.5 mM of b-WT1-pTj showed predominantly membrane and cytoplasmic staining. After 1 and 24 h the peptide was completely distributed in the cells, particularly in the nucleus where it colocalized with nuclear DNA (Fig. 2B).
3.3. WT1-pTj inhibits the proliferation of WT1-expressing tumor cells
The effect of WT1-derived peptide on cell viability was determined by Trypan blue exclusion staining. Assuming the potential oncogenic role of WT1 in tumor cells [25], the peptide could modify this phenotype by structural interference. WT1-pTj did not induce cell death, but inhibited the proliferation of different WT1-expressing cancer cell lines (Table 2). As indicated by EC50 values, WT1-pTj induced growth inhibition of B16F10-Nex2 murine melanoma cells, A2058 and SK-MEL-28 human melanoma cells, MCF-7 and MDA-MB231 human breast cancer cells and OVCAR-3 human ovarian cancer cells, and was less effective against HL-60 human leukemia cells. Treatment with the peptide blocked cell proliferation but did not induce a death mechanism in vitro. No effect on the proliferation of normal murine or human fibroblasts was detected, showing a selective activity of WT1-pTj on tumor cells.
Table 2.
Antiproliferative activity of WT1-pTj on WT1-expressing tumor cell lines and nontumor forming cell lines.
| Cell lines | EC50a (mol/l x 10−3) |
|---|---|
| A2058 human melanoma | 0.455 ± 0.032 |
| SK-MEL-28 human melanoma | 0.680 ± 0.020 |
| B16F10-Nex2 murine melanoma | 0.466 ± 0.067 |
| MCF-7 human breast cancer | 0.247 ± 0.058 |
| MDA-MB231 human breast cancer | 0.759 ± 1.40 |
| OVCAR-3 human ovarian cancer | 0.208 ± 0.02 |
| HL-60 human acute leukemia | >1 |
| HFF human foreskin fibroblast | >1 |
| MEF murine embryonic fibroblast | >1 |
EC50 is the concentration that decreases viability by 50% in a dose-dependent survival curve.
3.4. WT1-pTj but not C PEP displays strong antiproliferative effects on human melanoma cells
Since WT1 gene is essential for melanoma cells proliferation and metastasis [26,27,8] we examined whether WT1-pTj and more specifically, the truncated ZF-motif of WT1-pTj, displayed anti-melanoma proliferative effects. Human A2058 melanoma cells were incubated with WT1-pTj or C PEP (A3C, A16H) at 0.125 to 0.5 mM and the cell number of viable cells determined every 24 h for 4 days. We found that treatment with WT1-pTj resulted in a dose-dependent inhibition of proliferation of melanoma cells when compared to untreated cells. The number of cells remained stable from 24 to 96 h when treated with WT1-pTj at 0.5 mM, suggesting that the peptide had a sustained cytostatic effect on these cells. No significant changes on proliferation were observed in C PEP-treated cells, except at the highest dose after 96 h of incubation (Fig. 3A).
Fig. 3.
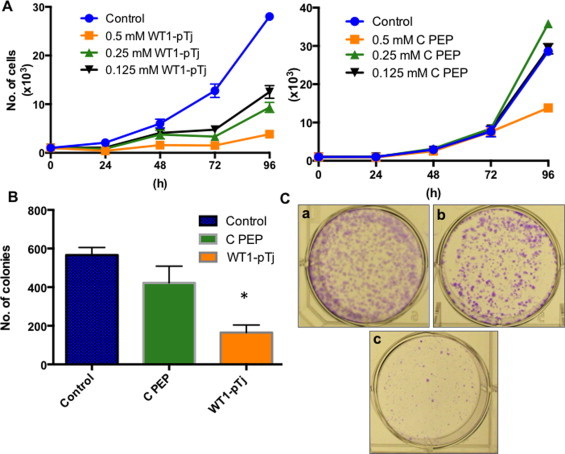
Effects of WT1-pTj on proliferation and clonogenic ability of human melanoma cells. (A) A2058 cells (103/well) were treated with WT1-pTj (left) or C PEP (right) at the indicated concentrations for 96 h, and cell proliferation was daily monitored by Trypan blue exclusion assay; (B) A2058 cells were suspended in 300 μl of 0.5 mM WT1-pTj, 0.5 mM C PEP, or unsupplemented culture medium (Control) and cultivated on soft-agar medium. The number of colonies was determined 11 days after the first treatment; *p < 0.02. (C) Representative photomicrographs of colony formation assay. A2058 cells were plated on (a) untreated culture medium (Control), (b) medium treated with 0.5 mM C PEP, or (c) treated with 0.5 mM WT1-pTj. After 5 days, culture media were aspirated and fresh medium with the respective peptides at 0.5 mM was added. After 10 days incubation, colonies were stained with crystal violet and counted on a stereomicroscope. Data represent means ± SD of three independent experiments. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Han et al. [28] reported on the transfection of MCF-7 human breast cancer cells with constructs containing only the ZF domains of WT1 and consequent reduction in the expression of proteins important for the survival and tumor cell proliferation, such as c-myc, Bcl-2 and amphregulin, also interfering with the clonogenic ability of breast cancer cells. Based on these findings we tested the influence of WT1-pTj on tumor cell clonogenicity. As observed, treatment with 0.5 mM WT1-pTj drastically decreased the number of A2058 colonies both in semi-solid soft-agar medium (Fig. 3B) and directly in culture plates (Fig. 3C), when compared to untreated or C PEP-treated cells. In the anchorage-independent growth assay in soft-agar, treatment with WT1-pTj reduced colony formation by 70.8%, as compared to 25.4% with C PEP. Quantification of cell growth on plates showed only 20 colonies after WT1-pTj treatment for 9 days, whereas cell incubation in unsupplemented complete culture medium or with 0.5 mM PEP C, resulted in 300 and 248 colonies, respectively.
3.5. WT1-pTj enhances the transcriptional activity of p53 in melanoma cells and competes with WT1 protein for binding to p53
WT1 binds to DNA and can interact also with several intracellular proteins [29]. A physical and functional association between WT1 and the product of p53 tumor suppressor gene [30] has been demonstrated. Such interaction was shown to cross-modulate transactivation properties. WT1 interacts with p53 through zinc fingers 1 and 2, stabilizes p53, enhances p53-mediated transcriptional activation, and antagonizes p53-mediated apoptosis triggered by ultraviolet radiation [31].
Since the sequence of WT1-pTj covers part of a ZF motif involved in the p53 interaction [31] we considered the possibility that the peptide might also bind to p53. As shown in Fig. 4A, CL-ELISA using recombinant p53 and biotinylated WT1-pTj confirmed the association between the peptide and the protein. In addition, using competitive ELISA we verified that WT1-pTj dose-dependently inhibited WT1-p53 complex formation (Fig. 4B). Further, in A2058 melanoma cells transfected with a p53-responsive luciferase reporter construct, WT1-pTj (but not C PEP) enhanced p53 transcriptional activation (Fig. 4C). Together, these results suggest that WT1-pTj interacts with p53, and may decrease the association between p53 and WT1. In parallel, it caused p53 access to specific transcriptional response elements on DNA.
Fig. 4.
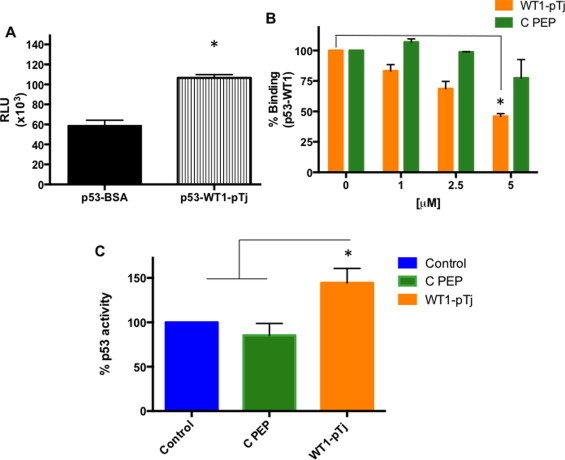
Binding of WT1-pTj to p53, displacement of p53 bound to WT1, and WT1-pTj modulation of transcriptional activity of p53. (A) The interaction between p53 and WT1-pTj was determined using biotinylated peptide and CL-ELISA; *p < 0.01. (B) Competitive inhibition of WT1 association to p53 was evaluated in wells coated with recombinant p53, incubated with WT1 and then exposed to WT1-pTj or C PEP at indicated concentrations. Binding of p53 to WT1 was quantified with anti-WT1 monoclonal antibody; *p < 0.02 compared to C PEP effect. (C) A2058 cells were transfected with the p53-responsive luciferase reporter construct, and then treated with 0.5 mM WT1-pTj or 0.5 mM C PEP for 24 h. The ratio of firefly to Renilla luciferase was determined and normalized to the value obtained for untreated cells (=100%). Data are representative of two independent experiments. * p < 0.02 compared to Control (untreated) and C PEP systems.
3.6. WT1-pTj induces G2/M cell cycle arrest and senescence features in human melanoma cells
The cell cycle of melanoma cells was examined by flow cytometry to better characterize the mechanism by which WT1-pTj suppresses tumor cell proliferation. Treatment with WT1-pTj effectively arrested the cell-cycle progression in A2058 cells, depleting the S-phase compartment (from 20.2% to 6% when compared to Control) and increasing the G2/M (from 10.5% to 27% when compared to Control) phase compartment (Fig. 5A). A2058 cells treated with C PEP showed no significant differences regarding the cell cycle profile when compared to the Control system, suggesting that the ZF-coordinating region is essential for the antiproliferative effects.
Fig. 5.
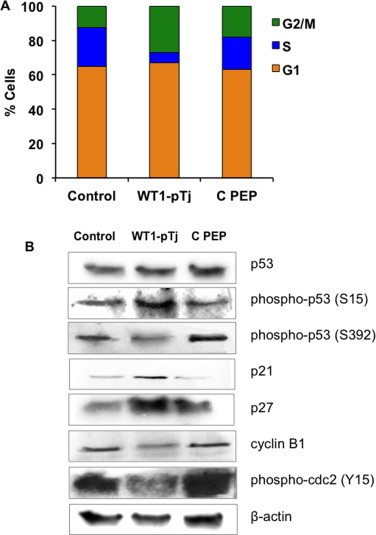
Effect of WT1-pTj on cell cycle progression and modulation of p53, and downstream mediators, in human melanoma cells. (A) A2058 cells were treated either with 0.5 mM WT1-pTj or 0.5 mM C PEP, for 24 h, and the cell cycle profile was examined by flow cytometry. Control, unsupplemented culture medium. Results are representative of three independent experiments. (B) Cellular extracts from A2058 cells exposed to 0.5 mM WT1-pTj or 0.5 mM C PEP were subjected to immunoblotting with antibodies specific for p53, phospho-p53, p27, p21, phospho-cdc2 and cyclin B1. Protein load was normalized to β-actin.
In addition, we evaluated the predominant signal pathways arbitrating cell cycle arrest in human melanoma cells under peptide treatment. As shown in Fig. 5B, by Western blotting analysis we determined that total p53 levels were not significantly affected in cells treated with WT1-pTj. Nevertheless, a decrease in serine 392 phosphorylation and an increase in serine 15 phosphorylation were observed upon WT1-pTj treatment. Additionally, increased levels of both p27Kip1 and p21Cip1, known to regulate both G1–S and G2–M transitions, were detected in WT1-pTj-treated cells. Furthermore, in order to elucidate the nature of the cell-cycle arrest caused by WT1-pTj, we examined the expression of cell cycle-regulating factors at the G2/M phase. We found that the protein levels of cyclin B1 and phospho-cdc2 (Tyr15) were markedly down regulated in WT1-pTj-treated A2058 cells. These results associate the inhibitory effects of WT1-pTj to blocking of the cell cycle progression at G2/M phase and modulation of p53 and downstream signaling cascades.
Several reports have shown the central role of p53 in controlling senescence [32], which is characterized by stable and irreversible loss of cell proliferation [33]. Particularly in melanoma, telomere dysfunction [34] oncogene activation [35] and anticancer agents [36] can activate p53 and its signaling partners (e.g. p16Ink4A, Rb and p21Cip1), which are thus implicated in the suppression of tumor initiation and progression.
Owing to the up-regulation of p53 activity, along with the morphological changes (e.g. large cells and cytoplasmic vacuolization) and the sustained inhibition of cellular proliferation observed in WT1-pTj-treated cells, we hypothesized that the peptide could induce cellular senescence. A2058 cells were incubated with WT1-pTj, C PEP or TMZ (positive control) or in unsupplemented culture medium (Control), for 4 days. Cells were washed to remove the peptides and incubated for additional 4 days in fresh medium. Melanoma cells barely resumed proliferation after removing WT1-pTj (Fig. 6A). Further, as depicted in Fig. 6B, the remaining cells treated with WT1-pTj exhibited altered morphology and stained positively for SA-β-galactosidase (72%), a widely used senescence biomarker [36]. In contrast, C PEP caused little reduction in the number of colonies as compared to untreated cells, and a minor effect on senescence induction (16%) when compared to WT1-pTj. Thus, the proliferative capacity of WT1-pTj-treated cells was lost even upon removal of the peptide.
Fig. 6.
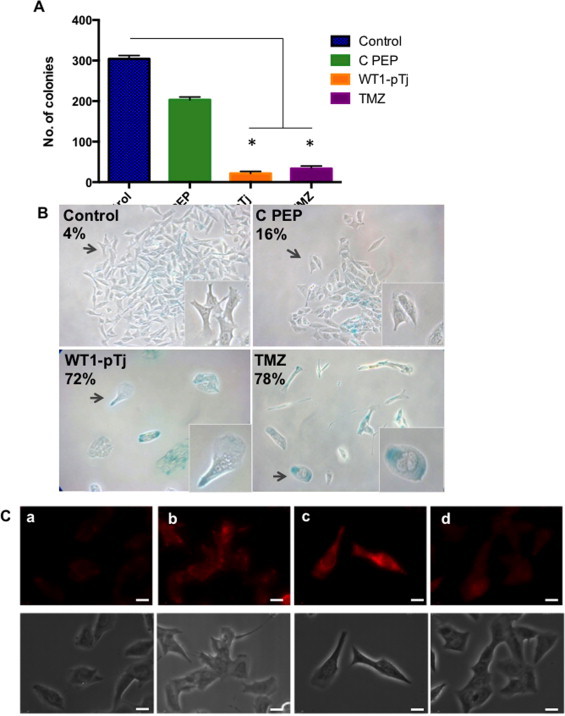
Induction by WT1-pTj of cellular senescence in human melanoma cells. (A) Quantification of colonies formed after removal of the peptides and TMZ. A2058 cells were treated with WT1-pTj, C PEP and TMZ for 4 days. Cells were detached and equal numbers of untreated and treated cells were seeded and cultivated in fresh media for additional 4 days. The number of colonies was scored in three independent experiments performed in duplicates; *p < 0.01 compared to Control and C PEP. (B) Representative photomicrographs (original magnification 400×) of A2058 cells treated with 0.5 mM WT1-pTj, C PEP or TMZ (positive control) for 4 days. Thereafter the peptides and TMZ were washed out and cells were stained for SA-β-Gal after incubation for additional 3 days in fresh media. Positive cells for SA-β-Gal were visualized under a phase contrast microscope and expressed as percentage of total cells counted at random. (C) Acridine orange was used to stain acidic vesicular organelles in untreated control cells (a); serum starved cells for 24 h (positive control) (b); cells treated with 0.5 mM WT1-pTj (c); cells treated with C PEP (d), for 4 days. Images are representative of two independent experiments performed in duplicates. Bars = 10 μM.
The SA-β-galactosidase has been shown to be a manifestation of residual lysosomal activity after alkalinization, only detectable due to the increased lysosomal content in senescent cells [37]. Therefore, we examined the lysosomal content in melanoma cells treated either with WT1-pTj or C PEP, using the lysosomotropic agent acridine orange. Staining of A2058 cells with acridine orange showed a marked increase in the number and size of lysosomes in senescent cells after treatment with 0.5 mM WT1-pTj for 4 days as visualized by fluorescence microscopy (Fig. 6C). Most of the C PEP-treated cells exhibited minimal red fluorescence. These results, along with the molecular mechanism underlying WT1-pTj-induced growth arrest, strongly suggest that WT1-pTj-treated cells undergo cellular senescence.
4. Discussion
Peptide-based antitumor therapy explores the high affinity and specificity of peptides for particular targets, the low toxicity and good tissue penetration [38]. Our laboratory has focused on antitumor peptides derived from internal sequences of immunoglobulins and transcription factors [22]. In the current study, we report on a peptide derived from WT1, endowed with anti-melanoma properties. Both human melanoma and B16F10 murine melanoma were sensitive to the WT1-pTj Trojan peptide.
WT1 called our attention due to its high expression and immunogenicity in B16F10 murine melanoma (unpublished results). In comparison with human cancer, WT1 has emerged as an important target expressed in hematologic and solid tumors. There is, indeed, plenty of evidence supporting the role of WT1 in the oncogenic process of melanoma. The function of WT1 in transcriptional regulation, as well in RNA metabolism and translation, has been extensively reviewed [25,39]. WT1 overexpression has been detected in melanoma patients’ samples and melanoma cells, whereas no WT1 staining has been observed in the majority of benign melanocytic nevi, epidermal keratinocytes and melanocytes [4,40]. RNAi silencing of WT1 inhibited melanoma proliferation associated with down-regulation of nestin and zyxin [6], sensitized B16F10 cells to conventional chemotherapeutic agents (e.g. doxorubicin and cisplatin) and reduced lung metastases, emphasizing WT1 function in melanoma progression [8].
The WT1-pTj peptide contains part of a particularly reactive zinc-finger domain, and owing to its abundant basic amino acid composition, exhibits cell-penetrating properties. The peptide was able to arrest tumor cell growth and induce senescence in human melanoma cells. A significant antimelanoma activity in vivo was also observed using subcutaneous and metastatic models in mice. Increased levels of active p53 and enhanced transcriptional activation were associated with WT1-pTj-induced senescence in A2058 human melanoma cells. Cyclin-dependent kinase inhibitors p27Kip1 and p21Cip1 mediated the sustained cell cycle arrest. The typical senescent phenotype in WT1-pTj-treated cells included enlarged cytoplasm, and cellular increase of both SA-β-galactosidase activity and lysosomal content. To exert its activities, peptide WT1-pTj, like WT1, does not bind to p53 promoter but physically interacts with the p53 protein itself. In fact, we have shown that the peptide competes with WT1 for protein binding to p53, supporting the hypothesis that the peptide may prevent important interactions of WT1 with its partners, including p53, and therefore disturb relevant pro-tumor signaling of the original protein via the ZF domain. Protein-protein interaction of WT1 with p53 modulates binding to different promoters determining gene expression or repression. As with WT1, we demonstrated that peptide WT1-pTj activated p53 to act as a transcription factor in a luciferase gene expression system.
Therapy-induced senescence is a novel therapeutic approach to treat cancers [41]. Standard chemotherapy and radiotherapy, besides inducing DNA damage and cytotoxicity, may trigger a robust senescence response in vitro and in vivo [42,43]. Pro-senescence therapy may also be achieved with greatly reduced toxicity.
The molecular mechanism underlying therapy-induced senescence conferred by WT1-pTj treatment depends on p53 transcriptional activation and is correlated with p53 post-transcriptional phosphorylation at Ser15 and Ser392. Consistent with previous findings that have reported on specific changes in p53, a sustained treatment triggers a p53-dependent senescence program. Prolonged β-interferon stimulation switched on p53 in two steps involving first, dephosphorylation at serine 392, and then, phosphorylation of p53 at serine 15 leading to its transcriptional activity [44]. Similar posttranslational modifications of p53 were observed in replicative senescence in human fibroblasts [45].
Likewise in WT1-pTj induced senescence of melanoma cells, reduced phosphorylation at S392 and increased phosphorylation at S15, were observed. Activation of p53 by phosphorylation, rendering an important effector of senescence [46,47] leads to the transcriptional activation of target genes. In agreement, we observed the induction of the senescence molecular markers p21Cip1 and p27Kip1 [41] in A2058 cells exposed to WT1-pTj. Our results excluded p16Ink4A-Rb signal activation in WT1-pTj-induced cellular senescence (data not shown), in line with previous data showing that p16 is frequently inactivated in melanoma [48,49].
Cell cycle analysis revealed that WT1-pTj inhibition of melanoma cell proliferation was caused by cell cycle arrest at G2/M phase, accompanied by a decrease in the number of cells in the S phase. The G2–M transition is positively regulated by Cdc2/cyclin B complex [50], which is controlled by phosphorylation at various sites, including the inhibitory phosphorylation at tyrosine-15 and threonine 14 by Wee1 and Myt1 [51,52]. A decrease in the levels of phospho-cdc2 (Tyr15), cdc2, cyclin A and cyclin B1 has been associated with cell cycle arrest at G2/M [53]. Similarly, we found a significant decrease in cyclin B1 and phospho-cdc2 (Tyr15) expression after treatment with WT1-pTj, in contrast with C PEP-treated cells. In conclusion, modulation of p53 activity and activation of downstream signaling is essential for senescence response in WT1-pTj-treated cells. These events depend on the ZF-coordinating region because C PEP elicited no similar response.
Given the limited expression of WT1 in adult animals, restricted to few cell types of the urogenital system [3], this factor might otherwise be an optimal target for treatment of WT1-expressing malignancies. In the field of targeted therapies, CPPs (cationic cell-penetrating peptides) have been used to overcome permeability barriers in the tumor and to mediate cargo delivery into cancer cells, leading to the development of tumor-specific molecular therapeutics. CPP-mediated transduction has been used to inhibit nuclear oncoprotein translocation [54], modulate oncoprotein signaling [10], enable apoptotic cell-death [16,55], and deliver oligonucleotides [56] to cancer cells. Presently, we show that peptide WT1-pTj exhibits trojan properties and exerts protective effects in experimental melanoma therapy. In addition to antiproliferative effects mediated by WT1-pTj treatment in vitro, the peptide inhibited human melanoma progression, in nude mice.
Conventional chemotherapeutic drugs act mainly through induction of apoptosis, and their reduced efficacy in melanoma patients is related to the high resistance of melanoma cells [57]. Therapy-induced senescence may represent an alternate functional approach to improve cancer therapy [58,59].
To conclude, our present study reports on the senescence-inducing peptide WT1-pTj as a promising candidate to eradicate tumor cell progression, including those that fail to respond to conventional antitumor therapies. The WT1-pTj is unique in that it derives from an oncoprotein, is cell-penetrating by itself, induces cellular senescence and effectively protects in a cancer preclinical model.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Acknowledgments
The authors thank the Ludwig Institute for Cancer Research, São Paulo branch, Dr. O. Keith Okamoto from the University of São Paulo, and Dr. Luiz F. L. Reis from Sirio-Libanez Hospital for the cell lines used in the present study. The State of São Paulo Research Support Foundation (FAPESP), Brazil, supported this work through Grants 2010/51423–0 and 2012/19476–2. L.R.T. is a research fellow from the Brazilian National Research Council (CNPq).
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Yang L. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007;21:868–876. doi: 10.1038/sj.leu.2404624. [DOI] [PubMed] [Google Scholar]
- 2.Call K.M. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumor locus. Cell. 1990;60:509–520. doi: 10.1016/0092-8674(90)90601-a. [DOI] [PubMed] [Google Scholar]
- 3.Pelletier J. WT1 mutations contribute to abnormal genital system development and hereditary Wilms’ tumour. Nature. 1991;353:431–434. doi: 10.1038/353431a0. [DOI] [PubMed] [Google Scholar]
- 4.Nakatsuka S. Immunohistochemical detection of WT1 protein in a variety of cancer cells. Mod. Pathol. 2006;19:804–814. doi: 10.1038/modpathol.3800588. [DOI] [PubMed] [Google Scholar]
- 5.Oji Y. Expression of the Wilms’ tumor gene WT1 in solid tumors and its involvement in tumor cell growth. Jpn. J. Cancer Res. 1999;90:194–204. [Google Scholar]
- 6.Wagner N. The Wilms’ tumor suppressor WT1 is associated with melanoma proliferation. Pflugers Arch. 2008;455:839–847. doi: 10.1007/s00424-007-0340-1. [DOI] [PubMed] [Google Scholar]
- 7.Zamora-Avila D.E. RNAi silencing of the WT1 gene inhibits cell proliferation and induces apoptosis in the B16F10 murine melanoma cell line. Melanoma Res. 2007;17:341–348. doi: 10.1097/CMR.0b013e3282efd3ae. [DOI] [PubMed] [Google Scholar]
- 8.Zamora-Avila D.E. WT1 gene silencing by aerosol delivery of PEI-RNAi complexes inhibits B16-F10 lung metastases growth. Cancer Gene Ther. 2009;16:892–899. doi: 10.1038/cgt.2009.35. [DOI] [PubMed] [Google Scholar]
- 9.Thundimadathil J. Cancer treatment using peptides: current therapies and future prospects. J. Amino Acids. 2012;2012:967347. doi: 10.1155/2012/967347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giorello L. Inhibition of cancer cell growth and c-Myc transcriptional activity by a c-Myc helix 1-type peptide fused to an internalization sequence. Cancer Res. 1998;58:3654–3659. [PubMed] [Google Scholar]
- 11.Bonfanti M. p21WAF1-derived peptides linked to an internalization peptide inhibit human cancer cell growth. Cancer Res. 1997;57:1442–1446. [PubMed] [Google Scholar]
- 12.Rosca E.V. Anti-angiogenic peptides for cancer therapeutics. Curr. Pharm. Biotechnol. 2011;12:1101–1116. doi: 10.2174/138920111796117300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan M. Selective inhibition of ErbB2-overexpressing breast cancer in vivo by a novel TAT-based ErbB2-targeting signal transducers and activators of transcription 3-blocking peptide. Cancer Res. 2006;66:3764–3772. doi: 10.1158/0008-5472.CAN-05-2747. [DOI] [PubMed] [Google Scholar]
- 14.Cardo-Vila M. From combinatorial peptide selection to drug prototype (II): targeting the epidermal growth factor receptor pathway. Proc. Natl. Acad. Sci. U.S.A. 2010;107:5118–5123. doi: 10.1073/pnas.0915146107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuo A.L. A new phage-display tumor-homing peptide fused to antiangiogenic peptide generates a novel bioactive molecule with antimelanoma activity. Mol. Cancer Res. 2011;9:1471–1478. doi: 10.1158/1541-7786.MCR-10-0501. [DOI] [PubMed] [Google Scholar]
- 16.Johansson H.J. Characterization of a novel cytotoxic cell-penetrating peptide derived from p14ARF protein. Mol. Ther. 2008;16:115–123. doi: 10.1038/sj.mt.6300346. [DOI] [PubMed] [Google Scholar]
- 17.Niesner U. Quantitation of the tumor-targeting properties of antibody fragments conjugated to cell-permeating HIV-1 TAT peptides. Bioconjug. Chem. 2002;13:729–736. doi: 10.1021/bc025517+. [DOI] [PubMed] [Google Scholar]
- 18.Massodi I. Inhibition of ovarian cancer cell metastasis by a fusion polypeptide Tat-ELP. Clin. Exp. Metastasis. 2009;26:251–260. doi: 10.1007/s10585-009-9237-z. [DOI] [PubMed] [Google Scholar]
- 19.Caino M.C., Meshki J., Kazanietz M.G. Hallmarks for senescence in carcinogenesis: novel signaling players. Apoptosis. 2009;14:392–408. doi: 10.1007/s10495-009-0316-z. [DOI] [PubMed] [Google Scholar]
- 20.Bringold F., Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp. Gerontol. 2000;35:317–329. doi: 10.1016/s0531-5565(00)00083-8. [DOI] [PubMed] [Google Scholar]
- 21.Arruda D.C. beta-Actin-binding complementarity-determining region 2 of variable heavy chain from monoclonal antibody C7 induces apoptosis in several human tumor cells and is protective against metastatic melanoma. J. Biol. Chem. 2012;287:14912–14922. doi: 10.1074/jbc.M111.322362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Massaoka M.H. Melanoma: perspectives of a vaccine based on peptides. In: Giese M., editor. Molecular Vaccines: From Prophylaxis to Therapy. Springer-Verlag; Wien: 2013. pp. 397–412. [Google Scholar]
- 23.Dobroff A.S. Protective, anti-tumor monoclonal antibody recognizes a conformational epitope similar to melibiose at the surface of invasive murine melanoma cells. Hybrid Hybridomics. 2002;21:321–331. doi: 10.1089/153685902761022661. [DOI] [PubMed] [Google Scholar]
- 24.Mhaidat N.M. Temozolomide induces senescence but not apoptosis in human melanoma cells. Br. J. Cancer. 2007;97:1225–1233. doi: 10.1038/sj.bjc.6604017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hohenstein P., Hastie N.D. The many facets of the Wilms’ tumour gene, WT1. Hum. Mol. Genet. 2006;15(Spec No 2):R196–R201. doi: 10.1093/hmg/ddl196. [DOI] [PubMed] [Google Scholar]
- 26.Zapata-Benavides P. WT1 silencing by RNAi synergizes with chemotherapeutic agents and induces chemosensitization to doxorubicin and cisplatin in B16F10 murine melanoma cells. Oncol. Lett. 2012;3:751–755. doi: 10.3892/ol.2012.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michiels J.F. PPARbeta activation inhibits melanoma cell proliferation involving repression of the Wilms’ tumour suppressor WT1. Pflugers Arch. 2010;459:689–703. doi: 10.1007/s00424-009-0776-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Han Y. The zinc finger domain of Wilms’ tumor 1 suppressor gene (WT1) behaves as a dominant negative, leading to abrogation of WT1 oncogenic potential in breast cancer cells. Breast Cancer Res. 2007;9:R43. doi: 10.1186/bcr1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Englert C. WT1–more than a transcription factor? Trends Biochem. Sci. 1998;23:389–393. doi: 10.1016/s0968-0004(98)01277-8. [DOI] [PubMed] [Google Scholar]
- 30.Maheswaran S. Physical and functional interaction between WT1 and p53 proteins. Proc. Natl. Acad. Sci. U.S.A. 1993;90:5100–5104. doi: 10.1073/pnas.90.11.5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maheswaran S. The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev. 1995;9:2143–2156. doi: 10.1101/gad.9.17.2143. [DOI] [PubMed] [Google Scholar]
- 32.Itahana K., Dimri G., Campisi J. Regulation of cellular senescence by p53. Eur. J. Biochem. 2001;268:2784–2791. doi: 10.1046/j.1432-1327.2001.02228.x. [DOI] [PubMed] [Google Scholar]
- 33.Larsson L.G. Oncogene- and tumor suppressor gene-mediated suppression of cellular senescence. Semin. Cancer Biol. 2011;21:367–376. doi: 10.1016/j.semcancer.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Sviderskaya E.V. p16/cyclin-dependent kinase inhibitor 2A deficiency in human melanocyte senescence, apoptosis, and immortalization: possible implications for melanoma progression. J. Natl. Cancer Inst. 2003;95:723–732. doi: 10.1093/jnci/95.10.723. [DOI] [PubMed] [Google Scholar]
- 35.Wajapeyee N. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cozzi SJ. Induction of senescence in diterpene ester-treated melanoma cells via protein kinase C-dependent hyperactivation of the mitogen-activated protein kinase pathway. Cancer Res. 2006;66:10083–10091. doi: 10.1158/0008-5472.CAN-06-0348. [DOI] [PubMed] [Google Scholar]
- 37.Kurz D.J. Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell. Sci. 2000;113:3613–3622. doi: 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- 38.Bitler B.G., Schroeder J.A. Anti-cancer therapies that utilize cell penetrating peptides. Recent Pat. Anticancer Drug Discov. 2010;5:99–108. doi: 10.2174/157489210790936252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts S.G. Transcriptional regulation by WT1 in development. Curr. Opin. Genet. Dev. 2005;15:542–547. doi: 10.1016/j.gde.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 40.Henderson E.J., Pentland B. Home pass assessment in neurorehabilitation practice. J. Adv. Nurs. 1991;16:1439–1443. doi: 10.1111/j.1365-2648.1991.tb01591.x. [DOI] [PubMed] [Google Scholar]
- 41.Nardella C. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer. 2011;11:503–511. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- 42.te Poele R.H. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–1883. [PubMed] [Google Scholar]
- 43.Sidi R. Induction of senescence markers after neo-adjuvant chemotherapy of malignant pleural mesothelioma and association with clinical outcome: an exploratory analysis. Eur. J. Cancer. 2011;47:326–332. doi: 10.1016/j.ejca.2010.09.044. [DOI] [PubMed] [Google Scholar]
- 44.Moiseeva O. DNA damage signaling and p53-dependent senescence after prolonged beta-interferon stimulation. Mol. Biol. Cell. 2006;17:1583–1592. doi: 10.1091/mbc.E05-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webley K. Posttranslational modifications of p53 in replicative senescence overlapping but distinct from those induced by DNA damage. Mol. Cell. Biol. 2000;20:2803–2808. doi: 10.1128/mcb.20.8.2803-2808.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Herbig U. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a) Mol. Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 47.Di Micco R. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 48.Castellano M. CDKN2A/p16 is inactivated in most melanoma cell lines. Cancer Res. 1997;57:4868–4875. [PubMed] [Google Scholar]
- 49.FitzGerald M.G. Prevalence of germ-line mutations in p16, p19ARF, and CDK4 in familial melanoma: analysis of a clinic-based population. Proc. Natl. Acad. Sci. U.S.A. 1996;93:8541–8545. doi: 10.1073/pnas.93.16.8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Innocente S.A. p53 regulates a G2 checkpoint through cyclin B1. Proc. Natl. Acad. Sci. U.S.A. 1999;96:2147–2152. doi: 10.1073/pnas.96.5.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McGowan C.H., Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993;12:75–85. doi: 10.1002/j.1460-2075.1993.tb05633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wells N.J. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J. Cell. Sci. 1999;112:3361–3371. doi: 10.1242/jcs.112.19.3361. [DOI] [PubMed] [Google Scholar]
- 53.Deep G. Silymarin and silibinin cause G1 and G2-M cell cycle arrest via distinct circuitries in human prostate cancer PC3 cells: a comparison of flavanone silibinin with flavanolignan mixture silymarin. Oncogene. 2006;25:1053–1069. doi: 10.1038/sj.onc.1209146. [DOI] [PubMed] [Google Scholar]
- 54.Lin Y.Z. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- 55.Selivanova G. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 1997;3:632–638. doi: 10.1038/nm0697-632. [DOI] [PubMed] [Google Scholar]
- 56.Kanazawa T. Suppression of tumor growth by systemic delivery of anti-VEGF siRNA with cell-penetrating peptide-modified MPEG-PCL nanomicelles. Eur. J. Pharm. Biopharm. 2012;81:470–477. doi: 10.1016/j.ejpb.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 57.Grossman D., Altieri D.C. Drug resistance in melanoma: mechanisms, apoptosis, and new potential therapeutic targets. Cancer Metastasis Rev. 2001;20:3–11. doi: 10.1023/a:1013123532723. [DOI] [PubMed] [Google Scholar]
- 58.Jing H. Opposing roles of NF-kappaB in anti-cancer treatment outcome unveiled by cross-species investigations. Genes Dev. 2011;25:2137–2146. doi: 10.1101/gad.17620611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schmitt C.A. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell. 2002;109:335–346. doi: 10.1016/s0092-8674(02)00734-1. [DOI] [PubMed] [Google Scholar]