

Abstract
Mcl-1 is a member of the Bcl-2 family protein; its degradation is required for the initiation of apoptosis. The mechanism, however, is not yet clearly known. Previously, it was reported that Mcl-1 is degraded through the ubiquitination-mediated pathway and the PEST domain is the motif responsible for promoting this degradation. We found evidence that this may not be true. We generated several Mcl-1 deletion mutants and examined their effects on protein stability. Deletion of the PEST domain did not prevent the degradation of Mcl-1 during apoptosis. The BH1 domain, but not the PEST, BH3 or BH2 domain, exhibited a short half-life. A peptide named “F3” (VTLISFG) in the C-terminus of the BH1 domain appears to be critical for the rapid turnover of Mcl-1. Deletion of F3 from GFP-Mcl-1-ΔPEST retarded the degradation of this mutant. F3 appeared to be the minimum functional sequence of the degradation motif, since deletion of a single residue was sufficient to abrogate its short half-life. Fusion of F3 with p32 resulted in the degradation of p32 during UV-induced apoptosis, while wild type p32 was not affected. Taken together, these findings suggest that F3 (VTLISFG), instead of PEST, is the major motif responsible for the degradation of Mcl-1 during apoptosis.
Keywords: Apoptosis, Mcl-1, Degradation motif, PEST domain
Abbreviations: β-TrCP, β-transducin repeat-containing protein; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma-2; BH domain, Bcl-2 homology domain; Bim, Bcl-2-interacting mediator; BSA, bovine serum albumin; Caspase, cysteine aspartase; CCD, charge-coupled device; EGFP, enhanced green fluorescent protein; EIF2, eukaryotic translation initiation factor 2; EYFP, enhanced yellow fluorescent protein; GCN2, general control nonrepressed 2; GSK-3β, glycogen synthase kinase-3β; HECT, homologous to E6-AP carboxylterminus; h, hour; HRP, horseradish peroxidase; kD, kilodaltons; Mcl-1, myeloid cell leukaemia sequence 1; MEM, minimum essential medium; Mule, Mcl-1 ubiquitin ligase E3; PBS, phosphate-buffered saline; PCR, polymerase chain reaction; pDNA, plasmid DNA; PERK, PKR-like ER kinase; SDS–PAGE, sodium dodecyl sulphate–polyacrylamide gel electrophoresis; TM domain, transmembrane domain; UV, ultraviolet light
Graphical abstract
Highlights
-
•
The PEST domain may not be responsible for the short half-life of Mcl-1 during apoptosis.
-
•
A short peptide (F3) inside the BH1 domain was found to have a short half-life.
-
•
Fusion of F3 with p32 impairs the stability of p32 during apoptosis.
-
•
Deletion of F3 increases the stability of GFP-Mcl-1-ΔPEST.
1. Introduction
Apoptosis is a tightly controlled process of cell suicide that is of fundamental importance in maintaining the normal physiological function of a biological organism. The Bcl-2 family proteins are known to play a crucial role in regulating the apoptotic progression. The Bcl-2 family proteins include both pro- and anti-apoptotic members. The ratio between these two subgroups plays an important role in determining the fate of cells [1–3].
Among the anti-apoptotic Bcl-2 family proteins, Mcl-1 has an unusual short half-life. Mcl-1 was originally identified in differentiating myeloid cells in 1993 [4]. The human MCL1 gene is located on chromosome 1q21. During the apoptotic process, the Mcl-1 protein level decreases dramatically in contrast to the other anti-apoptotic proteins Bcl-2 and Bcl-xL [5–7]. The down regulation of Mcl-1 is thought to result from a suppression of Mcl-1 synthesis as well as an enhancement of Mcl-1 protein degradation. The mRNA level of Mcl-1 decreases in response to various apoptotic stimuli such as UV irradiation and staurosporine [6]. In particular, during the apoptosis induced by DNA damage agents or hydrogen peroxide, transcription is blocked due to ubiquitination and this is subsequently followed by the degradation of RNA polymerase II [8–11].
The degradation of the Mcl-1 protein is believed to be mediated through the ubiquitination pathway, since applying the proteasome inhibitor MG132 is able to stabilize the protein level of Mcl-1 [6,7]. Moreover, Mcl-1 can be ubiquitinated at five lysines (5, 40, 136, 194 and 197) and a 482 KDa HECT-domain-containing ubiquitin ligase named Mule was identified as the E3 ubiquitin ligase [12]. Another E3 ligase, β-TrCP, which is a Skp1-CUL1-F box protein (SCF) family member, was also found to recognize the phosphorylated Mcl-1 mediated by GSK3 [13]. In addition, a deubiquitinylase called “ubiquitin-specific peptidase 9 X-linked” (USP9X) is reported to be a regulator of Mcl-1 degradation, as silencing of USP9X by RNA interference (RNAi) led to the loss of Mcl-1 at the protein but not mRNA level [14].
In addition to poly-ubiquitination, Mcl-1 may be cleaved by caspase-3 to disrupt the Mcl-1-Bim interaction, allowing the release of Bim to exert the pro-apoptotic function and lead to Bax activation and cytochrome c release [15,16]. On the other hand, a short form of Mcl-1, Mcl-1S/ΔTM, was generated by caspase-3 cleavage [15,17,18]. This short form resembles pro-apoptotic “BH3 only” proteins and promotes apoptosis. Therefore, the cleavage of Mcl-1 may contribute to the feed-forward amplification of apoptotic signals once caspase-3 is activated.
In addition to their role in down regulation, what is the functional domain that plays a crucial role in facilitating the degradation of Mcl-1? The Mcl-1 protein is comprised of 350 amino-acid residues and contains the BH domains 1–3. In the C-terminus, Mcl-1 contains a transmembrane (TM) domain that is involved in localization to the outer mitochondrial membrane [19]. In the N-terminus, Mcl-1 contains two PEST regions (rich in proline, glutamic acid, serine and threonine amino-acid residues), which are often found in rapidly turn-over proteins. Previously the PEST regions were suggested to be responsible for the short half-life of Mcl-1 [20]. This suggestion, however, was questioned by other investigators [21,22]. Thus, we would like to conduct a series of experiments to directly examine which domain is responsible for the degradation of Mcl-1 during apoptosis.
2. Materials and methods
2.1. Chemicals
Anti-GFP and anti-cdc2 monoclonal antibodies (sc-9996) were obtained from Santa Cruz Biotechnology, Inc. Anti-GFP polyclonal antibody (A-6455) was purchased from Molecular Probes. Anti-β-tubulin mouse monoclonal antibody (T4026) was from Sigma–Aldrich, Inc. MG132 (474781) was from Calbiochem.
2.2. Mammalian cell culture and gene transfection
HeLa cells, which were obtained from American Type Culture Collection (ATCC), were cultured in minimum essential medium (MEM) containing 10% fetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin in 5% CO2 at 37 °C. The fusion genes were transfected into cells with Lipofectamine™ 2000 (Invitrogen) using the standard protocol provided by Invitrogen.
2.3. Apoptosis induction
UV irradiation was used as the inducer of apoptosis in this study. The light source was originated from the UV light equipped inside a biological safety cabinet. To induce apoptosis using UV irradiation, cells that were grown as a monolayer in a petri dish were washed with PBS once, covered with PBS and then exposed to UV light (300 mW) for 3 min. The PBS was then replaced with MEM.
2.4. Plasmid construction
The human Mcl-1 gene was kindly provided by Dr. Steven W. Edwards from the University of Liverpool [21]. Mcl-1 was amplified with primers: Forward 5′-CCGGAATTCCGATGTTTGGCCTCAAAAGAAACG-3′ and Reverse 5′-CGCG-GATCCCGCTATCTTATTAGATATGCCAAAC-3′. Then the amplified Mcl-1 was cloned into a pEGFP-C3 vector (Clontech) using EcoR I and BamH I (Roche).
The human P32 gene construct pYW59, encoding the Flag-tagged P32/TAP (1–282) gene, was kindly provided by Dr. S. Diane Hayward from the Johns Hopkins School of Medicine [23]. P32 was then cloned into a pEYFP-N1 vector (Clontech) with primers: Forward 5′-CCGCTCGAGATGCTGCCTCTGCTGCGCTG-3′ and Reverse 5′-GGAATTCCCTGGCTCT-TGACAAAACTCTTG-3′. The truncation mutants of GFP-Mcl-1 and F3 fused P32-YFP were generated by the same method as described above (Table 1).
Table 1.
List of the plasmid DNA constructs.
| Gene construction | Primers & restriction sites |
|---|---|
| GFP-Mcl-1-1-78 | Forward: CGGAATTCCGATGTTTGGCCTCAAAAGAAACG |
| Reverse: CGCGGATCCCCGCGCGACCCTCCGGG | |
| EcoR I/BamH I | |
| GFP-Mcl-1-PEST (79–183) | Forward: CGGAATTCCGCCGCCGCCCATTGGCGC |
| Reverse: CGGGATCCCGAGAGATAATCTCCAGCGACTG | |
| EcoR I/BamH I | |
| GFP-Mcl-1-BH3 (208–233) | Forward: CCCAAGCTTGGGGCGCTGGAGACCTTACGAC |
| Reverse: GAAGATCTTCCCGAAGCATGCCTTGGAAGG | |
| Hind III/Bgl II | |
| GFP-Mcl-1-BH1-BH2 (251–329) | Forward: CGGAATTCCGATCCATGTTTTCAGCGACGGC |
| Reverse: GCGGATCCCGTCACCTGATGCCACCTTCTAGG | |
| EcoR I/BamH I | |
| GFP-Mcl-1-ΔPEST | Overlapping PCR primers |
| Forward: GAAGATCTTCCCGCGCGACCCTCCGGG | |
| Reverse: GAAGATCTTCGGTACCTTCGGGAGCAGG | |
| GFP-BH1 | Forward: GGAATTCCGATCCATGTTTTCAGCGACGG |
| Reverse:CGGGATCCCGACCAAAAGAAATGAGAGTCACAATC | |
| GFP-BH2 | Forward:CGGAATTCCGGACTGGCTAGTTAAACAAAGAG |
| Reverse: CGGGATCCCGGAAGAACTCCACAAACCCATC | |
| GFP-Linker | Forward: CGGAATTCCGGCCTTTGTGGCTAAACACTTG |
| Reverse: CGGGATCCCGTTTTGTCCTTACGAGAACG | |
| GFP-Mcl-1-BH1-F1 | Forward: CCCAAGCTTGGATCCATGTTTTCAGCGACGG |
| Reverse: CCGGAATTCCGCTAGCCGTCGCTGAAAACAT | |
| GFP-Mcl-1-BH1-F2 | Forward: CCCAAGCTTGGGTAACAAACTGGGGCAGGAT |
| Reverse: CCGGAATTCCGCTAAATCCTGCCCCAGTTTG | |
| GFP-Mcl-1-BH1-F3 | Forward: CCCAAGCTTGGGTGACTCTCATTTCTTTTGG |
| Reverse: CCGGAATTCCGCTAACCAAAAGAAATGAGAG | |
| F3-P32ΔN-YFP | First PCR Primer: TTCTTTTGGTGGGGGGGGGGGGCTGC-ACACCGACGGAGAC |
| Second PCR Primer: CCGCTCGAGATGGTGACTCTCATTT-CTTTTGGTGGGGGGGGG | |
| Xho I/EcoR I |
2.5. Western blotting analysis
HeLa cells were cultured in 60 mm petri-dishes. Cells at different time points after UV treatment or gene expression were collected and lysed in NP-40 lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl and 1% NP-40) in the presence of 1× protease inhibitor cocktail. Whole cell lysates (80–100 mg/lane) were separated on 10–12% SDS–PAGE and transferred onto a Hybond ECL nitrocellulose membrane (Amersham). After blocking, the membranes were incubated for 3 h at room temperature or overnight at 4 °C with antibodies at a dilution of 1:500 or 1:1000. Then, the membranes were washed three times with 1× PBS with 0.1% of Tween-20 for 10 min, incubated with horseradish peroxidase-conjugated secondary antibody at a dilution of 1:5000 for 1 h, and ultimately developed using the ECL™ Western-blotting analysis system.
2.6. The living cell imaging system and image analysis
The GFP fusion protein over-expressed HeLa cells were examined under a fluorescent microscope equipped with a CCD camera. The image data acquired were further processed and analyzed by MetaMorph (Universal Imaging Corp.). Then the refined images were obtained using Confocal Assistant v4.0 (Bio-Rad) and Adobe Photoshop (Adobe Systems).
3. Results
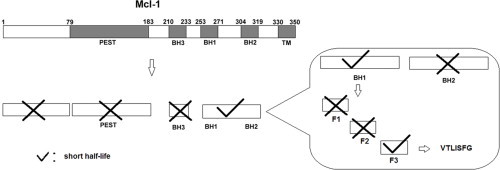
3.1. It is the BH1-BH2 domain instead of the PEST domain that has a short half-life
In order to determine which domains of Mcl-1 are responsible for its rapid turnover, we generated a series of mutants by fusing GFP with the different functional domains of Mcl-1 (Fig. 1A).
Fig. 1.
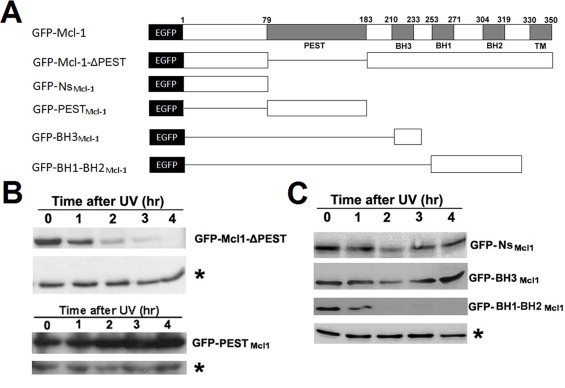
BH1-BH2 but not the PEST domain has a short half-life. (A) Schematic diagram of GFP-Mcl-1-ΔPEST, GFP-NsMcl-1, GFP-PESTMcl-1, GFP-BH3Mcl-1 and GFP-BH1-BH2Mcl-1. (B) Western blotting showing the protein amount of over-expressed GFP-Mcl-1-ΔPEST and GFP-PESTMcl-1 at different time points after UV irradiation in HeLa cells. *The protein amount of tubulin served as the loading control. (C) Western blotting showing the protein amount of over-expressed GFP-NsMcl-1, GFP-BH3Mcl-1 and GFP-BH1-BH2Mcl-1 at different time points after UV irradiation in HeLa cells. *The protein amount of tubulin served as the loading control.
It has been reported that the PEST domain, in which proline (P), glutamic acid (E), serine (S) and threonine (T) are enriched, is responsible for the short half-life of certain proteins [20]. However, researchers have also argued that the PEST domain is not responsible for the rapid turnover of Mcl-1 [21,22]. Therefore, we wanted to first test whether or not the PEST domain is responsible for its rapid turnover of Mcl-1. We examined the stability of two mutants, GFP-Mcl-1-ΔPEST and GFP-PESTMcl-1, by Western blotting analysis (see Fig. 1B). Surprisingly, the protein level of GFP-Mcl-1-ΔPEST was found to decrease rapidly, during UV-induced apoptosis, just like endogenous Mcl-1. The protein level of GFP-PESTMcl-1, on the other hand, remained largely unchanged. Fluorescence imaging also showed a high expression level of GFP-PESTMcl-1 (Fig. 2A). These findings suggest that the PEST domain is not responsible for the rapid turnover of Mcl-1.
Fig. 2.
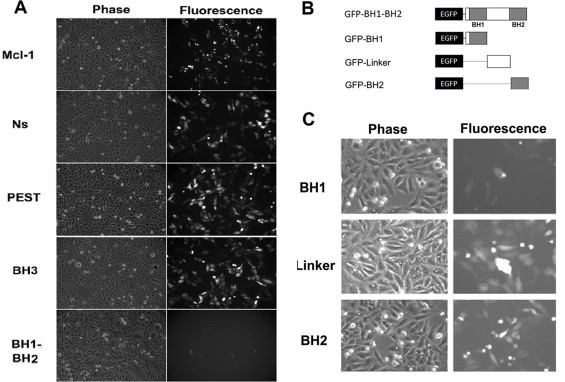
The expression of GFP fused Mcl-1 deletion mutants in HeLa cells. (A) Cells were transfected with GFP-Mcl-1 (Mcl-1), GFP-NsMcl-1 (Ns), GFP-PESTMcl-1 (PEST), GFP-BH3Mcl-1 (BH3) or GFP-BH1-BH2Mcl-1 (BH1-BH2) by lipofectamine. (B) Schematic diagram of GFP fused BH1-BH2 deletion mutants: GFP-BH1, GFP-Linker and GFP-BH2. (C) Phase contrast and fluorescent images of GFP-BH1, GFP-Linker and GFP-BH2 over-expressed HeLa cells.
Next, we examined the stability of other domains, including N-terminus (short N-terminus without PEST, Ns), BH3 and BH1-BH2. Interestingly, both the proportion of fluorescence-positive cells and the fluorescence intensity in the GFP-BH1-BH2Mcl-1 over-expressed HeLa cells were much lower than the other mutants (Fig. 2A). Also, as shown by Western blotting analysis, the protein level of GFP-BH1-BH2Mcl-1 in the HeLa cells decreased rapidly during UV-induced apoptosis, while GFP-BH3Mcl-1 and GFP-NsMcl-1 did not (Fig. 1C). These results suggest that the BH1-BH2 domain is responsible for the rapid turnover of Mcl-1.
3.2. VTLISFG was found to be the degradation motif
Our next question is to identify the essential degradation motif within the BH1-BH2 domain. As shown in Fig. 2B, we generated three GFP fused mutants by dissecting the BH1-BH2 domain into three distinct parts: GFP-BH1Mcl-1, GFP-BH2Mcl-1 and GFP-LinkerMcl-1 (the peptide between the BH1 and BH2 domains). It was found that GFP-BH1Mcl-1, but not GFP-BH2Mcl-1 or GFP-LinkerMcl-1 had a short half-life (Fig. 2C). In addition, MG132, a proteasome inhibitor, increased the expression level of GFP-BH1Mcl-1 (data not shown), suggesting that the degradation motif is inside the BH1 domain.
We further dissected the BH1 domain into three fragments: GFP-F1 (251–257), GFP-F2 (258–264) and GFP-F3 (265–271). As shown in Fig. 3, GFP-F3 (VTLISFG) but not the other two mutants displayed a short half-life and MG132 increased the expression level of GFP-F3 (data not shown). Therefore, we generated GFP fused deletion mutants of F3 to identify the minimal motifs: GFP-TLISFG and GFP-VTLISF. We found that the expression level of both mutants was significantly higher than F3, so F3 is apparently the minimal motif necessary for a short half-life (Fig. 4).
Fig. 3.
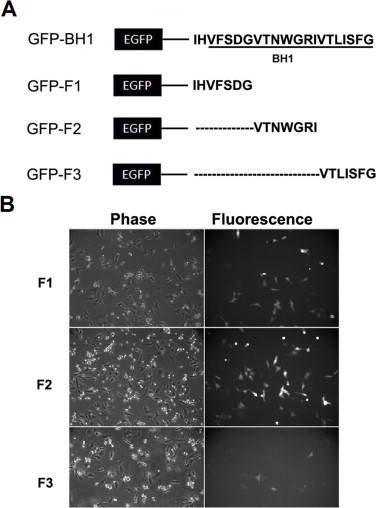
The expression of GFP fused BH1 deletion mutants in HeLa cells. (A) Schematic diagram of GFP fused BH1 deletion mutants: GFP-F1, GFP-F2 and GFP-F3. (B) Phase contrast and fluorescent images of GFP-F1, GFP-F2 and GFP-F3 over-expressed HeLa cells.
Fig. 4.
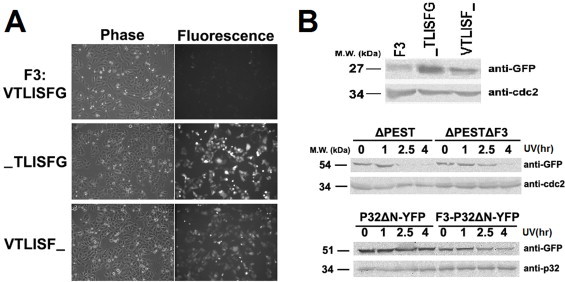
F3 is the putative degradation motif. (A) The expressions of GFP fused F3 and its single residue deletion mutants are shown by the phase contrast and fluorescent images of GFP-F3 (VTLISFG), GFP-TLISFG and GFP-VTLISF in HeLa cells. (B) Upper panel: The expression levels of GFP fused F3 and its mutants were determined by Western blotting analysis in HeLa cells. The endogenous cdc2 served as the loading control. Middle and lower panels: The dynamic protein level of over-expressed GFP-Mcl-1-ΔPEST and P32-ΔN-YFP with or without the fusion of F3 in HeLa cells after UV treatment were determined by Western blotting analysis. The endogenous cdc2 and p32 levels served as the loading controls, respectively.
The over-expression of GFP-Mcl-1 is able to significantly prevent apoptosis as well as the degradation of endogenous Mcl-1, and even the deletion of F3 did not show any significant difference from wild-type GFP-Mcl-1 (data not shown), so GFP-Mcl-1 cannot serve as the comparison control in studies on the stability of deletion mutants. Alternatively, we utilized the mutant GFP-Mcl-1-ΔPEST as the control, which displayed a similar degradation pattern as the endogenous Mcl-1 (Fig. 4B), but did not prevent apoptosis. Then we generated GFP-Mcl-1-ΔPESTΔF3 by deleting VTLISFG from GFP-Mcl-1-ΔPEST. We found that the degradation of this mutant was much slower than GFP-Mcl-1-ΔPEST in UV-induced apoptosis (Fig. 4B). This result suggests that F3 is responsible for the degradation of Mcl-1.
We also performed alignment of different F3 variants within some of the Bcl-2 family proteins. The second residue of F3 in Mcl-1 is threonine, but the second residue of other variants is alanine. Therefore, we mutated this threonine residue to alanine (VALISFG) and examined its effect. Interestingly, when we mutated serine to alanine (VTLIAFG), it made F3 much more unstable (Fig. 5B). We suspect that these point mutations may induce a conformational change in F3 or alter its capacity for post-translational modification, thus resulting in the change in the stability of F3. Further investigation will be required to elucidate why F3 is so unstable.
Fig. 5.
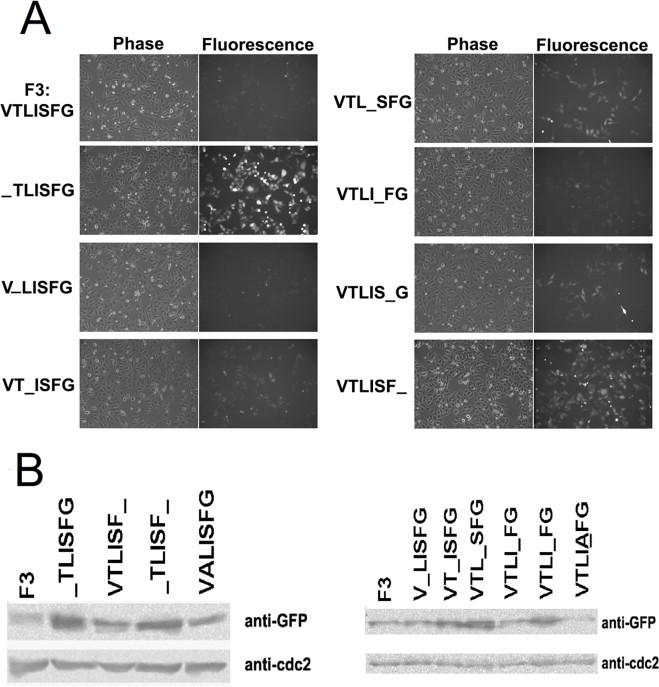
The expression of GFP fused F3 single residue deletion mutants in HeLa cells. (A) Phase contrast and fluorescent images of GFP-F3 (VTLISFG), GFP-TLISFG, GFP-VLISFG, GFP-VTISFG, GFP-VTLSFG, GFP-VTLIFG, GFP-VTLISG, and GFP-VTLISF in HeLa cells. (B) The expression of F3 mutants determined by Western blotting analysis in HeLa cells. The endogenous cdc2 served as the loading control.
3.3. F3 (VTLISFG) is a general degradation signal in apoptosis
If F3 is the degradation motif responsible for Mcl-1 degradation, inserting this motif into other proteins may also cause them to be degraded during apoptosis. Thus, we fused F3 to the N-terminus of the mature form of p32, which showed a stable protein level during UV-induced apoptosis, to generate F3-P32ΔN-YFP and examined the stability of this mutant during UV-induced apoptosis. The results are shown in the lower panel of Fig. 4B. While the protein level of P32-ΔN-YFP remained unchanged in UV-induced apoptosis, the protein level of F3-P32ΔN-YFP decreased with time. This result suggests that F3 does destabilize mature p32 and may thus be a general degradation signal for other proteins.
4. Discussion
In normal cells, anti-apoptotic Bcl-2 proteins prevent cells from undergoing accidental suicide. Upon the presence of certain apoptotic stimuli, these anti-apoptotic proteins thus need to be inactivated to allow the activation of key pro-apoptotic proteins required for apoptotic progression. The mechanisms underlying this process are highly complex. Among the anti-apoptotic Bcl-2 family proteins, Mcl-1 is unique in that it has a very short half-life and the degradation of Mcl-1 seems to be a common event in response to apoptotic stimuli [7,24,25]. Previously, the rapid turnover of Mcl-1 was attributed to its PEST region, which is reportedly responsible for the rapid turnover of many proteins with a short half-life [20]. Two pathways for the turnover of the PEST motif containing proteins have been identified: ubiquitination/proteasome mediated degradation and caspase cleavage dependent proteolysis. A lysine residue (K136) residing in the PEST region of Mcl-1 has been identified as an important poly-ubiquitination site [12]. Mcl-1 has also been reported as a substrate of caspase and its proteolysis may serve as a positive feedback for amplifying apoptotic signals after the activation of caspase [15,22,26]. However, this seems not to be the case in UV-induced apoptosis in HeLa cells, because the degradation of Mcl-1 occurs within 1 h of UV irradiation exposure, but caspase activation occurs at a much later stage. Moreover, there is evidence suggesting that the PEST domain is not responsible for the rapid turnover of Mcl-1, since the deletion of PEST does not affect the half-life of Mcl-1 [21,22]. Our findings, that GFP-Mcl-1-ΔPEST undergoes rapid turnover and GFP-PESTMcl-1 remains stable during UV-induced apoptosis (Fig. 1B), support the argument that the PEST domain is not responsible for the rapid turnover of Mcl-1.
At this point, the role of the other domains, including the short N-terminus, BH3 domain, BH1-BH2 domain and TM domain, in terms of regulating the degradation of Mcl-1 is not yet clear. In addition to the PEST region, the BH1 domain was also reported to be partially responsible for the degradation of Mcl-1 [27], but there are few reports concerning the role of other domains in regulating the degradation of Mcl-1. Therefore, we examined the stability of all these domains and identified the BH1-BH2 domain as the putative degradation motif. We speculated that the short half-life of BH1-BH2 is due to poly-ubiquitination meditated degradation, so we generated a non-ubiquitinated BH1-BH2 mutant (GFP-BH1-BH2-4KR) in which all the lysine residues were mutated to arginine. This mutant still exhibited a short half-life (data not shown). Then, we further dissected the BH1-BH2 domain into BH1, Linker and BH2, and found that the BH1 domain, but not the Linker or BH2 domain, exhibited a short half-life. Since there are no lysine residues inside the BH1 domain, this further demonstrated that poly-ubiquitination meditated degradation does not account for the short half-life of the BH1-BH2 domain.
A peptide named F3 (VTLISFG) residing in the C-terminus of the BH1 domain was identified as the putative degradation motif. Even the deletion of a single amino acid residue increased the expression of F3, so VTLISFG seemed to obviously be the minimum degradation motif. Notably, the deletion of hydrophobic residues (including V, L, I, F and G) almost completely abrogated the short half-life of F3, but deletion of hydrophilic residues (including T and S) resulted in a slightly promotive effect on the expression of F3. Therefore, the significance of each amino acid residue in determining the half-life of F3 varies and the hydrophobicity may in part determine the short half-life (Fig. 5).
The rapid turn-over property of F3 does not seem to be limited to the short half-life of Mcl-1 and it may in fact be a general degradation signal, since it is able to cause the degradation of ostensibly non-degradable mature p32 during UV-induced apoptosis. Deletion of F3 retarded, but did not completely prevent the degradation of Mcl-1 mutants, so F3 seems not to be the only degradation signal. Other mechanisms (such as poly-ubiquitination) may still be involved in regulating rapid turnover of Mcl-1. It has been reported recently that poly-ubiquitination by itself is not sufficient for the rapid degradation of tightly folded proteins and an additional unstructured region is required to traffic folded proteins into 26S/20S proteasomes [28]. Therefore, it is possible that F3 functions as an unstructured region that engages proteins for trafficking into proteasomes where they are unfolded and degraded. However, the detailed mechanisms are not well elucidated and further investigations will be needed to determine the specific role of F3 in regulating the degradation of Mcl-1 in the course of apoptosis.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
We thank Dr. Steven W. Edwards in University of Liverpool for providing pEGFP-C3-Mcl-1 gene and Dr. S. Diane Hayward in Johns Hopkins School of Medicine for providing the P32/TAP (1–282) gene. We also thank Mr. Mu Li, Director of Joint Biology Laboratory of BGI and Shenzhen Middle School, for his firm support in the data summarizing, manuscript writing and submitting. This work was supported by the grants from the Research Grants Council of Hong Kong (HKUST6466/05M, N_HKUST616/05 and 660207) as well as the grants from Shenzhen Middle School.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Oltvai Z.N., Milliman C.L., Korsmeyer S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 2.Adams J.M., Cory S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 2001;26:61–66. doi: 10.1016/s0968-0004(00)01740-0. [DOI] [PubMed] [Google Scholar]
- 3.Borner C. The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol. Immunol. 2003;39:615–647. doi: 10.1016/s0161-5890(02)00252-3. [DOI] [PubMed] [Google Scholar]
- 4.Kozopas K.M., Yang T., Buchan H.L., Zhou P., Craig R.W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craig R.W. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 2002;16:444–454. doi: 10.1038/sj.leu.2402416. [DOI] [PubMed] [Google Scholar]
- 6.Iglesias-Serret D., Pique M., Gil J., Pons G., Lopez J.M. Transcriptional and translational control of Mcl-1 during apoptosis. Arch. Biochem. Biophys. 2003;417:141–152. doi: 10.1016/s0003-9861(03)00345-x. [DOI] [PubMed] [Google Scholar]
- 7.Nijhawan D., Fang M., Traer E., Zhong Q., Gao W., Du F., Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inukai N., Yamaguchi Y., Kuraoka I., Yamada T., Kamijo S., Kato J., Tanaka K., Handa H. A novel hydrogen peroxide-induced phosphorylation and ubiquitination pathway leading to RNA polymerase II proteolysis. J. Biol. Chem. 2004;279:8190–8195. doi: 10.1074/jbc.M311412200. [DOI] [PubMed] [Google Scholar]
- 9.Lee K.B., Wang D., Lippard S.J., Sharp P.A. Transcription-coupled and DNA damage-dependent ubiquitination of RNA polymerase II in vitro. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4239–4244. doi: 10.1073/pnas.072068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y., Luo Z., Bregman D.B. RNA polymerase II large subunit is cleaved by caspases during DNA damage-induced apoptosis. Biochem. Biophys. Res. Commun. 2002;296:954–961. doi: 10.1016/s0006-291x(02)02028-4. [DOI] [PubMed] [Google Scholar]
- 11.Yamada A., Masutani C., Hanaoka F. Detection of reduced RNA synthesis in UV-irradiated Cockayne syndrome group B cells using an isolated nuclear system. Biochim. Biophys. Acta. 2002;1592:129–134. doi: 10.1016/s0167-4889(02)00292-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhong Q., Gao W., Du F., Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell. 2005;121:1085–1095. doi: 10.1016/j.cell.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Ding Q., He X., Hsu J.M., Xia W., Chen C.T., Li L.Y., Lee D.F., Liu J.C., Zhong Q., Wang X., Hung M.C. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol. Cell. Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwickart M., Huang X., Lill J.R., Liu J., Ferrando R., French D.M., Maecker H., O’Rourke K., Bazan F., Eastham-Anderson J., Yue P., Dornan D., Huang D.C., Dixit V.M. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010;463:103–107. doi: 10.1038/nature08646. [DOI] [PubMed] [Google Scholar]
- 15.Herrant M., Jacquel A., Marchetti S., Belhacene N., Colosetti P., Luciano F., Auberger P. Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene. 2004;23:7863–7873. doi: 10.1038/sj.onc.1208069. [DOI] [PubMed] [Google Scholar]
- 16.Gomez-Bougie P., Oliver L., Le Gouill S., Bataille R., Amiot M. Melphalan-induced apoptosis in multiple myeloma cells is associated with a cleavage of Mcl-1 and Bim and a decrease in the Mcl-1/Bim complex. Oncogene. 2005;24:8076–8079. doi: 10.1038/sj.onc.1208949. [DOI] [PubMed] [Google Scholar]
- 17.Snowden R.T., Sun X.M., Dyer M.J., Cohen G.M. Bisindolylmaleimide IX is a potent inducer of apoptosis in chronic lymphocytic leukaemic cells and activates cleavage of Mcl-1. Leukemia. 2003;17:1981–1989. doi: 10.1038/sj.leu.2403088. [DOI] [PubMed] [Google Scholar]
- 18.Michels J., O’Neill J.W., Dallman C.L., Mouzakiti A., Habens F., Brimmell M., Zhang K.Y., Craig R.W., Marcusson E.G., Johnson P.W., Packham G. Mcl-1 is required for Akata6 B-lymphoma cell survival and is converted to a cell death molecule by efficient caspase-mediated cleavage. Oncogene. 2004;23:4818–4827. doi: 10.1038/sj.onc.1207648. [DOI] [PubMed] [Google Scholar]
- 19.Yang T., Kozopas K.M., Craig R.W. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J. Cell Biol. 1995;128:1173–1184. doi: 10.1083/jcb.128.6.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rechsteiner M., Rogers S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- 21.Akgul C., Moulding D.A., White M.R., Edwards S.W. In vivo localisation and stability of human Mcl-1 using green fluorescent protein (GFP) fusion proteins. FEBS Lett. 2000;478:72–76. doi: 10.1016/s0014-5793(00)01809-3. [DOI] [PubMed] [Google Scholar]
- 22.Clohessy J.G., Zhuang J., Brady H.J. Characterisation of Mcl-1 cleavage during apoptosis of haematopoietic cells. Br. J. Haematol. 2004;125:655–665. doi: 10.1111/j.1365-2141.2004.04949.x. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y., Finan J.E., Middeldorp J.M., Hayward S.D. P32/TAP, a cellular protein that interacts with EBNA-1 of Epstein–Barr virus. Virology. 1997;236:18–29. doi: 10.1006/viro.1997.8739. [DOI] [PubMed] [Google Scholar]
- 24.Chao J.R., Wang J.M., Lee S.F., Peng H.W., Lin Y.H., Chou C.H., Li J.C., Huang H.M., Chou C.K., Kuo M.L., Yen J.J., Yang-Yen H.F. mcl-1 is an immediate-early gene activated by the granulocyte-macrophage colony-stimulating factor (GM-CSF) signaling pathway and is one component of the GM-CSF viability response. Mol. Cell. Biol. 1998;18:4883–4898. doi: 10.1128/mcb.18.8.4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H., Peng H.W., Cheng Y.S., Yuan H.S., Yang-Yen H.F. Stabilization and enhancement of the antiapoptotic activity of mcl-1 by TCTP. Mol. Cell. Biol. 2005;25:3117–3126. doi: 10.1128/MCB.25.8.3117-3126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weng C., Li Y., Xu D., Shi Y., Tang H. Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Jurkat leukemia T cells. J. Biol. Chem. 2005;280:10491–10500. doi: 10.1074/jbc.M412819200. [DOI] [PubMed] [Google Scholar]
- 27.Mei Y., Du W., Yang Y., Wu M. Puma(*)Mcl-1 interaction is not sufficient to prevent rapid degradation of Mcl-1. Oncogene. 2005;24:7224–7237. doi: 10.1038/sj.onc.1208873. [DOI] [PubMed] [Google Scholar]
- 28.Prakash S., Tian L., Ratliff K.S., Lehotzky R.E., Matouschek A. An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat. Struct. Mol. Biol. 2004;11:830–837. doi: 10.1038/nsmb814. [DOI] [PubMed] [Google Scholar]