

Abstract
Donohue syndrome (DS) is a rare and lethal autosomal recessive disease caused by mutations in the insulin receptor (INSR) gene, manifesting marked insulin resistance, severe growth retardation, hypertrichosis, and characteristic dysmorphic features. We report the clinical, molecular, and biochemical characterization of three new patients with DS, and address genotype–phenotype issues playing a role in the pathophysiology of DS. A female infant born to first-degree cousins Muslim Arab parents and two brothers born to first-degree cousins Druze parents presented classical features of DS with hypertrophic cardiomyopathy and died in infancy. Each patient was found homozygous for one missense mutation within the extracellular domain of the INSR gene. Western blot analysis identified the proreceptor of INSR, but not its mature subunits alpha and beta. Of 95 healthy Muslims, no heterozygous was found and of 52 healthy Druze from the same village, one was heterozygous. This study presents two novel familial mutations in the alpha subunit of the INSR which appear to impair post-translational processing of the INSR, resulting loss of its function. Both mutations cause DS with hypertrophic cardiomyopathy and early death. Identification of the causative mutation enables prevention of this devastating disease.
Keywords: Cardiomyopathy, Donohue syndrome, genotype–phenotype, insulin receptor.
Introduction
Single-gene defects are responsible for hyperglycemia in only a minority of individuals. Mutations affecting both insulin production and insulin sensitivity have been identified. Maturity onset diabetes in the young is a monogenic type of diabetes in which a mutation in an autosomal dominant gene disrupts insulin production (Winckler et al. 2007). Donohue syndrome (DS, OMIM#246200), also known as Leprechaunism, Rabson–Mendenhall syndrome, and type A insulin resistance are autosomal recessive (AR) disorders caused by biallelic mutations in the gene encoding the insulin receptor (INSR, OMIM#147670). These syndromes, sharing phenotype and genotype heterogeneity, are distinguished from one another based on the severity of symptoms, age of onset, and age of death (Porter and Barrett 2005). DS is considered the most severe syndrome of the group, and is usually lethal within 2 years of life (Musso et al. 2004; Semple et al. 2011; Grasso et al. 2013).
Donohue syndrome is characterized by markedly delayed linear growth and failure to thrive (FTT), loss of glucose homeostasis, hyperinsulinemia, thick skin with lack of subcutaneous fat, acanthosis nigricans (AN), distended abdomen, enlarged genitalia in the male and cystic ovaries in the female, and dysmorphic facial features: elfin faces with prominent eyes, thick lips, upturned nostrils, and low-set posterior rotated ears (Geffner et al. 1987; al-Gazali et al. 1993).
A wide spectrum of disorders caused by diverse monogenic etiologies resembles DS. Specifically, Berardinelli–Seip congenital lipodystrophy is a condition associating insulin resistance, absence of subcutaneous fat, AN, and muscular hypertrophy caused by mutation in either AGPAT2 (OMIM#603100) or BSCL2 (OMIM# 606158) (Friguls et al. 2009; Miranda et al. 2009), AN associating with severe skeletal dysplasias due to activating mutations in FGFR3 (OMIM#134934) (Alatzoglou et al. 2009) are just a few examples.
The association between diabetes and cardiovascular disease is well recognized (Kannel and McGee 1979). Furthermore, evidence for insulin resistance has been shown to associate with cardiovascular disease, and specifically, with hypertrophic cardiomyopathy (HCM), also in individuals without diabetes (Murakami et al. 2004; Bonora et al. 2007; Verhagen et al. 2011). Recently, a large meta-analysis substantiated the association between metabolic syndrome and cardiovascular disease (Mottillo et al. 2010). Insulin resistance is a central component of both metabolic syndrome and DS. Several reports (Baykan et al. 2008; Nobile et al. 2012; Hovnik et al. 2013) argue that the underlying mechanisms for cardiomyopathy in DS and metabolic syndrome involve excess insulin activation of Insulin-like growth factor 1 (IGF1) receptors [6].
In this study we characterized two novel missense mutations in the INSR causing DS with HCM.
Material and Methods
Patients
The patients were three newborns from two unrelated families who were hospitalized in neonatal intensive care units: a female born to first-degree cousins Muslim Arab parents (named patient ISR1) and two brothers with first-degree cousins Druze parents (named patients ISR2 and ISR3). The IRB of Nahariya Medical Center and the Israeli Ministry of Health approved the study.
Clinical examination
Family history was taken and physical examinations conducted. Imaging studies of the brain, abdomen, and heart were performed. Biochemical workup included renal and liver function tests, glucose, insulin, C-peptide, and glucagon levels. Blood was drawn for molecular studies, and fibroblast cells cultures were established from skin biopsies of patients ISR1 and ISR2 as previously described (Falik-Zaccai et al. 2008b).
Mutation analysis in the insulin receptor gene
For patient ISR 1
Studies were carried to locate the INSR underlying mutation by denaturing high-performance liquid chromatography (dHPLC) of each of the 22 INSR exons, including exon-intron boundaries. Exons demonstrating abnormal pattern, compared with control, in the dHPLC screening were further sequenced for determining possible DNA polymorphism. Results were confirmed using Allele-specific oligonucleotide (ASO) using radiolabelled oligoprobes: 5′-TCAGCTTCTGCCAGGACC-3′ (wild type) and 5′-TCAGCTTCTACCAGGACC-3′ (mutant). Population screening of 95 Arab Muslims was carried out using Sanger sequencing of a 347 bp amplicon containing the p.C286Y mutation (Macrogen Inc, Amsterdam, Netherlands).
For patients 2 and 3
The 22 exons of the INSR (NM_000208.2) were Sanger sequenced and analyzed using the ABI PRISM 3130 Genetic Analyzer (Applied Biosystems, Warrington, U.K.) according to the manufacturer's instructions.
For the Druze patients, sequencing results were confirmed and healthy controls were examined by chain reaction (PCR) amplification of exon 2 using the primers:
INSR_ex2 (1) F: 5′- GAT GAA AAC ACA GGG CCC AG- 3′
INSR_ex2(1) R: 5′- CTC CAC GGA ATC CAG GAT AC -3′
The reaction was followed by enzymatic digestion using the restriction enzyme TaqI (New England Biolabs, Ipswich, MA).
Immunoprecipitation and immunoblotting analysis
Protein lysate was prepared according to standard protocols and studies were carried out as detailed elsewhere (Wertheimer et al. 1993). Antibodies used for immunoblotting and immunoprecipitation procedures included monoclonal antibody recognizing phosphotyrosine residues (Upstate Biotechnology, Inc., Lake Placid, NY) and rabbit polyclonal antibodies against the insulin receptor beta subunit (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Prenatal diagnosis
Prenatal diagnosis (PND) was performed for the Arab Muslim family. After comprehensive genetic counseling, chorionic villous sampling (CVS) was performed. DNA was extracted from the villi and exon 3 of the INSR gene sequenced.
Results
Patient characteristics
All three patients presented with the classical elfin features characteristic of DS: coarse face, bulging eyes, and thick lips, absence of subcutaneous fat, hirsutism, and nipple hypertrophy. Laboratory tests revealed direct hyperbilirubinemia, and elevated Gamma Glutamyltransferase. Other liver and kidney function tests were normal. All patients suffered from intrauterine growth restriction (IUGR), FTT, and HCM.
The diagnosis of DS was established based on the above clinical characteristics, and determination of the INSR mutation. The particular features and disease course, and the biochemical tests of the patients, are depicted in Figure 1 and summarized in Table 1, respectively.
Figure 1.
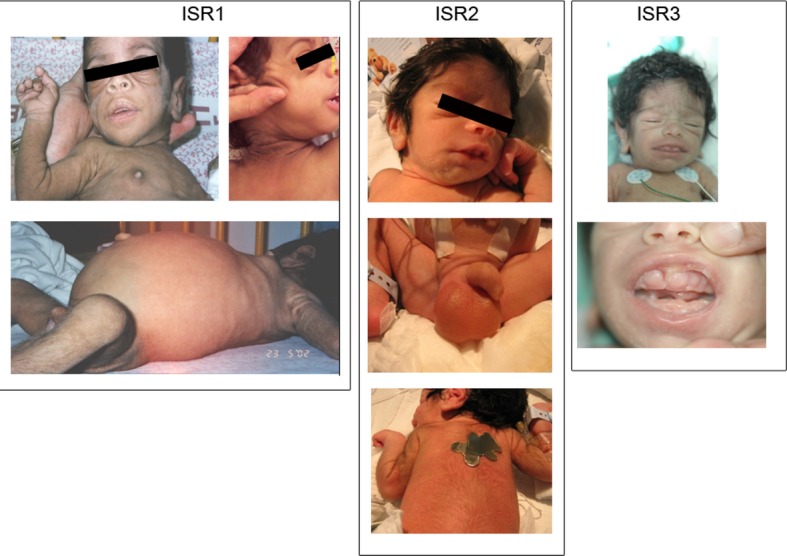
Clinical features of patients. See description in text.
Table 1.
Clinical and laboratory characteristics of the three patients.
| Patient number and sex (M/F) | Birth weight and gestational age | Head circumference | Insulin levels N = 5–25 | C-peptide levels N = 298–1324 | Age at death |
|---|---|---|---|---|---|
| ISR1 – F | 1700 g (−2.2 SD); 36 weeks | 32.5 cm (−2 SD) at 14 days | 3560 IU/mL (age: 1 month) | 15900 pmol/L (age: 1 month) | 18 months |
| ISR2 – M | 1540 g (−2 SD); 34 weeks | 31 cm (−3 SD) at birth | 4500 IU/mL (at birth) | Not available | 40 days |
| ISR3 – M | 1770 g (−2.5 SD); 38 weeks | 32.6 cm (−2 SD) at birth | 2761, 4300, 21525 IU/mL (at birth) | 24825 pmol/L | 12 months |
N, normal.
Family 1
Patient ISR1, the second child of first-degree cousins Muslim Arab parents, presented at age 14 days with abdominal distention and restlessness. Her weight was 1.900 g (−2.2 SD). She presented with classical physical signs of DS including hypertrophy of the labia majora and clitoromegaly. Ultrasound of the abdomen showed ovaries with bilateral multicystic masses (10 × 10 × 20 mm; 7 × 13 × 15 mm) and enlarged kidneys.
The clinical course showed no weight gain, episodes of hypoglycemia, AN, distended abdomen, and rectal prolapse. Echocardiography revealed hypertrophy of the left ventricle. Abdominal ultrasound demonstrated enlarged kidneys with medullary calcinosis and further enlargement of the ovaries: L – 50 × 70 × 75 mm, and R – 20 × 40 × 45 mm. The following months were typified by recurrent infections including urosepsis, bilateral otitis media, and pneumonia, and failure to reach neurological milestones. At age 18 months the patient died due to aspiration pneumonia.
Family 2
Patient ISR2, the third child of first-degree cousins Druze parents, was born at 34 weeks gestation. He presented with IUGR, dysmorphic features, and hirsutism (Table 1), hepatosplenomegaly, and hypotonicity. Ultrasound examination during pregnancy revealed polyhydramnion and enlarged kidneys, bladder, and stomach. Ultrasound imaging of the abdomen revealed hepatosplenomegaly and enlarged kidneys.
The clinical course showed FTT, and episodes of alternating hypoglycemia and hyperglycemia. At age 3 days a 2–3/6 systolic heart murmur was heard. Echocardiogram revealed severe HCM associated especially with hypertrophy of the left ventricle and septum. DS was diagnosed based on the clinical characteristics, and the identified causative mutation in INSR. At 2 weeks Klebsiella sepsis was diagnosed and treated. At age 40 days the infant died due to respiratory failure and cardiac arrest.
Patient ISR3, the brother of patient 2 (Fig. 2E), was born after an uneventful 38-week pregnancy, presenting symmetric IUGR. Ultrasound examination during pregnancy revealed an enlarged heart. He resembled his deceased brother both clinically and biochemically (Table 1). Echocardiography revealed hypertrophy of the left ventricle and mild pulmonic stenosis.
Figure 2.

Mutation analysis. Genomic DNA sequence analysis revealed a dHPLC abnormal pattern of exon 3 of patient ISR1; the novel c.858G>A mutation was detected (A). The mutation was further confirmed by allele-specific oligonucleotide hybridization (B). High conservation of p.C286 throughout the phylogenetic tree (C). 3 genomic DNA sequence analysis of the 22 exons of INSR gene revealed a homozygous T > C transition at nucleotide 167 in exon 2 (D). The family pedigree was drawn according to restriction digestion with TaqI enzyme (E). Fragmented polymerase chain reaction (PCR) products were visualized by ethidium bromide-stained acrylamide gel. P, PCR product (E). The amino acid residues Cysteine at position 286 (C) and Isoleucine at position 56 (F) in the INSR are shown to be conserved throughout the phylogenetic tree. Amino acid conservation was analyzed by NCBI Basic Local Alignment Search Tool, using protein blast.
At age 3 months, the patient suffered from FTT (weight 3.1 kg), AN, distended abdomen, rectal prolapse, and a right inguinal hernia. Hypothyroidism was diagnosed at the age of 2 months and 50 μg of eltroxin were administered daily. At age 7 months FTT was severe, and bilateral inguinal hernias hypotonia and severe cardiac hypertrophy were present. Liver enzymes were elevated. Clotting functions were abnormal, with factor 7 deficiency. MRI of the brain failed to demonstrate the neuronal pituitary gland. The stalk and the anterior part of the pituitary were normal. Maturation of white matter was described to be slow. At age 10 months urosepsis was diagnosed, and ampicillin and gentamicin were administered. Body weight was 4125 g (−6 SD) and head circumference 42 cm (−3 SD). At age 11 months right upper lobe pneumonia was detected. At age 12 months, the patient died following an episode of atrial fibrillation.
Mutation analysis
Patient ISR1 was found to be homozygous for a novel missense mutation c.858G>A, substituting cysteine to tyrosine at position 286 (p.C286Y) in exon 3 of the INSR gene within the extracellular alpha subunit (Fig. 2A). This was confirmed by allele-specific oligonucleotide (Fig. 2B). p.C286 is found to be highly conserved throughout the phylogenetic tree, suggesting a strong functional value for this amino acid position (Fig. 2C).
Patient ISR3 was found to be homozygous for a novel missense mutation, T > C transition at nucleotide 167 in exon 2 (c.167T>C) of the INSR (Fig. 2D), resulting in isoleucine substitution to threonine at position 56 (p.I56T) within the extracellular alpha subunit. p.I56 is also found to be conserved throughout the phylogenetic tree, suggesting a strong functional value for this amino acid as well (Fig. 2F). Restriction enzyme analysis showed the mutation to be in full segregation in the patient's family. Patient ISR2 was found to be homozygous for the same mutation. Parents are heterozygous carriers, a healthy brother and sister are heterozygous carriers, and another healthy sister did not carry the mutation (Fig. 2E).
Prenatal diagnosis
Prenatal diagnosis was performed for family 1 three times via CVS using sequencing analysis. One fetus was found to be homozygous for the causative mutation and the pregnancy was terminated. The two other fetuses were found to be heterozygous and two healthy babies were born as predicted.
Population screening
Screening of 95 healthy Muslims revealed no heterozygous carriers for the c.858G>A mutation. Of 52 healthy individuals from the Druze patient's village, one carrier for the novel mutation c.167T>C was found.
In vitro characterization of the insulin receptor
To study the effect of the detected genetic alterations on INSR function, we established fibroblast cultures from skin biopsies taken from patients 1 and 3. Figure 3 shows absence of tyrosine phosphorylation at endogenous state (Fig. 3A), and following induction of INSR precipitation by insulin (Fig. 3B), indicating a lack of functionality of the receptor in the two DS fibroblasts compared to normal fibroblast. In comparison, it appears that the expression and phosphorylation of the closely related IGF-1 receptor in response to IGF-1 was normal (Fig. 3C).
Figure 3.
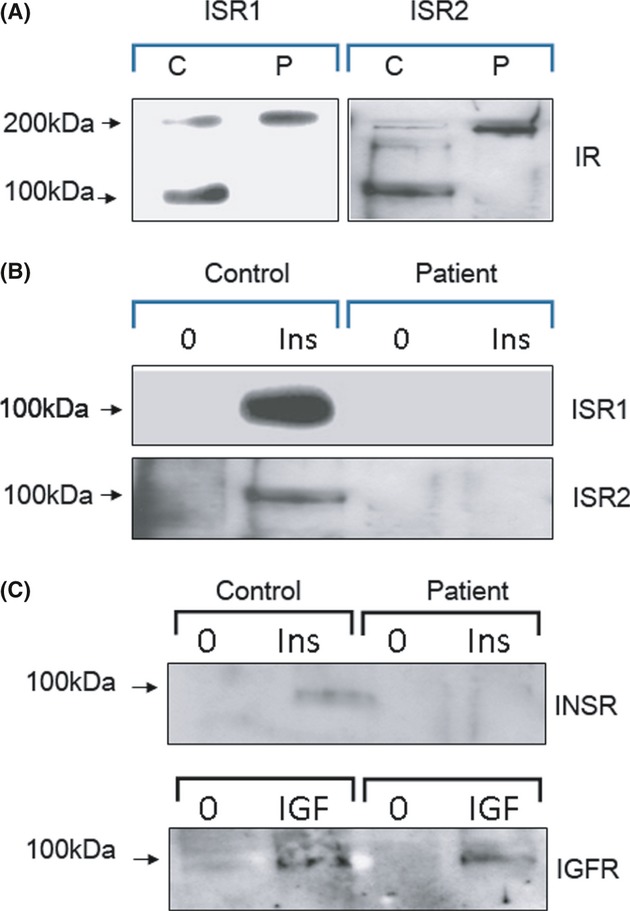
INSR determination and functionality. The proreceptor but not the mature subunits in the ISR1 and ISR2 fibroblasts (labeled P) is detected, compared with both forms identified in the healthy control (labeled C) (A). Fibroblasts were treated with 1 μmol/L of insulin (B) or IGF1(C), immunoprecipitated with an antibody against phosphorylated tyrosine, and immunoblotted with an antibody against INSR or IGF receptor, respectively.
To identify the reason for lack of INSR phosphorylation, we investigated INSR intracellular processing. Western blot analysis of INSR, revealed the proreceptor at position 210 kDa, but not the mature alpha and beta subunits (∼130 and 95–100 kDa, respectively), providing strong evidence that the two mutated amino acids disrupted the normal processing of INSR.
Discussion
We have ascertained three new patients with DS based on their clinical appearance and the identification of two familial causative novel missense mutations in INSR. The phenotype of all three was similar including HCM, and survival did not exceed 18 months.
The INSR is transcribed as a single glycosylated precursor, which after transport from the endoplasmic reticulum to the Golgi apparatus is further glycosylated and then cleaved into extracellular alpha domains comprising ligand-binding activity and beta subunits, encompassing intracellular tyrosine kinase activity. These subunits are subsequently transported to the plasma membrane as a α2β2 heterotetramer (Seino et al. 1989).
The two novel missense mutations described herein are located in the extracellular domain of INSR, and led to impair processing of the receptor. According to the tertiary structure of the ectodomain of INSR, the mutation presenting in patients ISR2 and ISR3 (p.I56T) is located within the L1 domain, and the mutation presenting in patient ISR1 (p.C286Y) in the cysteine-rich domain (Desbois-Mouthon et al. 1997; McKern et al. 2006). The severe biochemical and clinical consequences of this latter mutation highlights the crucial role of the cysteine residues in the INSR ligand-binding domain, not only in ligand binding, but also in stabilizing the three-dimensional IR structure affecting intracellular INSR processing.
Phenotypic heterogeneity has been demonstrated in a number of documentations of DS (Semple et al. 2011). For example; One patient who carried a homozygous deletion of the entire INSR gene, and thus absolute lack of insulin receptor activity, survived for 3.5 years before dying from postoperative complications (Wertheimer et al. 1993), contrasting with another infant with almost no insulin receptor activity consequent to a nonsense mutation at position 121, who failed to thrive, and died at 16 weeks of age (Krook et al. 1993). These two patients present a most striking discrepancy between genotype and phenotype in DS. Both carried what seemed to be complete inactivation of the INSR protein; however, one survived longer than all expectations, whereas the other died after a very short period. Maassen et al. found that the degree of insulin binding among five patients with defects in the INSR did not correspond to the severity of the clinical phenotype (Maassen et al. 2003). These data suggest that the absence of the INSR protein is not identical to absence of the gene itself, and that activity of transcriptional factors may also have an important effect.
Among the 100 missense mutations reported to date (Stenson et al. 2009), phenotypic variability has been shown to occur. Both novel mutations described here lie in the extracellular region and contain the insulin-binding region and a cysteine-rich domain, presenting severe type of DS. In contrast, Ahmad et al. (2013) reported a c.659C>T substitution in exon 3, which is in the same domain of the INSR, presenting a mild phenotype of AN with normal fasting and postprandial blood glucose levels. The same mutation was reported by Carrera et al. (1993) to cause Rabson–Mendenhall syndrome.
The Phenotypic variability caused by single missense mutation has been suggested to be due to the coexistence of an additional sequence variation in a modifier gene. Candidate genes might be BSCL2, AGPAT2, CAV1 (OMIM#601047), and PTRF (OMIM#603198), known to cause primarily generalized lipodystrophy with similar clinical manifestations of insulin resistance and hyperinsulinism, AN, and more (Rahman et al. 2013). Moreover, the phenotypic variability might result from various degrees of inactivation of INSR transcription, leading to diverse activation of compensatory pathways, as has been shown in many cases of protein inactivation in transgenic animals. If such a scenario exists in DS, the most likely compensatory pathway might be through the closely related IGF-1 receptor.
DS appears to result from either homozygous or compound heterozygous mutations. Approximately 130 mutations have been reported to be causative of DS so far (Stenson et al. 2009). As a classic AR trait homozygous or compound heterozygous mutations are expected to cause the phenotype while heterozygous individuals are healthy carriers and free of symptoms. This is the case in the two families reported here. However, there are reports of individuals heterozygous for other mutations in INSR who were symptomatic with hyperandrogenism, AN, hyperinsulinemia, and insulin resistance, as well as Rabson–Mendenhall and type A syndrome (Takahashi et al. 2010; Kadowaki et al. 1990; Wertheimer et al. 1994), suggesting again the possibility of a modifier gene involved in the phenotype.
In this study, all three patients presented cardiomyopathy with hypertrophy of the left ventricle and septum. The families did not consent to a histopathology analysis. HCM was previously documented as a manifestation of DS (Baykan et al. 2008); it has also been associated with less severe states of elevated insulin levels, such as in infants of mothers with diabetes (Russell et al. 2008). Furthermore, insulin resistance has been associated with left ventricular diastolic dysfunction (LVDD) in adults without diabetes (Dinh et al. 2010). Accumulating evidence suggests that reactive oxygen species (ROS) may have a role in insulin-resistant cardiomyopathy (Mellor et al. 2010) and that ROS may explain some of the pathophysiology of DS (Park et al. 2010). It is yet unclear if cardiomyopathy develops due to insulin resistance or to hyperinsulinemia. Investigation of congenital syndromes of impaired function in the insulin receptor may elucidate effects of insulin that are not known to be related to obesity or diabetes. We present here an example of HCM associated with mutations within the extracellular domain of the INSR gene. We do not know the degree to which the cardiomyopathy presenting in all three patients resulted in their early death. The congruence of insulin resistance and cardiomyopathy in the two novel mutations described here further supports a common genetic basis for insulin resistance and cardiovascular disease.
Parents of the three patients presented here are first-degree cousins who reside in villages with high rates of consanguineous marriages. Prevalence rates of rare AR diseases are high among the Druze and Arab Muslim populations (Falik-Zaccai et al. 2008a,b2008b, 2010). Each patient might be the tip of an iceberg indicating the presence of high-risk population for a devastating disease. Therefore, following identification of a new mutation in an isolated village we perform small-scale screening to identify high-risk populations for severe AR rare diseases (Falik-Zaccai et al. 2008a,b2008b, 2010). The fact that DS does not occur at elevated frequency in the Muslim and Druze population studied here suggests the identification of “private, familial” mutations.
High dosages of insulin (Casati et al. 2010) and recombinant human insulin-like growth factor-1 (rhIGF-1) (Kitamei et al. 2005) have been used to treat DS; the latter was considered to have played a role in the development of diabetes retinopathy (Kitamei et al. 2005). When the clinical course is fatal and treatment is not effective, as with our patients, then identification of the causal mutation and PND is of particularly importance. Characterization of the causative mutations enabled accurate genetic counseling and PND for the DS described.
Elucidation of correlations between genotypes and phenotypes of congenital syndromes of impaired function of the INSR may contribute to the understanding of the process of insulin action in both healthy and pathological states.
Conflict of Interest
None declared.
References
- Ahmad S, Mahmoudi H, Naeem M, Betz RC. Autosomal recessive isolated familial acanthosis nigricans in a Pakistani family due to a homozygous mutation in the insulin receptor gene. Br. J. Dermatol. 2013;169:476–478. doi: 10.1111/bjd.12293. [DOI] [PubMed] [Google Scholar]
- Alatzoglou KS, Hindmarsh PC, Brain C, Torpiano J, Dattani MT. Acanthosis nigricans and insulin sensitivity in patients with achondroplasia and hypochodroplasia due to FGFR3 mutations. J. Clin. Endocrinol. Metab. 2009;94:3959–3963. doi: 10.1210/jc.2009-0322. [DOI] [PubMed] [Google Scholar]
- Baykan A, Cansever M, Konuskan B, Nihal H, Kazim U, Nazmi N. Hypertrophic cardiomyopathy with leprechaunism. J. Pediatr. Endocrinol. Metab. 2008;21:317–318. doi: 10.1515/jpem.2008.21.4.317. [DOI] [PubMed] [Google Scholar]
- Bonora E, Kiechl S, Willeit J, Oberhollenzer F, Egger G, Meigs JB, et al. Insulin resistance as estimated by homeostasis model assessment predicts incident symptomatic cardiovascular disease in caucasian subjects from the general population: the Bruneck study. Diabetes Care. 2007;30:318–324. doi: 10.2337/dc06-0919. [DOI] [PubMed] [Google Scholar]
- Carrera P, Cordera R, Ferrari M, et al. Substitution of Leu for Pro-193 in the insulin receptor in a patient with a genetic form of severe insulin resistance. Hum. Mol. Genet. 1993;2:1437–1441. doi: 10.1093/hmg/2.9.1437. [DOI] [PubMed] [Google Scholar]
- Casati S, Zoppini G, Muggeo M, Marchini G. Sustained regression of florid diabetic retinopathy in a patient with Donohue syndrome (leprechaunism) Eur. J. Ophthalmol. 2010;20:224–247. doi: 10.1177/112067211002000133. [DOI] [PubMed] [Google Scholar]
- Desbois-Mouthon C, Magré J, Duprey J, et al. Major circadian variations of glucose homeostasis in a patient with Rabson-Mendenhall syndrome and primary insulin resistance due to a mutation (Cys284–>Tyr) in the insulin receptor alpha-subunit. Pediatr. Res. 1997;42:72–77. doi: 10.1203/00006450-199707000-00012. [DOI] [PubMed] [Google Scholar]
- Dinh W, Lankisch M, Nickl W, et al. Insulin resistance and glycemic abnormalities are associated with deterioration of left ventricular diastolic function: a cross-sectional study. Cardiovasc. Diabetol. 2010;9:63. doi: 10.1186/1475-2840-9-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falik-Zaccai TC, Laskar M, Kfir N, Nasser W, Slor H, Khayat M. Cockayne syndrome type II in a Druze isolate in Northern Israel in association with an insertion mutation in ERCC6. Am. J. Med. Genet. A. 2008a;146A:1423–1429. doi: 10.1002/ajmg.a.32309. [DOI] [PubMed] [Google Scholar]
- Falik-Zaccai TC, Kfir N, Frenkel P, et al. Population screening in a Druze community: the challenge and the reward. Genet. Med. 2008b;10:903–909. doi: 10.1097/GIM.0b013e31818d0e0f. [DOI] [PubMed] [Google Scholar]
- Falik-Zaccai TC, Khayat M, Luder A, et al. A broad spectrum of developmental delay in a large cohort of prolidase deficiency patients demonstrates marked interfamilial and intrafamilial phenotypic variability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010;153B:46–56. doi: 10.1002/ajmg.b.30945. [DOI] [PubMed] [Google Scholar]
- Friguls B, Coroleu W, Hilbert R, del Alcazar P, Pintos-Morell L, Van Maldergem G. Severe cardiac phenotype of Berardinelli-Seip congenital lipodystrophy in an infant with homozygous E189X BSCL2 mutation. Eur. J. Med. Genet. 2009;52:14–16. doi: 10.1016/j.ejmg.2008.10.006. [DOI] [PubMed] [Google Scholar]
- al-Gazali LI, Khalil M, Devadas K. A syndrome of insulin resistance resembling leprechaunism in five sibs of consanguineous parents. J. Med. Genet. 1993;30:470–475. doi: 10.1136/jmg.30.6.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geffner ME, Kaplan SA, Bersch N, et al. Leprechaunism: in vitro insulin action despite genetic insulin resistance. Pediatr. Res. 1987;22:286–291. doi: 10.1203/00006450-198709000-00010. [DOI] [PubMed] [Google Scholar]
- Grasso V, Colombo C, Favalli V, et al. Six cases with severe insulin resistance (SIR) associated with mutations of insulin receptor: is a Bartter-like syndrome a feature of congenital SIR? Acta Diabetol. 2013 doi: 10.1007/s00592-013-0490-x. PMID: 23824322 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Hovnik T, Bratanič N, Podkrajšek KT, Kovač J, Paro D, Podnar T, et al. Severe progressive obstructive cardiomyopathy and renal tubular dysfunction in Donohue syndrome with decreased insulin receptor autophosphorylation due to a novel INSR mutation. Eur. J. Pediatr. 2013;172:1125–1129. doi: 10.1007/s00431-012-1901-7. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Kadowaki H, Rechler MM, Serrano-Rios M, Roth J, Gorden P, et al. Five mutant alleles of the insulin receptor gene in patients with genetic forms of insulin resistance. J. Clin. Invest. 1990;86:254–264. doi: 10.1172/JCI114693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel WB, McGee DL. Diabetes and cardiovascular disease: the Framingham study. JAMA. 1979;241:2035–2038. doi: 10.1001/jama.241.19.2035. [DOI] [PubMed] [Google Scholar]
- Kitamei H, Yokoi M, Kase M, Ohno S. Retinal neovascularization during treatment with IGF-1 for insulin resistance syndrome. Graefes Arch. Clin. Exp. Ophthalmol. 2005;243:715–717. doi: 10.1007/s00417-004-1093-6. [DOI] [PubMed] [Google Scholar]
- Krook A, Brueton L, O'Rahilly S. Homozygous nonsense mutation in the insulin receptor gene in infant with leprechaunism. Lancet. 1993;342:277–278. doi: 10.1016/0140-6736(93)91820-c. [DOI] [PubMed] [Google Scholar]
- Maassen JA, Tobias ES, Kayserilli H, et al. Identification and functional assessment of novel and known insulin receptor mutations in five patients with syndromes of severe insulin resistance. J. Clin. Endocrinol. Metab. 2003;88:4251–4257. doi: 10.1210/jc.2003-030034. [DOI] [PubMed] [Google Scholar]
- McKern NM, Lawrence MC, Streltsov VA, et al. Structure of the insulin receptor ectodomain reveals a folded-over conformation. Nature. 2006;443:218–221. doi: 10.1038/nature05106. [DOI] [PubMed] [Google Scholar]
- Mellor KM, Ritchie RH, Delbridge LM. Reactive oxygen species and insulin-resistant cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2010;37:222–228. doi: 10.1111/j.1440-1681.2009.05274.x. [DOI] [PubMed] [Google Scholar]
- Miranda DM, Wajchenberg BL, Calsolari MR, et al. Novel mutations of the BSCL2 and AGPAT2 genes in 10 families with Berardinelli-Seip congenital generalized lipodystrophy syndrome. Clin. Endocrinol. (Oxf.) 2009;71:512–517. doi: 10.1111/j.1365-2265.2009.03532.x. [DOI] [PubMed] [Google Scholar]
- Mottillo S, Filion KB, Genest J, et al. The metabolic syndrome and cardiovascular risk. A systematic review and meta-analysis. J. Am. Coll. Cardiol. 2010;56:1113–1132. doi: 10.1016/j.jacc.2010.05.034. [DOI] [PubMed] [Google Scholar]
- Murakami K, Shigematsu Y, Hamada M, Higaki J. Insulin resistance in patients with hypertrophic cardiomyopathy. Circ. J. 2004;68:650–655. doi: 10.1253/circj.68.650. [DOI] [PubMed] [Google Scholar]
- Musso C, Cochran E, Moran SA, et al. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore) 2004;83:209–222. doi: 10.1097/01.md.0000133625.73570.54. [DOI] [PubMed] [Google Scholar]
- Nobile S, Semple RK, Carnielli VP. A novel mutation of the insulin receptor gene in a preterm infant with Donohue syndrome and heart failure. J. Pediatr. Endocrinol. Metab. 2012;25:363–366. doi: 10.1515/jpem-2011-0448. [DOI] [PubMed] [Google Scholar]
- Park JW, Kuehn HS, Kim SY, et al. Downregulation of Wnt-mediated ROS generation is causally implicated in leprechaunism. Mol. Cells. 2010;29:63–69. doi: 10.1007/s10059-010-0017-z. [DOI] [PubMed] [Google Scholar]
- Porter JR, Barrett TG. Monogenic syndromes of abnormal glucose homeostasis: clinical review and relevance to the understanding of the pathology of insulin resistance and beta cell failure. J. Med. Genet. 2005;42:893–902. doi: 10.1136/jmg.2005.030791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman OU, Khawar N, Khan MA, Ahmed J, Khattak K, Al-Aama JY, et al. Deletion mutation in BSCL2 gene underlies congenital generalized lipodystrophy in a Pakistani family. Diagn. Pathol. 2013;8:78. doi: 10.1186/1746-1596-8-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell NE, Holloway P, Quinn S, Foley M, Kelehan P, McAuliffe FM. Cardiomyopathy and cardiomegaly in stillborn infants of diabetic mothers. Pediatr. Dev. Pathol. 2008;11:10–14. doi: 10.2350/07-05-0277.1. [DOI] [PubMed] [Google Scholar]
- Seino S, Seino M, Nishi S, Bell GI. Structure of the human insulin receptor gene and characterization of its promoter. Proc. Natl Acad. Sci. USA. 1989;86:114–118. doi: 10.1073/pnas.86.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple RK, Savage DB, Cochran EK, Gorden P, O'Rahilly S. Genetic syndromes of severe insulin resistance. Endocr. Rev. 2011;32:498–514. doi: 10.1210/er.2010-0020. [DOI] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Howells K, Phillips AD, Thomas NS, et al. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;22:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi I, Yamada Y, Kadowaki H, et al. Phenotypical variety of insulin resistance in a family with a novel mutation of the insulin receptor gene. Endocr. J. 2010;57:509–516. doi: 10.1507/endocrj.k09e-339. [DOI] [PubMed] [Google Scholar]
- Verhagen SN, Wassink AM, Gorter Y, Van der Graaf PM, Visseren FL. Study Group TS. Insulin resistance increases the occurrence of new cardiovascular events in patients with manifest arterial disease without known diabetes. The SMART study. Cardiovasc. Diabetol. 2011;10:100. doi: 10.1186/1475-2840-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertheimer E, Lu SP, Backeljauw PF, Davenport ML. Taylor SI Homozygous deletion of the human insulin receptor gene results in leprechaunism. Nat. Genet. 1993;5:71–73. doi: 10.1038/ng0993-71. [DOI] [PubMed] [Google Scholar]
- Wertheimer E, Litvin Y, Ebstein RP, et al. Deletion of exon 3 of the insulin receptor gene in a kindred with a familial form of insulin resistance. J. Clin. Endocrinol. Metab. 1994;78:1153–1158. doi: 10.1210/jcem.78.5.8175972. [DOI] [PubMed] [Google Scholar]
- Winckler W, Weedon MN, Graham RR, et al. Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes. 2007;56:685–693. doi: 10.2337/db06-0202. [DOI] [PubMed] [Google Scholar]