

Abstract
In addition to their classical antigen presenting functions, MHC class II molecules potentiate the TLR-triggered production of pro-inflammatory cytokines. Here, we have addressed the effect of Tollip and MARCH1 on the regulation of MHC II trafficking and TLR signaling. Our results show that MARCH1-deficient mice splenocytes are impaired in their capacity to produce pro-inflammatory cytokines in response to poly(I:C) and that TLR3 and MHC II molecules interact in the endocytic pathway. Knocking down Tollip expression in human CIITA+ HeLa cells increased expression of HLA-DR but reduced the proportion of MHC II molecules associated with the CLIP peptide. Truncation of the HLA-DR cytoplasmic tails abrogated the effect of Tollip on MHC class II expression. While overexpression of Tollip did not affect HLA-DR levels, it antagonized the function of co-transfected MARCH1. We found that Tollip strongly reduced MARCH1 protein levels and that the two molecules appear to compete for binding to MHC II molecules. Altogether, our results demonstrate that Tollip regulates MHC class II trafficking and that MARCH1 may represent a new Tollip target.
Keywords: Tollip, MARCH1, MHC II, TLR3, Antigen presentation
Abbreviations: TLR, toll-like receptor; PAMPs, pathogen-associated molecular patterns; TIR, Toll/IL-1 receptor; APCs, antigen presenting cells; SOCS1, suppressor of cytokine signaling 1; Tollip, Toll-interacting protein; TBD, Tom1-binding domain; CUE, coupling of ubiquitin to endoplasmic reticulum degradation domain; C2, internal protein kinase C conserved region 2; TGFBR1, TGF-beta type I receptor; IRAK, IL-1 receptor-associated kinase; MHC II, MHC class II; DCs, dendritic cells; iDCs, immature DCs; MARCH, membrane-associated RING-CH; MIR, modulator of immune recognition; CIITA, class II trans-activator; Tfr, transferrin receptor; Btk, Bruton tyrosine kinase; IL-1RI, IL-1 receptor; IL-1RAcP, IL-1R-associated protein; MFVs, mean fluorescence values
Highlights
-
•
Splenocytes from MARCH1- and Ii-deficient mice are impaired in cytokine production following TLR stimulation.
-
•
MHC class II molecules bind TLR3.
-
•
Tollip regulates endocytic trafficking of MHC II proteins and chaperones.
-
•
Tollip binds MHC II molecules
1. Introduction
In humans, 10 members of the toll-like receptor (TLR) family of proteins recognize different pathogen-associated molecular patterns (PAMPs) through their luminal leucine-rich repeats [1]. TLRs are type I trans-membrane proteins capable of forming homo- and heterodimers [2]. While their expression patterns often differ, some, like TLR1, are ubiquitously expressed [3]. They localize on cell surface (TLR1, 2, 4, 5, 6 and 11) or in endosomes (TLR3, 7, 8 and 9), in line with the subcellular accumulation of their specific ligands [4,5]. TLRs are essential in the early events of innate immunity as well as in the development of robust adaptive immune responses [6,7]. Microbial products, such as LPS and DNA, trigger signaling cascades through the cytoplasmic Toll/IL-1 receptor (TIR) domain and various adaptor proteins, which include MyD88, TIRAF, TRIM, TRAF and IRAK [7]. One exception is TLR3, which is MyD88-independent and thus signals through TRIF [8]. The recognition of PAMPs by TLRs ultimately leads to NF-κB and AP-1 activation and the production of many pro-inflammatory cytokines, such as TNF-α and IL-6 [9]. Additionally, type I interferons are induced through the phosphorylation of IRF3 and IRF7 [10]. Thus, TLRs are important in the early innate immune responses against pathogens. These initial mediators and the activation of antigen presenting cells (APCs) will also impact the ensuing adaptive immunity.
Many accessory molecules, which modulate the activity of TLRs, have been identified. Some are implicated, for instance, in the folding, trafficking and processing of the TLRs [11]. Other cofactors include CD14 and granulin, which have been shown to deliver specific ligands to TLR4 and 9 respectively. TLR signals are also regulated by molecules such as the suppressor of cytokine signaling 1 (SOCS1) and Toll-interacting protein (Tollip) [12–14]. While four isoforms of Tollip have been described in humans and mice, the canonical protein is composed of three domains [15] and is ubiquitously expressed [14]. A TBD (Tom1-binding domain) and a CUE (coupling of ubiquitin to endoplasmic reticulum degradation) domain, located on the N- and C-terminal regions respectively, confer a potential for multiple protein interactions [16]. Finally, a C2 (internal protein kinase C conserved region 2) domain binds phosphoinositides and is responsible for the intracellular trafficking of the protein to the endocytic pathway [17].
While experiments on deficient mice suggested that Tollip was needed for maximal cytokine production in response to low doses of TLR agonists, most studies imply a negative regulatory role for Tollip in various signaling pathways [14,18–22]. For instance, Tollip has been shown to participate in IL-1β signaling as well as in the intracellular sorting and degradation of the ubiquitinated IL-1RI receptor [14,23]. Similarly, Tollip was recently shown to modulate TGF-β signaling through its interaction with Smad7 and by regulating the degradation of the activated TGF-β type I receptor (TβRI) [20]. Also, Tollip associates with TLRs and attenuates signaling by suppressing the activity of IL-1 receptor-associated kinase (IRAK) and NF-κB activation [18,22,24]. A high throughput shRNA screen identified Tollip as a potential regulator of MHC class II (MHC II) trafficking [25]. If ubiquitin links MHC II and Tollip pathways remains to be addressed. Interestingly, ubiquitination of MHC II molecules can occur in many different physiological conditions and cell types, allowing, for instance, maturation-dependent fine-tuning of antigen presentation in dendritic cells (DCs) [26,27].
Up to now, only two E3 ubiquitin ligases have been shown to modify MHC class II molecules. These are the membrane-associated RING-CH (MARCH) 1 and 8, two close homologues of viral modulator of immune recognition (MIR) proteins [28,29]. MARCH1 is mostly expressed in the spleen and more specifically in follicular B cells [30,31]. Comparably to the class II trans-activator (CIITA), which is the master regulator of MHC II gene transcription, MARCH1 appears to be the master regulator of MHC II expression at the post-translational level [32]. Indeed, the increased MHC II surface expression following activation of immature DCs (iDCs) is accompanied by the down-regulation of MARCH1 expression [33,34]. On the other hand, the immunosuppressive cytokine IL-10 up-regulates MARCH1 in monocytes and DCs to decrease MHC II expression and antigen presentation [35,36]. MARCH1 also ubiquitinates the transferrin receptor (Tfr), CD86, HLA-DM and Fas to modulate their expression and antigen presentation [30,31,37–39].
The impact of MARCH1 on the trafficking of MHC II has consequences beyond antigen presentation. It has been shown recently that DCs from MARCH1-deficient mice are impaired in the production of the pro-inflammatory cytokines IL-12 and TNF-α in response to LPS, suggesting that this ubiquitin ligase might be another accessory molecule involved in TLR signalling [40]. This activity of MARCH1 is dependent on the ubiquitination of MHC II molecules since the same phenotype was observed in MARCH1-proficient mice expressing non-ubiquitinable I-Ab β chain [40]. The fact that MHC II molecules potentiate LPS-induced signaling in human monocytes and mouse cells has been known for many years [41]. It was postulated that a LPS-binding receptor may interact with MHC II molecules to up-regulate TNFα secretion. Then, using MHC II-deficient primary cells from human patients or knock-out mice as well as reconstituted in vitro systems, Lauener and collaborators showed that MHC II molecules enhance TLR-induced responses [42,43]. More recently, it has been shown that MHC class II molecules promote TLR signaling in antigen presenting cells by maintaining activation of the Bruton tyrosine kinase (Btk) [44].
The cooperation between MHC II and TLRs promotes the innate as well as the adaptive immune response. Direct interactions between the MHC II and the TLRs have been observed and the generation of peptide-MHC class II complexes depends on endosomal trafficking of LPS-associated antigens in a phagosome-autonomous fashion [43,45,46]. Considering these clear functional links between innate and adaptive immunity and the interplay between the TLR4 signaling and the antigen presentation pathway, we hypothesized, as proposed recently, that Tollip might regulate the trafficking of MHC II molecules [47]. Our results demonstrate a direct interaction between MHC II and Tollip, which is reduced in the presence of MARCH1. Also, Tollip impairs the expression of MHC II and of MARCH1, in line with its previously described inhibitory functions.
2. Materials and methods
2.1. Antibodies
L243 (HLA-DR), XD5.117 (HLA-DRβ), CerCLIP.1 (CLIP/HLA-DR complexes), BU45 (human invariant chain), MaP.DM1 (HLA-DM) mAbs have been previously described [48–50]. The rabbit antisera against denatured HLA-DRα and HLA-DRβ were a kind gift from Dr. Rafick Sékaly (Vaccine and Gene Therapy Institute, Port St-Lucie, FL, USA). Rabbit polyclonal anti-GFP that recognizes both GFP and YFP, Alexa488- and Alexa633-fluor-coupled goat-anti mouse antibodies were purchased from Invitrogen (Laval, QC, Canada). The mouse anti-flag antibody was bought from Sigma (St-Louis, MS, USA). The rabbit anti-human Tollip was purchased from the cell signaling technology (Pickering, ON, Canada). The mouse anti-human Tfr OKT9 antibody was bought from ebioscience (San Diego, CA, USA). The mouse anti-MARCH1 (H1) was described previously [33]. The mouse anti-actin antibody was purchased from Millipore (Billerica, MA, USA).
2.2. Reagents
Poly(I:C) (Invivogen, San Diego, CA, USA) was used at a final concentration of 2 μg/mL. LPS was purchased from Sigma (St-Louis, MS, USA) and used at a concentration of 100 ng/mL. Benzyl Coelenterazine and luciferine were used at final concentrations of 5 uM and 20 μg/mL, respectively (Nanolight technology, Pinetop, AZ, USA).
2.3. Cell lines and mice
HeLa DR1, HeLa DR1 TM/TM, HeLa CIITA, HeLa CIITA/DO and HEK 293E CIITA stable transfectants were described previously [51,52]. Cells were cultured in DMEM supplemented with 5% FBS (Wisent, Saint-Jean-Baptiste, QC, Canada).
C57BL/6 (B6) mice were purchased from Charles River Laboratory (Wilmington, MA, USA). The M1K-O mice were described previously [31]. Xid mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Use of animals as described herein was approved by the University of Montreal's Institutional Animal Care and Use Committee (CDEA; protocol #12–042).
2.4. Plasmids and constructs
The flag, Rluc, EGFP2 or EYFP tags were fused by PCR overlap to the N-terminus of HLA-DRβ or TLR3 molecules using pcDNA3.1_flag_MCS, pcDNA3.1_Rluc_MCS, pcDNA3.1_EYFP_MCS or pcDNA3.1_EGFP2_MCS constructs. The cDNAs for the DRαTM and DRβTM chains include a stop codon immediately after the transmembrane coding regions, as described previously [53,54]. The GFP-SOCS1 and the GFP-Tollip constructs were obtained from Dr. Gerardo Ferbeyre (Université de Montréal, Montreal, QC, Canada) and Dr. Liwu Li (Virginia Polytechnic Institute and State University, Virginia, USA) respectively. The pcDNA3.1_MARCH1, pcDNA3.1_EYFP-MARCH1 and pcDNA-3.1_EYFP-MARCH1K-0 were described previously [48]. For the luciferase assay, we used the P2(2x)TK-pGL3_NF-κB reporter plasmid that was describe previously [55].
2.5. Transfections
For HeLa, 1×106 cells were plated 24 h prior to transfection in 10 cm petri dishes and transfected using lipofectamine LTX and Plus reagents according to the manufacturer's protocol (Invitrogen, Laval, QC, Canada). For HEK 293T and HEK 293E CIITA, 1.5×106 cells were plated and transfected 24 h later using 3 μg of polyethyleimine per μg of DNA (Polyscience, Warrington, PA, USA).
2.6. Flow cytometry
Cells were harvested, fixed, permeabilized using saponin and incubated with primary antibodies. After 45 min at 4 °C, cells were washed twice in PBS and incubated for another 45 min with the secondary antibody in PBS. Cells were analyzed on a FACS®calibur (Becton Dickinson, CA).
2.7. Immunoprecipitation and western-blot analysis
Cells were lysed on ice in 1% Triton X-100 lysis buffer supplemented with a complete protease inhibitor cocktail (Roche, Laval, QC) and centrifuged. The post-nuclear supernatants were pre-cleared for 1 h with protein G-coated sepharose beads (GE Healthcare, Mississauga, ON, Canada) and specific proteins were immunoprecipitated overnight using protein G-sepharose beads pre-coated with the selected antibody. The beads were washed and the samples were solubilized in reducing sample buffer, boiled and analyzed by immunoblotting following separation of proteins on 12% acrylamide gels, as described. Proteins were transferred to Hybond ECL membrane (GE Healthcare, Mississauga, ON, Canada) and analyzed with specific mAbs. Goat anti-mouse and anti-rabbit antibodies coupled to peroxydase (Bio/Can Scientific) were used as secondary antibodies and detected by chemiluminescence (BM Chemiluminescence Blotting Substrate (POD), Roche, Laval, QC, Canada). The films were scanned and analysed using Photoshop CS4 for signal quantification. Briefly, the colors were inverted and the mean intensity of the signal in a blank area was subtracted from that of different portions of the same size.
2.8. Bioluminescence resonance energy transfer (BRET) experiment
The pcDNA3.1_DRβ_Rluc (20 ng) and pcDNA3.1_DRα (20ng) were co-transfected in 1.25×105 HEK 293T cells with 0–500 ng of pcDNA3.1_TLR3-EYFP, resulting in different fluorescence/luminescence ratios [56]. Cells were harvested after 48 h and washed. For each sample, 1×105 cells were plated in duplicate into a 96-well plate. The background values of fluorescence were determined on a Mithras LB940 spectrofluorometer before the addition of coelenterazine by measuring the fluorescence emission at 538 nm after an excitation at 485 nm. After the addition of coelenterazine at a final concentration of 5 μM, the luminescence and fluorescence emissions in the 460–500 nm and 510–550 nm windows, respectively, were measured on a Mithras LB940 multidetector plate reader. The BRET ratio on the y-axis was calculated by dividing the acceptor-emitted fluorescence by the donor-emitted luminescence. BRET ratios were normalized by subtracting the background signal from cells transfected without YFP. The fluorescence over luminescence ratio on the x-axis is the ratio between the fluorescence of acceptor (YFP–YFP0, where YFP0 is the fluorescence value of cells expressing the BRET donor alone) and the luminescence of the acceptor.
2.9. Microscopy
For fluorescence resonance energy transfer (FRET) experiments, HeLa cells were used as they are highly adherent. Cells (6.25×104 per well) were plated on 35 mm glass bottom microwell dishes (MatTek Cultureware, Ashland, MA, USA) 24 h prior to transfection. Cells were transfected as described above and incubated for 48 h. Images of live cells were taken using a LSM 510 Meta Zeiss confocal microscope.
2.10. Luciferase assay
Cells were transfected with the NF-κB luciferase reporter plasmid and stimulated for 5 h with various amounts of poly(I:C). After stimulation, cells were washed and incubated with luciferine for 5 min prior to luminescence reading using a luminescence counter (Perkin–Elmer, Vaudreuil-Dorion, QC, Canada).
2.11. siRNA
Tollip specific and non-targeting control siRNAs were purchased from QIAGEN (Toronto, ON, Canada). The siRNAs were transfected using HiPerfect reagent according to the manufacturer's recommendations (QUIAGEN, Toronto, ON, Canada).
2.12. Real-time quantitative PCR
RNA/DNA were extracted using TRIzol (Sigma, St. Louis, MO, USA), DNA digestion (Ambion, Grand Island, NY, USA) and reverse transcription of RNA (Invitrogen, Laval, QC, Canada) were all performed as per the manufacturers’ instructions. Each sample was run in duplicates and a no-template control without cDNA was run for every primer set. Primer sequences are available upon request.
3. Results
3.1. Accessory molecules improve TLR signaling
Recent evidence suggest that MHC II molecules are pivotal in the innate immune response initiated by the TLRs [43]. Liu et al. have demonstrated that a multi-protein complex composed of TLR4, CD40, MHC II molecules and Btk is responsible for transducing signals from TLR4 [44,57]. Interestingly, it appears that the complex forms in the endocytic pathway and originates from the intracellular pool of MHC II. To further support the role of MHC II in the TLR signaling complex and the importance of MHC II trafficking to endosomes, we tested the cellular response to LPS in Ii-deficient mice. This chaperone associates with MHC II in the ER and its cytosolic di-leucine signals direct newly synthesized complexes to endosomes [58]. We used whole splenocytes since the majority of MHC II+ cells are B lymphocytes, which respond to LPS [59]. Our results show that Ii-deficiency reduced the TNF-α mRNA up-regulation in splenocytes in response to LPS (Fig. 1A). As a control, we also tested Xid mice, which have a mutation in the Btk gene [60]. Btk KO mice have been shown very recently to be impaired in their TLR response but expression profiling of the Xid and the Btk KO mice demonstrated important differences, suggesting that their phenotypes are not entirely redundant [44,61]. Results in Fig. 1A confirmed the role of Btk in TLR4 signaling as TNF-α production was lower in LPS-treated splenocytes from Xid mice.
Fig. 1.
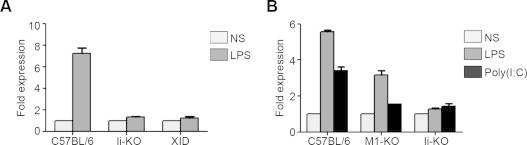
The response to poly(I:C) and LPS is impaired in the Ii KO and M1 KO mice. (A) Splenocytes from C57BL/6, Ii KO and Xid mice were isolated and treated ex vivo for 24 h with LPS prior to RNA extraction and qPCR analysis of TNFα mRNA expression. (B) Splenocytes from C57BL/6, Ii KO and M1 KO mice were isolated and treated ex vivo for 24 h with either LPS or poly(I:C) prior to RNA extraction and qPCR analysis of TNFα mRNA expression. Expression is illustrated as fold level compared to the value of untreated C57BL/6 cells, which was set at 1. Data is representative of at least two different experiments.
Mature MHC II molecules at the plasma membrane get ubiquitinated by MARCH1 and are sent to late compartments [28,29]. Pro-inflammatory cytokine production was found to be impaired in DCs from MARCH1-deficient mice and this phenotype was caused by the lack of I-Ab ubiquitination [40]. As MARCH1 is strongly expressed in B cells [31,62], we tested splenocytes from MARCH1-proficient and -deficient animals for the up-regulation of the TNF-α gene expression in response to LPS. Also, we extended these experiments to the study of poly(I:C) as MHC II deficiency also down-regulated TLR3 signaling [44]. Our results demonstrate that mouse cells deficient for either Ii or MARCH1 accessory molecules are impaired in their capacity to produce TNF-α in response to TLR3 or TLR4 ligands (Fig. 1B). These datas are in line with a generalized functional role in APCs of intracellular MHC II molecules and Btk for TLR signaling.
3.2. MHC II molecules interact with TLR3
The above-described results are in line with a role of MHC II molecules in the regulation of innate signals, including TLR3 ligands. TLR3 is the prototypical example of the TLR family members that reside in intracellular compartments [5]. Thus, we assessed by co-immunoprecipitation the capacity of TLR3 and human MHC II to associate. HEK 293E CIITA cells were co-transfected with a flag-tagged TLR3, lyzed and immunoprecipitated with a flag-specific mAb. Fig. 2A shows a co-precipitated HLA-DRα band on immunoblots (left panel). The interaction was specific as the control HLA-DM did not bind TLR3 in the same conditions (Fig. 2A, right panel). A similar type of experiment using transfected HEK 293 cells previously unveiled TLR2–HLA-DR interactions [43].
Fig. 2.
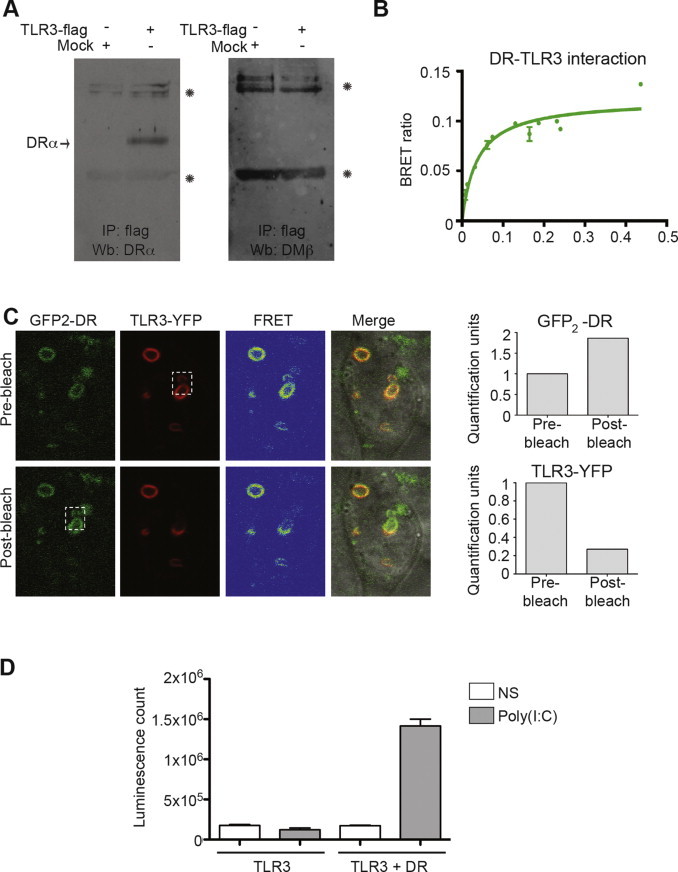
HLA-DR interacts with TLR3 in live cells. (A) HEK 293E CIITA cells were transfected with TLR3-flag. 48 h post-transfection, cells were lysed and immunoprecipitated with a flag specific antibody and blotted for HLA-DRα or HLA-DMβ. Asterisks represent the antibodies. (B) HEK 293T cells were transfected with HLA-DR–Rluc and increasing amounts of TLR3-EYFP. The BRET ratio was calculated by dividing the fluorescence with substrate, subtracted from the fluorescence without substrate, by the luminescence. Error bars represent standard deviation obtained for two different transfections. (C) FRET experiment performed in HeLa cells 48 h after transfection with TLR3-EYFP and HLA-DRα–EGFP2/β. One stack of living cells was observed by confocal microscopy. The dotted square shows the bleached area. The signal intensity for the bleached region was quantified for pre- and post-bleach. The signals were normalized for the ones of the corresponding regions prior to the beach and plotted in a bar chart. (D) Luciferase assay of HeLa or HeLa HLA-DR1 cells transfected or not with TLR3 and the NF-κB-luciferase reporter plasmid. The cells were stimulated for 5 h with poly(I:C) prior to the addition of luciferine. Error bars represent standard deviation obtained for two different transfections. Data is representative of a least three different experiments.
The interaction between MHC II molecules and TLR3 was confirmed by BRET. This technique offers great sensitivity and allows the monitoring of interactions in living cells, thereby avoiding possible artifacts resulting from cell lysis during co-immunoprecipitation experiments [56]. TLR3 was linked to EYFP, and co-transfected in HEK 293T cells along with an Rluc-fused HLA-DR molecule. After 48 h, luminescence and fluorescence emissions were measured. The BRET ratio reached a plateau, indicating a specific interaction rather than stochastic collisions between the two molecules (Fig. 2B).
To get insights into the localization of the HLA-DR–TLR3 complex, we performed a FRET experiment between EGFP2–HLA-DR and TLR3-EYFP (Fig. 2C). This technique allows to co-localize the signals of the two interacting partners in living cells and to visualize the energy transfer by the detection of YFP fluorescence emission following the excitation of the EGFP2 (FRET signal). FRET was detected in intracellular vesicles and the interaction was confirmed with the release of the EGFP2 signal after bleaching a distinct area (Fig. 2C, bottom panels). The relative fluorescence intensities of HLA-DR and TLR3 in the designated area (white square) were plotted as bar chart (Fig. 2C, right panels).
The fact that HLA-DR interacts with TLR3 suggests that MHC II molecules may also modulate the response to poly(I:C) in human cells. To test this hypothesis, we measured the activity of a reporter luciferase whose transcription is under the control of NF-κB-responsive promoter elements. Cells were transfected with the reporter construct together with combinations of HLA-DR and TLR3 before treatment with poly(I:C) for 5 h. Fig. 2D shows that co-expression of HLA-DR is needed for poly(I:C) to trigger TLR3 signaling in these conditions. Altogether, these results demonstrate the synergistic effect of MHC II molecules on the TLR3 response.
3.3. Tollip regulates MHC II trafficking
Very recently, using the human MelJuso cell line, a shRNA-based high throughput screen suggested a role for Tollip in the trafficking of MHC II molecules [25]. We further investigated this issue in HeLa cells stably transfected with CIITA and HLA-DO (HeLa-CIITA-DO). We used these cells because the overexpressed HLA-DO increases the amount of CLIP, which is otherwise almost undetectable in HeLa CIITA cells [51,52]. Also, the absence of endogenous MARCH1 in HeLa cells allowed us to uncouple the effect of Tollip and MARCH1. HeLa-CIITA-DO cells were treated with control or specific siRNAs and the efficient knockdown of Tollip mRNA expression was demonstrated by qPCR (Fig. 3A). Flow cytometry experiments demonstrated that the transfection of Tollip-specific siRNAs resulted in a 29% increase in the mean fluorescence value (MFV) for MHC II expression at the plasma membrane (Fig. 3B left and right panels). This increase does not appear to result from relocalization of some intracellular pools. Indeed, the total amount of MHC II molecules, as measured from the staining of permeabilized cells, was also increased (Fig. 3B, middle and right panels). These results suggest that Tollip decreases the turn-over of MHC II molecules. Interestingly, we found that knocking down Tollip reduced the amount of CLIP/MHC II complexes but increased the levels of unprocessed Ii (Fig. 3C). We performed the same experiments in HeLa CIITA cells and reached similar conclusions, showing that these results are independent of HLA-DO overexpression (data not shown). The impact of Tollip knockdown on CLIP and Ii suggests a somewhat slower trafficking of immature MHC II/Ii complexes to late endosomes. Altogether, these results confirm that Tollip plays a direct or indirect role in the maturation of MHC II/Ii complexes as well as in the degradation of MHC II molecules [25].
Fig. 3.
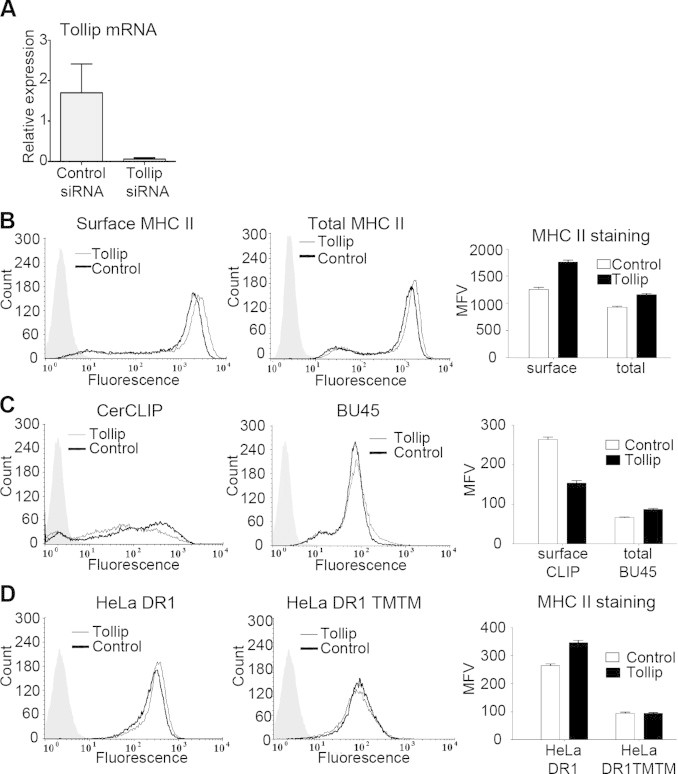
Tollip knockdown increases HLA-DR expression. (A) HeLa-CIITA-DO cells were transfected with control or TOLLIP-specific siRNAs, and cultured for 48 h at 37 °C. The bar chart represents the mRNA expression of Tollip for cells transfected with specific or control siRNA. (B) The cells were stained and analyzed by flow cytometry for cell surface and total expression of HLA-DR (L243 Ab). The mean fluorescence values (MFV) were plotted to account for variations in the levels of HLA-DR. Error bars represent standard deviation obtained for two different transfections. (C) Cells were stained for cell surface expression of CLIP (CerCLIP) and total expression of invariant chain (BU45). The mean fluorescence values (MFV) were plotted to account for variations in the levels of CLIP and invariant chain Error bars represent standard deviation obtained for two different transfections. (D) HeLa-DR1 and HeLa-DR1 TM/TM cells were transfected with control or Tollip-specific siRNAs, and cultured for 48 h in 37 °C. Cells were stained for cell surface expression of HLA-DR (L243 Ab). The mean fluorescence values (MFV) were plotted to account for variations in the levels of HLA-DR expression. Error bars represent standard deviation obtained for two different transfections. Data is representative of at least two different experiments.
To get insights into the mechanism of action of Tollip, we knocked down its expression in cells transfected exclusively with HLA-DR. Again, Tollip-specific siRNAs increased the plasma membrane expression of HLA-DR by 24% (Fig. 3D, left and right panels). This result demonstrated that the effect of Tollip on MHC II molecules is not dependent on the presence of Ii. We repeated this experiment using a HLA-DR1 molecule devoid of its α and β chains cytoplasmic regions (DR1 TMTM). Interestingly, there was no increase in the level of the truncated HLA-DR (Fig. 3D, middle and right panels). Altogether, these results confirm the role of Tollip on MHC class II trafficking and demonstrate that the cytoplasmic tails of MHC II are required for this effect.
3.4. Overexpressed Tollip interferes with MARCH1 activity
Tollip, through its ubiquitin-interacting CUE domain, is necessary for sorting of the activated IL-1 receptor (IL-1RI) at late endosomes [23]. Interestingly, MARCH8 was recently found to ubiquitinate and down-regulate the IL-1R-associated protein (IL-1RAcP), which is needed to form the membrane proximal signalosome [63]. The fact that the MHC II cytoplasmic tails were needed for Tollip to exert its effect suggested that ubiquitination-related events may be involved. This could explain the rather marginal effect of Tollip knockdown on MHC II expression in Hela cells as in the absence of overexpressed MARCH1 or MARCH8, ubiquitination of MHC II molecules is limited [31]. Thus, we postulated that overexpressing both Tollip and MARCH1 might have dramatic consequences and precipitate the degradation of MHC II molecules. To test this hypothesis, Tollip and MARCH1 were transiently transfected separately or together in HEK 293E CIITA cells. First, transfection of GFP-Tollip alone did not affect the surface expression of two of its targets, MHC II and Tfr (Fig. 4A, left and right panels). No difference was observed in the expression of HLA-DR or Tfr between the non-transfected cell population and the Tollip-expressing cells showing green fluorescence. This suggests that the basal endogenous levels of Tollip are sufficient to exert its effect on MHC II. On the other hand, cells co-transfected with MARCH1 and YFP as a tracer showed the well-characterized MHC II and Tfr down-regulation in fluorescent cells. Interestingly, to our surprise, co-expression of Tollip antagonized the effect of MARCH1 and reduced the magnitude of the MHC II and Tfr down-regulation (Fig. 4A, right panels). The mean fluorescence values (MFVs) obtained for HLA-DR and Tfr expression in the transfected populations were plotted in the right panels. To generalize the findings on MHC II molecules, these experiments were repeated in HeLa CIITA cells and similar results were obtained (Fig. 4B). Moreover, analysis of total cell lysates on immunoblots demonstrated that Tollip protects MHC II molecules from the MARCH1-induced degradation (Fig. 4C). Altogether, these results indicate that overexpression of Tollip antagonizes the activity of MARCH1 and suggest that both molecules compete for binding to the cytoplasmic tail of their targets.
Fig. 4.
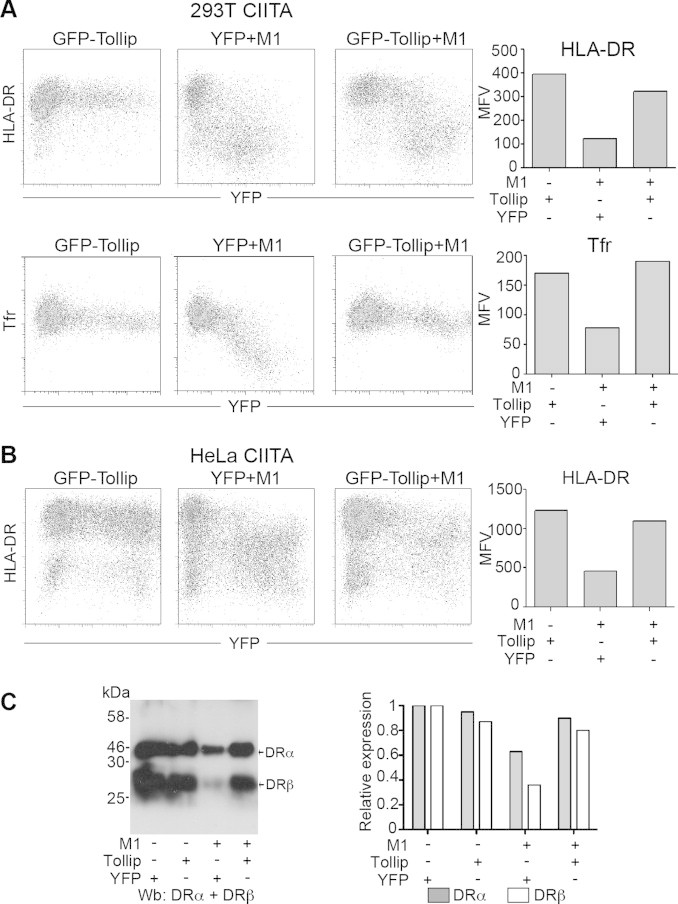
Tollip blocks MARCH1-mediated down-regulation of HLA-DR. (A) HEK 293E CIITA cells were transfected with GFP-Tollip, EYFP and MARCH1 or GFP-Tollip and MARCH1. Cells were stained for cell surface expression of HLA-DR and Tfr. Bar charts represent the mean fluorescence intensity of EYFP or GFP positive cells. (B) HeLa CIITA cells were transfected with GFP-Tollip, EYFP and MARCH1 or GFP-Tollip and MARCH1. Cells were stained for cell surface expression of HLA-DR. The bar chart represents the mean fluorescence intensity of EYFP or GFP positive cells. (C) Cells were lysed and blotted for HLA-DRα and HLA-DRβ. The intensity of the bands was quantified and divided by the one of cells transfected with the YFP control. Results are represented as a bar chart. Data is representative of at least two different experiments.
3.5. The expression of MARCH1 is reduced in the presence of Tollip
To get insights into the functional basis of MARCH1 dysfunction in the presence of overexpressed Tollip, we analyzed cell lysates on immunoblots. Fig. 5A shows that MARCH1 protein levels are greatly reduced in HeLa and HEK 293E cells co-transfected with Tollip. Actin was used as an internal control to confirm equal loading (asterisk). On the other hand, Tollip expression was not affected by the co-transfected MARCH1 (Fig. 5B). These results demonstrate that, at least in part, Tollip restores MHC II surface levels by reducing MARCH1 expression.
Fig. 5.
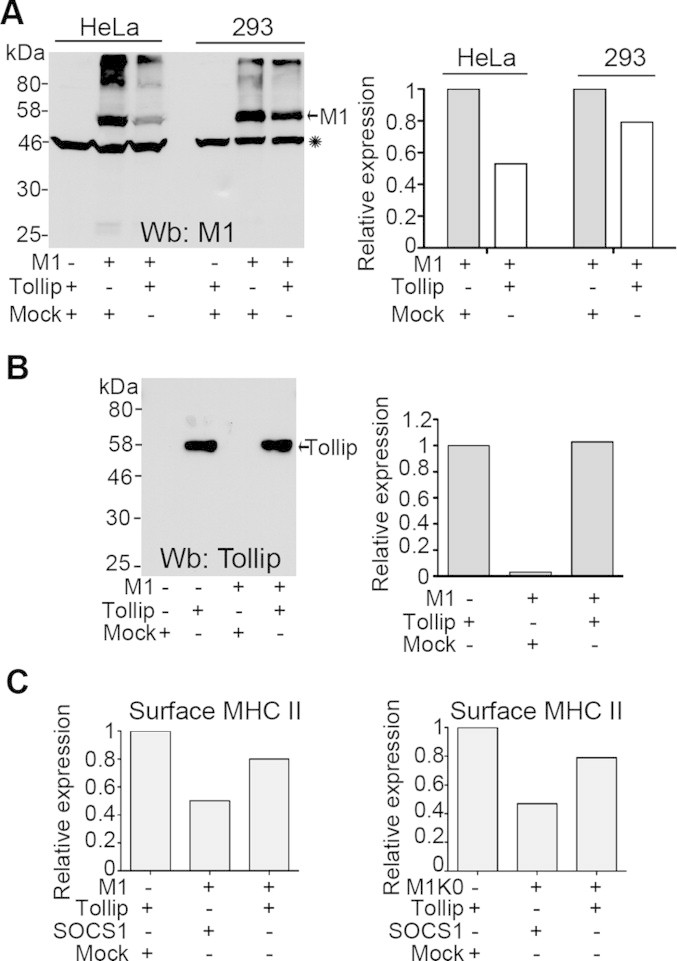
Tollip reduces the expression of MARCH1. (A) HeLa CIITA and HEK 293E CIITA cells were transfected with EYFP-MARCH1, GFP-Tollip or an empty vector (mock). Cell lysates were blotted for actin (asterisk) and MARCH1. The intensity of the bands was quantified, normalized to actin and divided by the one of cells transfected with MARCH1. Results are represented as a bar chart. (B) HeLa CIITA cells were transfected with EYFP-MARCH1, GFP-Tollip or an empty vector (mock). Cell lysates were blotted for Tollip. The intensity of the bands was quantified and the value obtained for cells expressing Tollip alone was set to 1. Results are represented as a bar chart. (C) HeLa CIITA or HEK 293E CIITA cells were transfected with EYFP-MARCH1 (left panel) of EYFP-MARCH1K-0 (right panel) with or without GFP-Tollip, GFP-SOCS1 and EYFP. Cells were stained for cell surface MHC II and analysed by flow cytometry. The mean fluorescence values for MHC II in cells expressing Tollip and EYFP was set to 1. Data is representative of a least two different experiments.
Tollip has been shown to regulate the trafficking of the ubiquitinated IL-1RI following activation [23]. We investigated the importance of ubiquitin in the regulation of MARCH1 by Tollip. Our results have demonstrated that MARCH1 is capable of auto-ubiquitination and that mutation of N- and C-terminal lysine residues (M1K0) reduces the overall level of ubiquitination without affecting functionality [48]. We compared the effect of overexpressed Tollip on the activity of M1K0 versus MARCH1 in HEK 293E CIITA cells. Cells were transfected with M1K0 or MARCH1 and either GFP-Tollip or an irrelevant GFP-tagged negative control protein (GFP-SOCS1) and the cell surface expression of HLA-DR was measured. Fig. 5C shows that Tollip restored MHC II levels in the presence of M1K0 as well, suggesting that the control of MARCH1 expression is independent of lysine ubiquitination in the N- and C-terminal regions of MARCH1.
3.6. Tollip interacts with HLA-DR
Despite the clear interplay described above between Tollip and MARCH1, we have not been able to co-immunoprecipitate the two molecules from transfected cells (data not shown). However, given the impact of Tollip siRNAs on the trafficking of HLA-DR (Fig. 3), we investigated the possible interaction between the two molecules. HEK 293E cells were co-transfected with Tollip and/or MARCH1 and HLA-DR was immunoprecipitated. Again, we found that Tollip restored the overall levels of the HLA-DRα chain (Fig. 6A). Interestingly, while HLA-DR co-immunoprecipitated GFP-Tollip, this interaction was abrogated upon co-expression of MARCH1 (Fig. 6B). The endogenous Tollip was not detected in these conditions, probably due to the weak expression of the protein. These results suggest that MARCH1 and Tollip compete for the binding to HLA-DR.
Fig. 6.
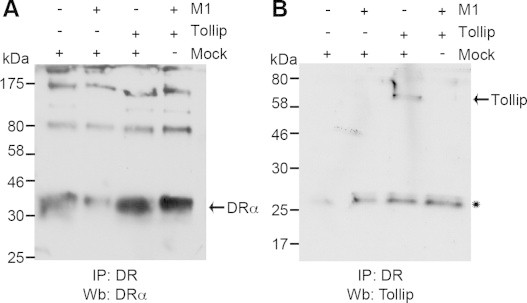
Tollip interacts with MHC II. HeLa cells were transfected with MARCH1 and/or GFP-Tollip and/or empty vector. Samples were immunoprecipitated with a HLA-DR-specific antibody and blotted for (A) Tollip and (B) DRα. The asterisk indicates the position of the immunoprecipitating mouse antibody light chain recognized by the goat secondary antibody. Data is representative of at least two different experiments.
4. Discussion
There are many different mechanisms that link the innate and adaptive arms of the immune response. The first phase of innate immunity signals danger to the beleaguered organism, setting the stage and potentiating the following, more specific, adaptive response. Indeed, signaling through the TLRs will up-regulate the expression of MHC II molecules in APCs and trigger important changes in the endocytic pathway [64]. Interestingly, this potentiation can work both ways. For instance, in professional APCs, the expression of MHC II molecules amplifies the innate immune response and increases the production of pro-inflammatory cytokines [42]. As such, the synergistic effect of IFN-γ on the LPS response is, at least in part, mediated by the up-regulation of MHC II molecules [41]. Moreover, both innate and adaptive responses can also synergize, with excessive physiological consequences, and lead to autoimmunity [65,66]. These findings warrant the reevaluation of the role of accessory proteins on not only their classical pathway, but on both arms of immunity.
As such, we looked at the role of the invariant chain on TLR signaling. Our results showing that Ii-deficiency impairs TLR responses are in line with a functional role of MHC II molecules in innate immunity. More specifically, given the well-described endosomal sorting signals of Ii, this data supports the assertion that the intracellular pool of MHC II molecules is needed for efficient LPS response [44]. However, we cannot rule out that the effect of Ii may be due to the lower trafficking of MHC II molecules from the ER in the C57BL/6 background [67]. It will be extremely interesting to test the impact of Ii deficiency on other mice backgrounds where surface MHC II, at least quantitatively, appears normal [67].
Additionally, we also tested the effect of MARCH1-deficiency on TLR signaling, as this ubiquitin ligase redirects mature MHC II molecules to late endosomes. Our results confirm and extend those of Ohmura-Hoshino et al. showing that MARCH1-deficient DCs produce less TNF-α in response to LPS [40]. Again, it appears that the presence of MHC II molecules in the endocytic pathway enhances innate responses. The exact nature of the compartments involved remains to be determined. Under the influence of MARCH1, ubiquitinated MHC II molecules are sent to late endosomes/lysosomes to be degraded [31]. Our results also confirmed the interaction between TLRs and MHC II molecules in the endocytic pathway. It remains to be addressed if a fraction of the molecules find themselves in more specialized vesicles, possibly on their way to lysosomes, to potentially increase the formation of the CD40/Btk/MHC II signaling complex [44]. Both Ii and MARCH1 are expressed in hematopoietic cells such as immature DCs and non-activated B cells. Thus, these resting APCs are equipped to maximally respond to TLR ligands. Our results suggest that both immature and mature MHC II molecules contribute to TLR signaling. Therefore, at least two different mechanisms are involved since Ii/MHC II complexes are not ubiquitinated, at least by MARCH proteins [27].
The implication of ubiquitination prompted us to investigate a possible role for Tollip in the regulation of MHC II transport. Our results demonstrated that Tollip decreased the total protein level of HLA-DR and the need for the MHC II cytoplasmic tails is certainly in line with a role of ubiquitination. Interestingly, even though the ubiquitinated cytoplasmic lysine is conserved, it was postulated that MHC II polymorphisms may affect TLR signaling [41]. Future studies should investigate this important issue and test the capacity of a panel of alleles and isotypes to associate with various TLRs. Our results showed that DR and Tollip could interact. Considering that Tollip is cytoplasmic, this interaction most likely requires the cytoplasmic tails of MHC II molecules. This is in line with the fact that MARCH1 ubiquitinates the cytoplasmic tails of HLA-DR [68] and that its expression reduced the number of Tollip/HLA-DR complexes (Fig. 6). Confirmation of these molecular interactions in primary mouse and/or human cells will be needed. However, such experiments will still rely on overexpression assays as the endogenous MARCH1 protein is barely detectable [69,48]. In this context, easily transfectable cells lines such as CIITA+ HeLa or HEK 293 represent suitable alternatives as they express endogenous Tollip and have been used in the past to characterize the MHC II pathway (see for example [70,71]).
The interplay between MARCH1 and Tollip on MHC II molecules is reminiscent of the recently described regulation of the IL-1β receptor complex in stimulated cells. Indeed, both Tollip and MARCH8 bind the accessory IL1RAcP protein and negatively regulate signalling [14,63]. It is not known if Tollip down-regulates MARCH8. Interestingly, the expression of Tollip increases in cells treated with LPS, possibly contributing to the loss of MARCH1 as part of a regulatory loop [24]. As MARCH1 and Tollip do not appear to physically interact, the effect of Tollip may be indirect and achieved through modulation of the endocytic pathway. Future studies will address the possible accelerated degradation of MARCH1 proteins in the presence of Tollip and the underlying molecular mechanisms.
That MARCH1 has a positive effect on TLR signalling is in line with a recent study indicating that MARCH5 affects the TLR7 response. Its mechanism of action would implicate the negative regulation of the TLR-inhibitor TANK and the localisation of the E3 in mitochondria [72]. However, MARCH8 has been shown to impair the TLR4-mediated induction of IL-6 and TNF-α in bone-marrow-derived macrophages and DCs [28,29]. Thus different MARCHs may play opposite effects on TLR signaling and further studies are required to support our initial suggestion for a role of MARCH1 in regulating TLR/MHC II interactions. The role of Tollip is also controversial as studies in mice have suggested that it may potentiate cytokine production in cells treated with low doses of LPS [19]. Our preliminary observations presented here of an interplay between MARCH1 and Tollip warrant a more in-depth mechanistic characterization of the molecular interactions taking place between TLRs, MHC II, Tollip, MARCH1 and the endocytic machinery. Also, future studies should address more in depth the effect of Tollip on MHC II trafficking in the absence of MARCH1.
Acknowledgements
We thank Dr. Gerardo Ferbeyre (Université de Montréal, Montreal, QC, Canada) and Dr. Liwu Li (Virginia Polytechnic Institute and State University, Virginia, USA) for providing the GFP-SOCS1 and GFP-Tollip, respectively. MCBD was supported by a fellowship from the Cole foundation. This project was supported by grants from the Roche Organ Transplantation Research Foundation/Juvenile Diabetes Research Foundation joint initiative (Project #665990157) to JT and AA and by the Canadian Institutes for Health Research (CIHR; Grant #93592) to JT.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Akira S., Takeda K. Toll-like receptor signalling. Nature Reviews Immunology. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 2.Botos I., Segal D.M., Davies D.R. The structural biology of Toll-like receptors. Structure. 2011;19:447–459. doi: 10.1016/j.str.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rock F.L. A family of human receptors structurally related to Drosophila Toll. Proceedings of the National Academy of Sciences. 1998;95:588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barton G.M., Kagan J.C. A cell biological view of Toll-like receptor function: regulation through compartmentalization. Nature Reviews Immunology. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blasius A.L., Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 6.Browne E.P. Regulation of B-cell responses by Toll-like receptors. Immunology. 2012;136:370–379. doi: 10.1111/j.1365-2567.2012.03587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda K., Akira S. TLR signaling pathways. Seminars in Immunology. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 8.Medzhitov R., Janeway C.A. An ancient system of host defense. Current Opinion in Immunology. 1998;10:12–15. doi: 10.1016/s0952-7915(98)80024-1. [DOI] [PubMed] [Google Scholar]
- 9.Kawai T., Akira S. TLR signaling. Seminars in immunology. 2007;19:24–32. doi: 10.1016/j.smim.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Colonna M. TLR pathways and IFN-regulatory factors: to each its own. European Journal of Immunology. 2007;37:306–309. doi: 10.1002/eji.200637009. [DOI] [PubMed] [Google Scholar]
- 11.Lee C.C., Avalos A.M., Ploegh H.L. Accessory molecules for Toll-like receptors and their function. Nature Reviews Immunology. 2012;12:168–179. doi: 10.1038/nri3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elliott J., Johnston J.A. SOCS: role in inflammation, allergy and homeostasis. Trends in Immunology. 2004;25:434–440. doi: 10.1016/j.it.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 13.Nakagawa R., Naka T., Tsutsui H., Fujimoto M., Kimura A., Abe T., Seki E., Sato S., Takeuchi O., Takeda K. SOCS-1 participates in negative regulation of LPS responses. Immunity. 2002;17:677–687. doi: 10.1016/s1074-7613(02)00449-1. [DOI] [PubMed] [Google Scholar]
- 14.Burns K., Clatworthy J., Martin L., Martinon F., Plumpton C., Maschera B., Lewis A., Ray K., Tschopp J., Volpe F. Tollip, a new component of the IL-1RI pathway, links IRAK to the IL-1 receptor. Nature Cell Biology. 2000;2:346–351. doi: 10.1038/35014038. [DOI] [PubMed] [Google Scholar]
- 15.Lo Y.-L.S., Beckhouse A.G., Boulus S.L., Wells C.A. Diversification of TOLLIP isoforms in mouse and man. Mammalian Genome: Official Journal of the International Mammalian Genome Society. 2009;20:305–314. doi: 10.1007/s00335-009-9188-3. [DOI] [PubMed] [Google Scholar]
- 16.Capelluto D.G.S. Tollip: a multitasking protein in innate immunity and protein trafficking. Microbes and Infection / Institut Pasteur. 2012;14:140–147. doi: 10.1016/j.micinf.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 17.Katoh Y., Shiba Y., Mitsuhashi H., Yanagida Y., Takatsu H., Nakayama K. Tollip and Tom1 form a complex and recruit ubiquitin-conjugated proteins onto early endosomes. Journal of Biological Chemistry. 2004;279:24435–24443. doi: 10.1074/jbc.M400059200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang G., Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. Journal of Biological Chemistry. 2002;277:7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 19.Didierlaurent A., Brissoni B., Velin D., Aebi N., Tardivel A., Käslin E., Sirard J.C., Angelov G., Tschopp K., Burns J. Tollip regulates proinflammatory responses to interleukin-1 and lipopolysaccharide. Molecular and Cellular biology. 2006;26:735–742. doi: 10.1128/MCB.26.3.735-742.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu L., Wang L., Luo X., Zhang Y., Ding Q., Jiang X., Wang X., Pan Y., Chen Y. Tollip, an intracellular trafficking protein, is a novel modulator of the transforming growth factor-beta signaling pathway. Journal of Biological Chemistry. 2012;287:39653–39663. doi: 10.1074/jbc.M112.388009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohnuma K., Yamochi T., Uchiyama M., Nishibashi K., Iwata S., Hosono O., Kawasaki H., Tanaka H., Dang N.H., Morimoto C. CD26 mediates dissociation of Tollip and IRAK-1 from caveolin-1 and induces upregulation of CD86 on antigen-presenting cells. Molecular and Cellular Biology. 2005;25:7743–7757. doi: 10.1128/MCB.25.17.7743-7757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bulut Y., Faure E., Thomas L., Equils O., Arditi M. Cooperation of Toll-Like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. Journal of Immunology. 2001;167:987–994. doi: 10.4049/jimmunol.167.2.987. [DOI] [PubMed] [Google Scholar]
- 23.Brissoni B., Agostini L., Kropf M., Martinon F., Swoboda V., Lippens S., Everett H., Aebi N., Janssens S., Meylan E. Intracellular trafficking of interleukin-1 receptor I requires Tollip. Current Biology. 2006;16:2265–2270. doi: 10.1016/j.cub.2006.09.062. [DOI] [PubMed] [Google Scholar]
- 24.Li T., Hu J., Li L. Characterization of Tollip protein upon lipopolysaccharide challenge. Molecular Immunology. 2004;41:85–92. doi: 10.1016/j.molimm.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 25.Paul P., Van den Hoorn T., Jongsma M.L.M., Bakker M.J., Hengeveld R., Janssen L., Cresswell P., Egan D.A., Van Ham M., Ten Brinke A. A Genome-wide multidimensional RNAi screen reveals pathways controlling MHC class II antigen presentation. Cell. 2011;145:268–283. doi: 10.1016/j.cell.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 26.Shin J.-S., Ebersold M., Pypaert M., Delamarre L., Hartley A., Mellman I. Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature. 2006;444:115–118. doi: 10.1038/nature05261. [DOI] [PubMed] [Google Scholar]
- 27.Van Niel G., Wubbolts R., Ten Broeke T., Buschow S.I., Ossendorp F.A., Melief C.J., Raposo G., Van Balkom B.W., Stoorvogel W. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity. 2006;25:885–894. doi: 10.1016/j.immuni.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Ohmura-Hoshino M., Goto E., Matsuki Y., Aoki M., Mito M., Uematsu M., Hotta H., Ishido S. A novel family of membrane-bound E3 ubiquitin ligases. Journal of Biochemistry. 2006;140:147–154. doi: 10.1093/jb/mvj160. [DOI] [PubMed] [Google Scholar]
- 29.Ohmura-Hoshino M., Matsuki Y., Aoki M., Goto E., Mito M., Uematsu M., Kakiuchi T., Hotta H., Ishido S. Inhibition of MHC Class II expression and immune responses by c-MIR. Journal of Immunology. 2006;177:341–354. doi: 10.4049/jimmunol.177.1.341. [DOI] [PubMed] [Google Scholar]
- 30.Bartee E., Mansouri M., Hovey Nerenberg B.T., Gouveia K., Früh K., Nerenberg B.T.H., Fru K., Irol J.V. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. Journal of Virology. 2004;78:1109–1120. doi: 10.1128/JVI.78.3.1109-1120.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuki Y., Ohmura-Hoshino M., Goto E., Aoki M., Mito-Yoshida M., Uematsu M., Hasegawa T., Koseki H., Ohara O., Nakayama M. Novel regulation of MHC class II function in B cells. The EMBO Journal. 2007;26:846–854. doi: 10.1038/sj.emboj.7601556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steimle V., Siegrist C., Mottet A., Lisowska-Grospierre B., Mach B. Regulation of MHC class II expression by interferon-gamma mediated by the transactivator gene CIITA. Science. 1994;265:106–109. doi: 10.1126/science.8016643. [DOI] [PubMed] [Google Scholar]
- 33.De Gassart A., Camosseto V., Thibodeau J., Ceppi M., Catalan N., Pierre P., Gatti E. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3491–3496. doi: 10.1073/pnas.0708874105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walseng E., Furuta K., Bosch B., Weih K.A., Matsuki Y., Bakke O., Ishido S., Roche P.A. Ubiquitination regulates MHC class II-peptide complex retention and degradation in dendritic cells. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20465–20470. doi: 10.1073/pnas.1010990107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thibodeau J., Bourgeois-Daigneault M.-C., Huppé G., Tremblay J., Aumont A., Houde M., Bartee E., Brunet A., Gauvreau M.-E., De Gassart A. Interleukin-10-induced MARCH1 mediates intracellular sequestration of MHC class II in monocytes. European Journal of Immunology. 2008;38:1225–1230. doi: 10.1002/eji.200737902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tze L.E., Horikawa K., Domaschenz H., Howard D.R., Roots C.M., Rigby R.J., Way D.A., Ohmura-Hoshino M., Ishido S., Andoniou C.E. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. Journal of Experimental Medicine. 2011;208:149–165. doi: 10.1084/jem.20092203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baravalle G., Park H., McSweeney M., Ohmura-Hoshino M., Matsuki Y., Ishido S., Shin J.-S. Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. Journal of Immunology. 2011;187:2966–2973. doi: 10.4049/jimmunol.1101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Corcoran K., Jabbour M., Bhagwandin C., Deymier M.J., Theisen D.L., Lybarger L. Ubiquitin-mediated regulation of CD86 protein expression by the ubiquitin ligase membrane-associated RING-CH-1 (MARCH1) Journal of Biological Chemistry. 2011;286:37168–37180. doi: 10.1074/jbc.M110.204040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jahnke M., Trowsdale J., Kelly A.P. Ubiquitination of human leukocyte antigen (HLA)-DM by different membrane-associated RING-CH (MARCH) protein family E3 ligases targets different endocytic pathways. Journal of Biological Chemistry. 2012;287:7256–7264. doi: 10.1074/jbc.M111.305961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohmura-Hoshino M., Matsuki Y., Mito-Yoshida M., Goto E., Aoki-Kawasumi M., Nakayama M., Ohara O., Ishido S. Cutting edge: requirement of MARCH-I-mediated MHC II ubiquitination for the maintenance of conventional dendritic cells. Journal of Immunology. 2009;183:6893–6897. doi: 10.4049/jimmunol.0902178. [DOI] [PubMed] [Google Scholar]
- 41.Santamaria P., Gehrz R., Bryan M., Barbosa J. Involvement of class II MHC molecules in the LPS-induction of IL-1/TNF secretions by human monocytes. Quantitative differences at the polymorphic level. Journal of Immunology. 1989;143:913–922. [PubMed] [Google Scholar]
- 42.Piani A., Hossle J.P., Birchler T., Siegrist C.A., Heumann D., Davies G., Loeliger S., Seger R., Lauener R.P. Expression of MHC class II molecules contributes to lipopolysaccharide responsiveness. European Journal of Immunology. 2000;30:3140–3146. doi: 10.1002/1521-4141(200011)30:11<3140::AID-IMMU3140>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 43.Frei R., Steinle J., Birchler T., Loeliger S., Roduit C., Steinhoff D., Seibl R., Büchner K., Seger R., Reith W. MHC class II molecules enhance Toll-like receptor mediated innate immune responses. PloS One. 2010;5:e8808. doi: 10.1371/journal.pone.0008808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X., Zhan Z., Li D., Xu L., Ma F., Zhang P., Yao H., Cao X. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase Btk. Nature Immunology. 2011;12:416–424. doi: 10.1038/ni.2015. [DOI] [PubMed] [Google Scholar]
- 45.Blander J.M., Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nature Immunology. 2006;7:1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 46.Husebye H., Halaas Ø., Stenmark H., Tunheim G., Sandanger Ø., Bogen B., Brech A., Latz E., Espevik T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. The EMBO Journal. 2006;25:683–692. doi: 10.1038/sj.emboj.7600991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neefjes J., Jongsma M.L.M., Paul P., Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nature Reviews Immunology. 2011;11:823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 48.Bourgeois-Daigneault M.-C., Thibodeau J. Autoregulation of MARCH1 expression by dimerization and autoubiquitination. Journal of Immunology. 2012;188:4959–4970. doi: 10.4049/jimmunol.1102708. [DOI] [PubMed] [Google Scholar]
- 49.Pezeshki A.M., Côté M.-H., Azar G.A., Routy J.-P., Boulassel M.-R., Thibodeau J. Forced expression of HLA-DM at the surface of dendritic cells increases loading of synthetic peptides on MHC class II molecules and modulates T cell responses. Journal of Immunology. 2011;187:74–81. doi: 10.4049/jimmunol.1002747. [DOI] [PubMed] [Google Scholar]
- 50.Genève L., Chemali M., Desjardins M., Labrecque N., Thibodeau J. Human invariant chain isoform p35 restores thymic selection and antigen presentation in CD74-deficient mice. Immunology and Cell Biology. 2012;90:896–902. doi: 10.1038/icb.2012.27. [DOI] [PubMed] [Google Scholar]
- 51.Khalil H., Deshaies F., Bellemare-Pelletier A., Brunet A., Faubert A., Azar G.A., Thibodeau J. Class II transactivator-induced expression of HLA-DObeta in HeLa cells. Tissue Antigens. 2002;60:372–382. doi: 10.1034/j.1399-0039.2002.600504.x. [DOI] [PubMed] [Google Scholar]
- 52.Gauvreau M.-E., Côté M.-H., Bourgeois-Daigneault M.-C., Rivard L., Xiu F., Brunet A., Shaw A., Steimle V., Thibodeau J. Sorting of MHC class II molecules into exosomes through a ubiquitin-independent pathway. Traffic. 2009;10:1518–1527. doi: 10.1111/j.1600-0854.2009.00948.x. [DOI] [PubMed] [Google Scholar]
- 53.Robadey C., Ammerlaan W., Muller C., Cloutier I., Sékaly R.P., Haefliger J.A., Demotz S. The processing routes determined by negatively charged residues in DR1-restricted T cell determinants. Journal of Immunology. 1997;159:3238–3246. [PubMed] [Google Scholar]
- 54.Bouillon M., El Fakhry Y., Girouard J., Khalil H., Thibodeau W., Mourad Lipid raft-dependent and -independent signaling through HLA-DR molecules. Journal of Biological Chemistry. 2003;278:7099–7107. doi: 10.1074/jbc.M211566200. [DOI] [PubMed] [Google Scholar]
- 55.Sharma S., tenOever B.R., Grandvaux N., Zhou G.-P., Lin R., Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 56.Perroy J., Pontier S., Charest P.G., Aubry M., Bouvier M. Real-time monitoring of ubiquitination in living cells by BRET. Nature Methods. 2004;1:203–208. doi: 10.1038/nmeth722. [DOI] [PubMed] [Google Scholar]
- 57.Ní Gabhann J., Jefferies C.A. TLR-induced activation of Btk—role for endosomal MHC class II molecules revealed. Cell Research. 2011;21:998–1001. doi: 10.1038/cr.2011.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Anderson M.S., Miller J. Invariant chain can function as a chaperone protein for class II major histocompatibility complex molecules. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:2282–2286. doi: 10.1073/pnas.89.6.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alexopoulou L., Holt A.C., Medzhitov R., Flavell R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 60.Rawlings D., Saffran D., Tsukada S., Largaespada D., Grimaldi J., Cohen L., Mohr R., Bazan J., Howard M., Copeland N. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 1993;261:358–361. doi: 10.1126/science.8332901. [DOI] [PubMed] [Google Scholar]
- 61.Lindvall J.M., Blomberg K.E.M., Berglöf A., Yang Q., Smith C.I.E., Islam T.C. Gene expression profile of B cells from Xid mice and Btk knockout mice. European Journal of Immunology. 2004;34:1981–1991. doi: 10.1002/eji.200324051. [DOI] [PubMed] [Google Scholar]
- 62.Galbas T., Steimle V., Lapointe R., Ishido S., Thibodeau J. MARCH1 down-regulation in IL-10-activated B cells increases MHC class II expression. Cytokine. 2012;59:27–30. doi: 10.1016/j.cyto.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 63.Chen R., Li M., Zhang Y., Zhou Q., Shu H.-B. The E3 ubiquitin ligase MARCH8 negatively regulates IL-1β-induced NF-κB activation by targeting the IL1RAP coreceptor for ubiquitination and degradation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:14128–14133. doi: 10.1073/pnas.1205246109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vyas J.M., Van der Veen A.G., Ploegh H.L. The known unknowns of antigen processing and presentation. Nature Reviews Immunology. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pasare C., Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 66.Walsh E.R., Pisitkun P., Voynova E., Deane J.A., Scott B.L., Caspi R.R., Bolland S. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell-mediated autoimmunity. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16276–16281. doi: 10.1073/pnas.1209372109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koonce C.H., Bikoff E.K. Dissecting MHC Class II export, B cell maturation, and DM stability defects in invariant chain mutant mice. Journal of Immunology. 2004;173:3271–3280. doi: 10.4049/jimmunol.173.5.3271. [DOI] [PubMed] [Google Scholar]
- 68.Lapaque N., Jahnke M., Trowsdale J., Kelly A.P. The HLA-DRalpha chain is modified by polyubiquitination. Journal of Biological Chemistry. 2009;284:7007–7016. doi: 10.1074/jbc.M805736200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jabbour M., Campbell E.M., Fares H., Lybarger L. Discrete domains of MARCH1 mediate its localization, functional interactions, and posttranscriptional control of expression. Journal of Immunology. 2009;183:6500–6512. doi: 10.4049/jimmunol.0901521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bania J., Gatti E., Lelouard H., David A., Cappello F., Weber E., Camosseto V., Pierre P. Human cathepsin S, but not cathepsin L, degrades efficiently MHC class II-associated invariant chain in nonprofessional APCs. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:6664–6669. doi: 10.1073/pnas.1131604100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zuo J., Thomas W.A., Haigh T.A., Fitzsimmons L., Long H.M., Hislop A.D., Taylor G.S., Rowe M. Epstein-Barr virus evades CD4+ T cell responses in lytic cycle through BZLF1-mediated downregulation of CD74 and the cooperation of vBcl-2. PLoS Pathogens. 2011;7:e1002455. doi: 10.1371/journal.ppat.1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shi H.-X., Liu X.X.-Y., Wang Q., Tang P.-P., Shan Y.-F., Wang C. Mitochondrial ubiquitin ligase MARCH5 promotes TLR7 signaling by attenuating TANK action. PLoS Pathogens. 2011;7:e1002057. doi: 10.1371/journal.ppat.1002057. [DOI] [PMC free article] [PubMed] [Google Scholar]