

Abstract
Zellweger syndrome (ZS) is a severe manifestation of disease within the spectrum of peroxisome biogenesis disorders that includes neonatal adrenoleukodystrophy, infantile Refsum disease, and rhizomelic chondroplasia punctata. Patients with ZS present in the neonatal period with a characteristic phenotype of distinctive facial stigmata, pronounced hypotonia, poor feeding, hepatic dysfunction, and often seizures and boney abnormalities. In patients with ZS, a mutation in one of the PEX genes coding for a peroxin (a peroxisome assembly protein) creates functionally incompetent organelles causing an accumulation of very long chain fatty acids (VLCFA), among other complications. Despite an absence of treatment options, prompt diagnosis of ZS is important for providing appropriate symptomatic care, definitive genetic testing, and counseling regarding family planning.
Zellweger syndrome (ZS) is a severe manifestation of disease within the spectrum of peroxisome biogenesis disorders that includes neonatal adrenoleukodystrophy, infantile Refsum disease, and rhizomelic chondroplasia punctata. Patients with ZS present in the neonatal period with a characteristic phenotype of distinctive facial stigmata, pronounced hypotonia, poor feeding, hepatic dysfunction, and often seizures and boney abnormalities. In patients with ZS, a mutation in one of the PEX genes coding for a peroxin (a peroxisome assembly protein) creates functionally incompetent organelles causing an accumulation of very long chain fatty acids (VLCFA), among other complications. Despite an absence of treatment options, prompt diagnosis of ZS is important for providing appropriate symptomatic care, definitive genetic testing, and counseling regarding family planning.
CLINICAL CASE: PART I
A male infant was born at term and weighed 2,948 g (<25th percentile). A fetal ultrasound at 28 weeks revealed oligohydramnios, ventriculomegaly, and club feet. Labor and delivery were uncomplicated. After birth, he was noted to be floppy and had Apgar scores of 5 and 7. He required oxygen supplementation and was transferred to the neonatal intensive care unit.
In the unit, he was persistently hypotonic with periods of apnea. He was noted to have a large anterior fontanelle, large forehead, and broad nasal bridge. His irides had concentric speckles (Brushfield spots); ophthalmologic examination was otherwise unremarkable. He had transverse palmar creases and bilateral club feet. Liver was palpable 2 cm below the costal margin. He exhibited minimal spontaneous movement. He withdrew to painful stimuli. He was areflexic. He required a nasogastric tube for feeding.
Laboratory studies revealed hyperbilirubinemia with a mild elevation in liver transaminases. Skeletal radiographs demonstrated punctate calcifications in the long bones. Head ultrasound revealed mild dilation of the lateral ventricles with a small germinal matrix hemorrhage. CT scan of the brain confirmed ventriculomegaly and a small right caudate hemorrhage.
DISCUSSION
The differential diagnosis of the hypotonic dysmorphic newborn with poor feeding includes ZS, trisomy 21 (Down syndrome), Prader-Willi syndrome, and congenital neuromuscular diseases (e.g., spinal muscular atrophy, congenital myotonic dystrophy type 1, X-linked myotubular myopathy, multiminicore myopathies). Patient appearance and physical examination can reveal some salient features that suggest ZS rather than other congenital conditions (figure, table).1,2 Patients presenting outside the newborn period may be evaluated for Usher syndrome types I or II, Leber congenital amaurosis, Cockayne syndrome, or congenital leukodystrophies (Krabbe disease, metachromatic leukodystrophy). Later age at presentation indicates one of the less severe forms of peroxisomal disorders, e.g., neonatal adrenoleukodystrophy or infantile Refsum disease.
Figure. Abnormal facial features associated with Zellweger syndrome.
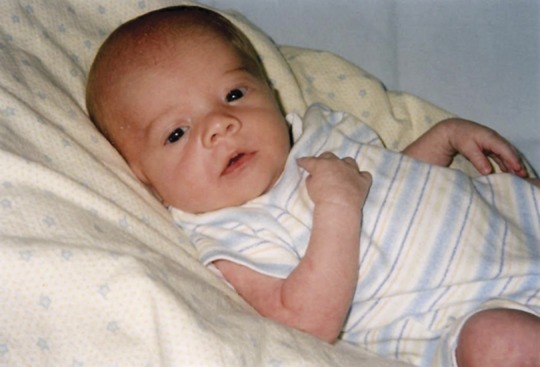
An example of a patient displaying stigmata found in Zellweger syndrome. Note the high forehead, widely spaced eyes, broad nasal bridge, and mildly upturned nose. Photograph used with permission from Shannon Butalla, The Global Foundation for Peroxisomal Disorders.
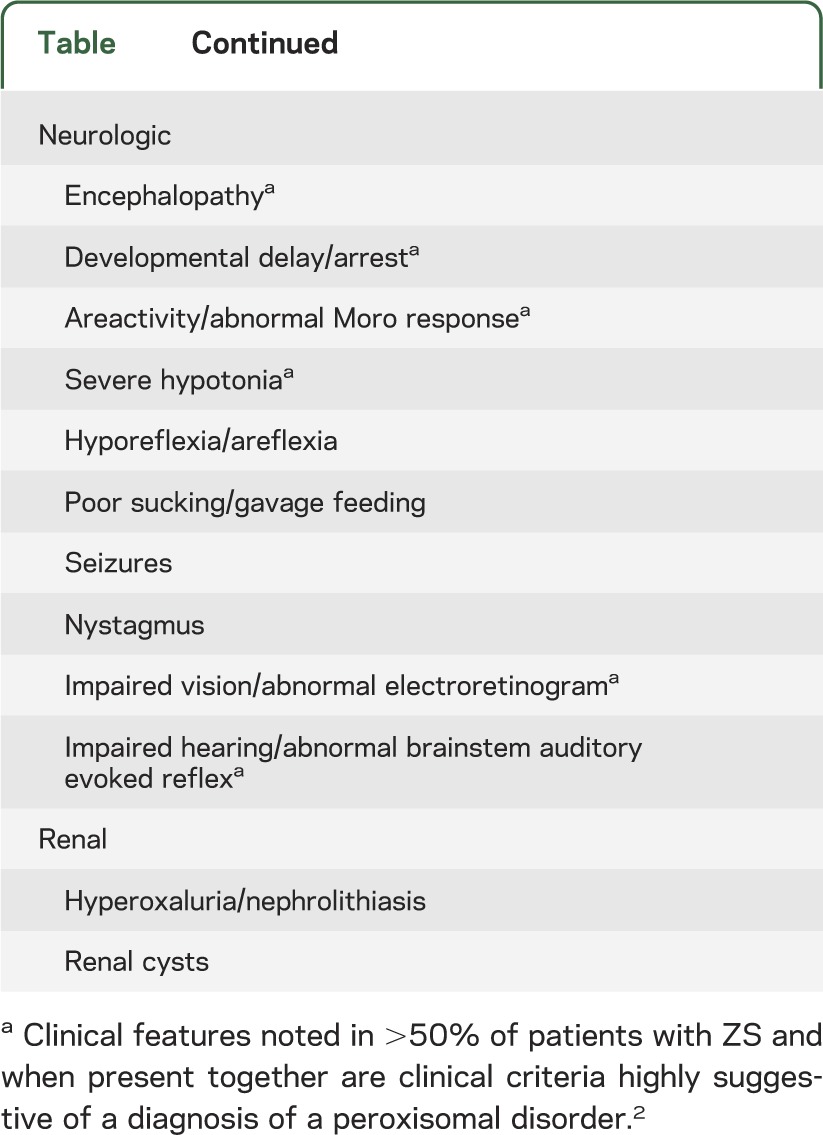
Table.
Abnormal clinical features associated with Zellweger syndrome1,2

The worldwide prevalence of ZS is estimated between 1:50,000 and 1:100,000, with reports of higher incidence of ZS in the Saguenay-Lac-St-Jean region of Quebec and a lower incidence in Japan.1,3,4
The initial published description of ZS described several members of a single family with multiple congenital anomalies involving the brain, liver, and kidneys; the authors aptly described this as a “cerebrohepatorenal” syndrome.5 While today more is known about the genetics of ZS, the clinical phenotype remains as initially described and is exemplified by the example case (figure). Reflecting the ubiquity of peroxisomes, infants with ZS have multiple congenital abnormalities evident at birth involving the eyes, bone, liver, kidneys, endocrine glands, and brain (table). Hypotonia is marked; feeding and respiratory issues continue throughout life. ZS patients make little developmental progress. ZS is fatal in early life.
ZS is an autosomal recessive inherited disorder of the peroxisome, an intracellular organelle composed of a single membrane containing a matrix embedded with over 50 enzymes for metabolism of fatty acids.6 The proper assembly of a peroxisome requires a unique set of proteins termed “peroxins.” Peroxins help incorporate enzymes into the forming peroxisome's matrix. A mutation in a peroxin, or “PEX,” gene yields a reduced or nonfunctioning peroxin. A defective peroxin means peroxisomes may not form or, if they assemble, have lower or undetectable levels of key internal enzymes. Incomplete peroxisomes fail to perform their metabolic duties including the β-oxidation of fatty acids with a chain length of more than 22 carbons, the α-oxidation of phytanic acid and similar compounds, pipecolic acid oxidation, and early plasmalogen synthesis.6 The intracellular accumulation of VLCFA damages developing organs (e.g. liver, bone, kidneys) and is especially deleterious to the organizing brain. There is a characteristic neuronal migration defect of cortical neurons failing to reach their destinations within the uppermost layers of the neocortex.7 Macroscopically and on neuroimaging, the consequences on brain morphology can include cortical gyral abnormalities (lissencephaly, pachygyria, polymicrogyria), generalized or focal leukoencephalopathy, and brain atrophy.7
There are 16 known human PEX genes, and disease-associated mutations have been identified in 13 of these genes.6 ZS is most commonly caused by mutations in the genes PEX1 (two-thirds of cases) and PEX66; PEX5 was the most common mutation in a Middle East cohort.8 A patient's specific PEX mutation cannot be predicted from serum biochemical abnormalities. While certain mutations may correlate with known phenotypes, variability occurs and only characterization of peroxisomal performance in patient-derived tissue culture can definitively establish the biochemical consequences of a given genetic mutation in vivo.6,7
While mitochondrial fatty acid disorders are included in newborn screening, peroxisomal disorders are not. The appropriate screening test for an infant with suspected ZS is a measurement of plasma VLCFA levels. Elevated plasma concentrations of the following VLCFAs—hexacosanoic acid (designated C26:0 for a fully saturated 26 carbon chain), the monounsaturated hexacosenoic acid (C26:1), and tetracosenoic acid (C24:0)—in addition to elevated ratios of C26:0 to docosanoic acid (C22:0) and C24:0 to C22:0 are consistent with peroxisomal disease. These findings do not implicate a specific biochemical abnormality or gene mutation. An abnormal value in VLCFA mandates further testing, including repeat VLCFA measurement and analysis of other peroxisomal markers such as plasma phytanic acid, pristanic acid, plasmalogens, plasma or urine pipecolic acid, and plasma or urine bile acids. Some laboratories routinely perform VLCFA testing concurrently with these other parameters, obviating the need for supplemental evaluation. The VLCFA profile of a patient with ZS will often be dramatically abnormal, but it is important to note the limitations of these assays. A ketogenic diet will elevate VLFCA levels. Phytanic and pristanic acid accumulate with dietary consumption and are normal in the newborn. Plasmalogen levels may be normal in infants older than 20 weeks. Therefore, a skin biopsy should be obtained from any suspected ZS patient to establish a cell line for future investigations.
Genetic testing for family planning purposes should be considered in potential carriers prior to pregnancy and in instances where a parent or first-degree relative is a known carrier or there is a sibling or relative with a peroxisomal biogenesis disorder. Preimplantation genetic diagnosis is possible, as is prenatal diagnosis using cultured cells derived from amniotic or placental cells.7
CLINICAL CASE: PART II
A plasma assay noted elevations in multiple VLCFA parameters consistent with ZS. Specific gene testing was declined. At home, the patient continued to have issues with hypotonia and feeding. A gastrostomy tube was placed. At 2 months of age, he developed seizures that were poorly controlled. His parents felt that he had visual attention, though he would not track consistently and had fine nystagmus. He developed a smile and some head control, but remained profoundly hypotonic. Testing revealed sensorineural hearing loss, and he received hearing aids. At 11 months of age, he became progressively apneic and died.
There is no curative treatment for ZS. Treatment should be focused on optimizing quality of life and congruent with the amount of intervention desired by the family.1 Children with ZS often require specialized services or testing at pediatric rehabilitation centers or institutions with large genetic/metabolic clinics. ZS affects multiple organs (table) but common complications are discussed below.
Patients with ZS will have special dietary needs and oral feeding difficulties. While theoretical concerns about accumulation of dietary-derived phytanic acid exist, this has never been demonstrated to be significant in ZS as it is in Refsum disease. Therefore, breast milk feeding (recommended given its known health benefits) or commercial formulas can be provided despite containing measurable amounts of phytanic acid. Providing diets depleted of VLCFA precursors have no benefit in ZS due to endogenous VLCFA production.9 Patients with ZS experience lipid malabsorption (elemental formulas may be better tolerated) and supplementation of the fat-soluble vitamins A, D, E, and K is recommended. Many patients with ZS will need gastrostomy tube placement, and surgical anesthesia can be managed safely despite hypotonia.1,10
ZS is associated with cholestasis and hepatic injury that can be ameliorated by oral bile acid replacement.1,7 Patients with ZS over age 12 months should be monitored for oxaluria and resultant nephrolithiasis, which can lead to renal failure.7 Adrenal insufficiency and consequential rapid deterioration may occur with physiologic stress.7
Early life intervention services should be provided with the expectation that any developmental progress will be minimal. Infants with ZS will have sensory impairments attributable to deficient myelination. In one study, three-quarters of patients with ZS had impaired hearing, so hearing aids can be considered.2 Electroretinograms performed on patients with ZS typically demonstrate no response to stimulation; the routine use of electroretinograms is not recommended. At least one-third of patients with ZS will have seizures, which can be managed with any currently available anticonvulsants.1,7 Though liver transaminases are elevated in ZS, in our experience the use of anticonvulsants with primary hepatic clearance like phenobarbital is safe and effective.
ZS is a serious disorder with multiple congenital anomalies that is progressive and fatal. While affected children may survive only 2 to 3 months, with improved care and genetic–phenotypic variability, there are reports of patients living up to 2 years; thus any estimated lifespan should take this longevity into account.7 Patients with ZS typically die from apnea, respiratory failure, or complications from infection.1,6,7
ACKNOWLEDGMENT
The authors thank Shannon Butalla of The Global Foundation for Peroxisomal Disorders for providing the example patient photograph.
AUTHOR CONTRIBUTIONS
Dr. Lee participated in the medical care of the patient discussed in the clinical vignette, proposed the review article, reviewed the relevant literature on this topic, and drafted and revised the manuscript. Dr. Raymond participated in the medical care of the patient discussed in the clinical vignette, drafted the clinical vignette, provided the source for the patient photograph, and reviewed and revised the manuscript.
STUDY FUNDING
Dr. Lee is supported by the intramural program of the National Institute of Neurological Disorders and Stroke.
DISCLOSURE
P.R. Lee reports no disclosures. G.V. Raymond has served as a consultant for BlueBird Bio (Cambridge, MA). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Grayer J. Recognition of Zellweger syndrome in infancy. Adv Neonatal Care 2005;5:5–13 [DOI] [PubMed] [Google Scholar]
- 2.Theil AC, Schutgens RB, Wanders RJ, Heymans HS. Clinical recognition of patients affected by a peroxisomal disorder: a retrospective study in 40 patients. Eur J Pediatr 1992;151:117–120 [DOI] [PubMed] [Google Scholar]
- 3.Shimozawa N, Nagase T, Takemoto Y, Suzuki Y, Kondo N. Genetic heterogeneity in Japanese patients with peroxisome biogenesis disorders and evidence for a founder haplotype for the most common mutation in PEX10 gene. Adv Exp Med Biol 2003;544:71. [DOI] [PubMed] [Google Scholar]
- 4.Levesque S, Morin C, Guay SP, et al. A founder mutation in the PEX6 gene is responsible for increased incidence of Zellweger syndrome in a French Canadian population. BMC Med Genet 2012;13:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowen P, Lee CS, Zellweger H, Lindenberg R. A familial syndrome of multiple congenital defects. Bull Johns Hopkins Hosp 1964;114:402–414 [PubMed] [Google Scholar]
- 6.Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta 2006;1763:1733–1748 [DOI] [PubMed] [Google Scholar]
- 7.Gould SJ, Raymond GV, Valle D. The peroxisome biogenesis disorders. In: Scriver CD, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease, 8th ed New York: McGraw-Hill; 2001:3181–3218 [Google Scholar]
- 8.Shaheen R, Al-Dirbashi OY, Al-Hassnan ZN, et al. Clinical, biochemical and molecular characterization of peroxisomal diseases in Arabs. Clin Genet 2011;79:60–70 [DOI] [PubMed] [Google Scholar]
- 9.Arai Y, Kitamura Y, Hayashi M, Oshida K, Shimizu T, Yamashiro Y. Effect of dietary Lorenzo's oil and docosahexaenoic acid treatment for Zellweger syndrome. Congenit Anom 2008;48:180–182 [DOI] [PubMed] [Google Scholar]
- 10.Platis CM, Kachko L, Peled E, Katz J. Anesthesia for the child with Zellweger syndrome: a case report. Paediatr Anaesth 2006;16:361–362 [DOI] [PubMed] [Google Scholar]