

Background: Dysregulated fibroblast-to-myofibroblast transitions cause fibrotic diseases.
Results: Aortic carboxypeptidase-like protein (ACLP) stimulates myofibroblast differentiation through activation of transforming growth factor β receptor signaling.
Conclusion: Eliminating ACLP activity reduces SMA expression and myofibroblast formation.
Significance: Reducing ACLP function in fibrotic tissue may provide a novel strategy to reduce the rate of fibrotic disease progression.
Keywords: Collagen, Myofibroblast, Pulmonary Fibrosis, SMAD Transcription Factor, Transforming Growth Factor β (TGFβ), ACLP
Abstract
Idiopathic pulmonary fibrosis (IPF) is a chronic and fatal lung disease characterized by the overgrowth, hardening, and scarring of lung tissue. The exact mechanisms of how IPF develops and progresses are unknown. IPF is characterized by extracellular matrix remodeling and accumulation of active TGFβ, which promotes collagen expression and the differentiation of smooth muscle α-actin (SMA)-positive myofibroblasts. Aortic carboxypeptidase-like protein (ACLP) is an extracellular matrix protein secreted by fibroblasts and myofibroblasts and is expressed in fibrotic human lung tissue and in mice with bleomycin-induced fibrosis. Importantly, ACLP knockout mice are significantly protected from bleomycin-induced fibrosis. The goal of this study was to identify the mechanisms of ACLP action on fibroblast differentiation. As primary lung fibroblasts differentiated into myofibroblasts, ACLP expression preceded SMA and collagen expression. Recombinant ACLP induced SMA and collagen expression in mouse and human lung fibroblasts. Knockdown of ACLP slowed the fibroblast-to-myofibroblast transition and partially reverted differentiated myofibroblasts by reducing SMA expression. We hypothesized that ACLP stimulates myofibroblast formation partly through activating TGFβ signaling. Treatment of fibroblasts with recombinant ACLP induced phosphorylation and nuclear translocation of Smad3. This phosphorylation and induction of SMA was dependent on TGFβ receptor binding and kinase activity. ACLP-induced collagen expression was independent of interaction with the TGFβ receptor. These findings indicate that ACLP stimulates the fibroblast-to-myofibroblast transition by promoting SMA expression via TGFβ signaling and promoting collagen expression through a TGFβ receptor-independent pathway.
Introduction
Idiopathic pulmonary fibrosis (IPF)3 is a chronic lung disease with no effective cure (1). It is characterized by sequential lung injury that results in epithelial and endothelial cell damage, inflammation, and progressive deposition of ECM molecules, including collagen. Fibrotic lungs contain large numbers of contractile, filament-laden parenchymal cells known as myofibroblasts and an increase in overall tissue contractility (2). These myofibroblasts characteristically express smooth muscle α-actin (SMA) and are known to be critical components of wound healing (2, 3). Myofibroblasts originate from different sources, including fibroblasts, epithelial cells, and bone marrow-derived cells (4–8). Myofibroblasts are spindle- or stellate-shaped cells that are similar to smooth muscle cells in that they are both contractile and contain SMA (2). Differentiated myofibroblasts are also responsible for increased collagen synthesis in the lung in IPF (2).
In the fibrotic lung, normal fibroblastic cells become activated by cytokines released from local inflammatory and resident cells after tissue injury to promote ECM component synthesis (9, 10). Mechanical challenges in the extracellular environment also stimulate the fibroblast-to-myofibroblast transition. In response to mechanical challenges, these cells develop stress fibers that connect the cell to ECM proteins (11). Along with extracellular remodeling, accumulation of active TGFβ and the presence of ECM proteins like the extra domain A (ED-A) splice variant of fibronectin are required events for the production of SMA-positive myofibroblasts (9). TGFβ is found at high levels in human IPF lungs (12) as well as in the lungs of mice and hamsters with bleomycin-induced fibrosis (13, 14). TGFβ overexpression is sufficient to induce fibrosis in rat lungs (15). Additionally, TGFβ promotes the formation of granulation tissue with abundant SMA-expressing myofibroblasts in rats and induces SMA expression in cultured fibroblasts (16). TGFβ has also been shown to activate the transcriptional regulator myocardin-related transcription factor A (MRTFA), which stimulates both SMA and collagen expression (17, 18).
Canonical TGFβ signaling is initiated when the latent TGFβ complex is secreted by the cell and is dissociated and activated in the extracellular environment (19, 20). TGFβ forms a dimer that binds to TGFβ receptor II (TβRII), a serine/threonine kinase that activates TGFβ receptor I (TβRI) (21). TβRI phosphorylates Smad1, Smad2, Smad3, or Smad5 (20, 22). The phosphorylated Smad forms a complex with Smad4 and translocates into the nucleus and acts as a transcription factor or DNA binding factor to promote the transcription of ECM genes such as fibrillar collagens (23).
Because ECM proteins and growth factors regulate the fibroblast-to-myofibroblast transition, our laboratory investigated the role of aortic carboxypeptidase-like protein (ACLP) in myofibroblast differentiation. ACLP is a secreted protein with more than 1100 amino acids that is associated with the ECM (24, 25). It contains an N-terminal signal sequence; a charged lysine, proline, and glutamic acid-rich domain; a discoidin domain; and a catalytically inactive metallocarboxypeptidase domain (24, 26, 27). ACLP is secreted by fibroblasts, myofibroblasts, and smooth muscle cells and is expressed in collagen-rich tissues (28). It contributes to both vascular smooth muscle cell proliferation and the wound healing process (29). Human lung tissue from patients with IPF and mouse lung tissue from mice with bleomycin-induced fibrosis exhibit abundant ACLP expression (25). Importantly, ACLP knockout mice are protected from bleomycin-induced fibrosis and have a decreased accumulation of lung myofibroblasts while exhibiting a normal inflammatory response. These findings indicate that ACLP potentially acts downstream of the inflammatory response (25). Additionally, the ACLP discoidin domain is at least partially responsible for mediating collagen matrix contraction (25).
The goal of this work is to elucidate the mechanisms by which ACLP influences myofibroblast formation. We found that ACLP promotes myofibroblast differentiation, in part, by stimulating the TGFβ signaling pathway. These studies uncovered a novel pathway that may serve as a therapeutic target to combat IPF and other types of fibrosis.
EXPERIMENTAL PROCEDURES
Cell Culture
Primary lung fibroblasts were isolated from wild-type C57Bl6 mice or from MRTFA+/+ and MRTF−/− mice (provided by Dr. Eric Olson) (30) harboring the Col1a1 promoter driving GFP (col3.6GFPtpz, provided by Dr. David Rowe) (31) as described (18). The Boston University School of Medicine Institutional Animal Care and Use Committee approved all animal experiments. Briefly, mice between 6 and 15 weeks of age were euthanized with CO2, and the lungs were perfused through the right ventricle with cold PBS containing calcium and magnesium. The lungs were minced finely, and the tissue was digested with 1× dispase (BD Falcon), 1 mg/ml type 1 filtered collagenase (Worthington), and 4.5 μg/ml DNase (Worthington). Lung fibroblasts were cultured in DMEM supplemented with 10% fetal bovine serum (Hyclone) and 1% penicillin/streptomycin and incubated in a 5% CO2 atmosphere at 37 °C. Cells were analyzed between passages 0 and 4. IMR90 fetal human lung fibroblasts (ATCC) were grown in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and incubated in a 5% CO2 atmosphere at 37 °C. Where indicated, IMR90 cells were serum-starved and treated in low-serum medium containing 0.5% fetal bovine serum and 1% penicillin/streptomycin. Mink lung epithelial cells stably transfected with a plasminogen activator inhibitor 1 promoter-luciferase reporter construct (MLEC-TGFβ) were cultured as described (25, 32). Luciferase activity was measured using luciferase assay substrate (Promega) on a BioTek Synergy HT plate reader.
SDS-PAGE and Western Blotting
Total protein lysates were harvested and analyzed by Western blot analysis as described previously (24). Nuclear and cytoplasmic protein fractionation was performed using a NE-PER kit (Thermo). Briefly, equal amounts of protein were diluted in SDS-PAGE sample buffer, boiled, run on 4–20% Novex SDS-polyacrylamide gels (Invitrogen) or 4-20% Mini-PROTEAN TGX gels (Bio-Rad), and transferred onto a nitrocellulose membrane according to standard procedures. Antibodies used include ACLP (24) (1:4000); SMA (Sigma, 1:4000); collagen α1(I) (Rockland, 1:4000); pan-actin (Sigma, 1:4000); phospho-Smad3, phospho-Smad1/5/8, total Smad2/3, total Smad1 (Cell Signaling Technology, 1:1000); TGFβ (Cell Signaling Technology, 1:1000); HDAC2 (Santa Cruz Biotechnology, 1:1000); smooth muscle myosin heavy chain (provided by Robert S. Adelstein, 1:1000); vimentin (BD Biosciences, 1:5000); and GAPDH (Sigma, 1:10000). Blots were imaged using either film or a Bio-Rad Chemidoc imaging system. Protein quantification relative to pan-actin or GAPDH was measured using Image Lab software.
Expression and Purification of Recombinant ACLP
To generate recombinant ACLP (rACLP), 293 cells were stably transfected with an ACLP construct with a BM40 (Sparc) signal sequence replacing the signal peptide and a C-terminal myc-His tag for detection and purification (Fig. 2A) (33). These cells were cultured in suspension in serum-free conditions. The conditioned medium was collected and dialyzed using 100,000 molecular weight cut off (MWCO) dialysis tubing into 300 mm KCl, 50 mm KH2PO4 (pH 8). The protein was bound to an immobilized metal affinity chromatography column (Bio-Rad Duo Flow) and washed exhaustively. During the final wash, the column was briefly washed with a sodium carbonate buffer (pH 11) as described (34) to remove remaining contaminating proteins, including, potentially, TGFβ, which was not detected in the final rACLP preparations (Fig. 2B). Protein was eluted from the column with 250 mm imidazole and concentrated and dialyzed into 1× PBS with calcium and magnesium. Protein purity was determined by SDS-PAGE followed by Coomassie staining (Fig. 2C). Protein concentration was determined via a Bradford assay, and accuracy was calibrated using amino acid analysis (Molecular Biology Core, Dana Farber Cancer Institute). The authenticity of the recombinant protein was also determined by mass spectrometry (Taplin Mass Spectrometry Facility, Harvard Medical School).
FIGURE 2.
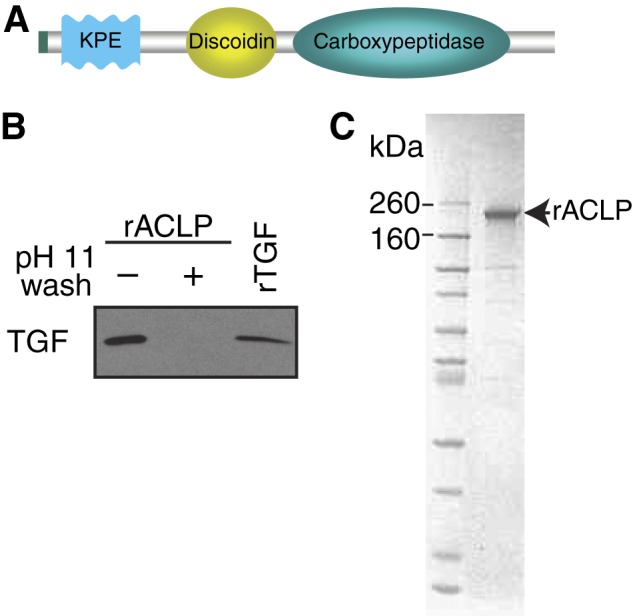
Purification of recombinant ACLP. Recombinant ACLP was purified from the conditioned medium of AD293 cells stably transfected with an ACLP construct containing a BM40 signal sequence and a His tag. A, schematic of the ACLP construct. B, ACLP (1 μg) prior to a sodium carbonate (pH 11) wash, 1 μg of recombinant ACLP after a sodium carbonate (pH 11) wash, and 0.4 ng of TGFβ were analyzed by Western blot analysis with anti-TGFβ. C. Protein gels were stained with Coomassie Blue.
Quantitative Real-time PCR
RNA was isolated from cells using RNeasy (Qiagen) according to the instructions of the manufacturer. cDNA was generated from 0.2–0.5 μg of mRNA using a Superscript III reverse transcript kit (Invitrogen). mRNA was quantified using an ABI7300 instrument with SYBR probes (Invitrogen). Intron-spanning primers were used as follows. 18 S, 5′-CGGCTACCACATCCAAGGAA-3′ (forward) and 5′-TTTTCGTCACTACCTCCCCG-3′ (reverse); Acta2, 5′-TGACGCTGAAGTATCCGATAGA-3′ (forward) and 5′-GTACGTCCAGAGGCATAGAGG-3′ (reverse); and Col1a2, 5′-GTAACTTCGTGCCTAGCAACA-3′ (forward) and 5′-CCTTTGTCAGAATACTGAGCAGC-3′ (reverse).
siRNA
Primary lung myofibroblasts were transfected with siRNA targeting ACLP (Dharmacon) using RNAiMAX (Invitrogen) either on day 1 post-isolation or between passages 2 and 4. IMR90 fibroblasts and day 2 primary lung fibroblasts were transfected with siRNA targeting Smad2 and Smad3 (siGenome SMARTpool Thermo) using RNAiMAX. Knockdown was measured by Western blot analysis. Non-targeting control siRNA was used as a control in all siRNA experiments (Dharmacon). Sequences are available upon request.
Immunofluorescence Staining
IMR90 cells were plated onto glass chamber slides and treated as described in the figure legends. At the time of harvest, cells were washed, fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and incubated with anti-Smad2/3 antibodies (Cell Signaling Technology, catalog no. 8685), followed by Alexa Fluor 488- or 568-conjugated secondary antibodies (Invitrogen). Samples were counterstained with DAPI and mounted. Images were taken at identical exposures using an Axiovert 200 M microscope (Carl Zeiss) and an ORCA-ER camera (Hamamatsu) or an Eclipse TE2000e microscope and Digital Sight DSQi1Mc camera (Nikon). Passaged primary lung myofibroblasts isolated from MRTFA+/+ and MRTF−/− mice were plated onto glass chamber slides and treated with 3.75 μg/ml rACLP or 1 nm TGFβ. At the time of harvest, Cells were then fixed in 1× PIPES-HEPES-EGTA-magnesium (PHEM) buffer, 3.7% paraformaldehyde and mounted with DAPI counterstain (35). Images were taken on a Zeiss LSM confocal microscope with constant exposure times.
TβR-I HA K232R Transfection
IMR90 cells were plated onto chamber slides coated with 0.1% gelatin and transfected with 500 ng of pCMV TβR-I HA K232R using Lipofectamine 2000 (Invitrogen) (21). The cells were serum-starved, treated as described, fixed, permeabilized, and stained with anti-HA (Roche) and anti-Smad2/3, followed by Alexa Fluor 568 goat anti-rabbit IgG and Alexa Fluor 488 goat anti-rat IgG (Invitrogen).
ACLP Binding Assays
rACLP or TGFβ (R&D Systems) were biotinylated using an EZ-Link Sulfo-NHS-LC-Biotin kit (Thermo). Approximately 1 μg of biotinylated rACLP or 0.5 μg of TGFβ was immobilized on streptavidin high binding capacity coated plates (Thermo) overnight at 4 °C in binding buffer (PBS containing magnesium, calcium, 0.1% BSA, and 0.05% Tween 20). The next day, the plates were washed with binding buffer and blocked with the same buffer containing 1% BSA at room temperature for 1 h, followed by incubation with increasing amounts of TβR-II Fc chimera (R&D Systems) at room temperature for 1 h. Wells were washed, and binding was detected with anti-human-IgG-HRP diluted 1:800, followed by incubation with 3,3′,5,5′-tetramethylbenzidine (eBioscience). The reaction was quenched with 2 m H2SO4, and absorbance at 450 nm was measured.
Statistical Analysis
Data are presented as mean ± S.E. Statistical significance was determined by Student's t test for comparisons between two groups or analysis of variance for groups of more than two and defined as p < 0.05.
RESULTS
ACLP Promotes the Fibroblast-to-Myofibroblast Transition in Differentiating Primary Lung Cells
Passaged primary lung fibroblasts express high levels of myofibroblast markers, including collagen I, SMA, and ACLP (data not shown), which suggests that these cells quickly differentiate into myofibroblasts when grown on plastic. To characterize the kinetics of the differentiation process and elucidate the role of ACLP in myofibroblast differentiation, protein was harvested from freshly isolated mouse lung fibroblasts 1, 2, 3, and 4 days post-isolation and analyzed by Western blot analysis (Fig. 1A). On day 1, the cells did not express detectable ACLP, SMA, or collagen. ACLP protein expression was first detected on day 2 after isolation, whereas SMA and collagen were first detected on day 3 post-isolation. By day 4, the cells expressed high levels of ACLP, SMA, and collagen. These data suggest that ACLP protein expression precedes SMA and collagen I protein expression as fibroblasts differentiate into myofibroblasts. On the basis of these results and our prior in vivo studies correlating a loss of ACLP with a reduction in lung fibrosis (25), we hypothesized that ACLP promotes the fibroblast-to-myofibroblast transition.
FIGURE 1.
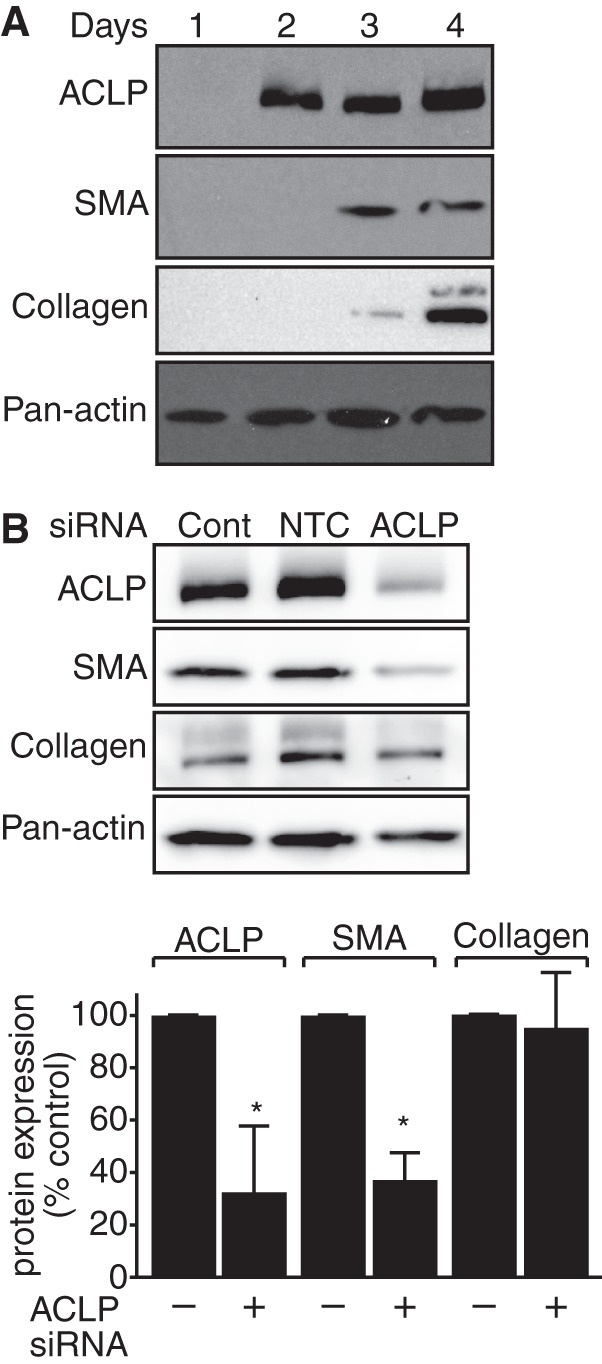
ACLP expression drives SMA and collagen expression in primary differentiating mouse lung fibroblasts. A, primary lung fibroblasts were isolated from wild-type mice and plated onto tissue culture plastic. Protein was harvested 1, 2, 3, and 4 days post-isolation and analyzed using SDS-PAGE and Western blot analysis with antibodies against ACLP, SMA, collagen, and pan-actin. B, differentiating mouse lung fibroblasts were transfected with siRNA targeting ACLP 1 day after isolation. Protein was harvested 2 days after transfection and analyzed by SDS-PAGE and Western blot analysis with antibodies against ACLP, SMA, collagen I, and pan-actin. Protein levels were quantified relative to pan-actin levels. Data presented are representative of three separate experiments. Cont., control. NTC., non-targeting control. *, p < 0.05 versus control treated cells.
To examine the role of ACLP in promoting fibroblast-to-myofibroblast differentiation, we used siRNA-mediated knockdown of ACLP during myofibroblast differentiation. Freshly isolated wild-type primary lung fibroblasts were transfected on day 1 post-isolation with siRNA targeting ACLP (Fig. 1B). Cells transfected with ACLP siRNA showed a 68% reduction in ACLP protein expression and exhibited a statistically significant 63% reduction in SMA protein expression as compared with cells transfected with control siRNA. Collagen levels were reduced by only 5%, suggesting that ACLP knockdown alone is insufficient to significantly regulate collagen at this time point.
Recombinant ACLP Enhances the Fibroblast-to-Myofibroblast Transition
We generated and purified recombinant ACLP from mammalian cells to perform gain of function studies in fibroblasts (Fig. 2). Trace amounts of TGFβ were removed from recombinant ACLP preparations by adding a sodium carbonate buffer (pH 11) wash as described (34) (Fig. 2B). Recombinant ACLP was glycosylated in a pattern consistent with native ACLP (not shown). Primary lung fibroblasts were isolated from wild-type mice and plated into medium containing rACLP or recombinant TGFβ (R&D Systems) as a positive control because TGFβ is a well known mediator of the fibroblast-to-myofibroblast transition (9, 16). RNA was harvested after 3 days and analyzed by RT quantitative PCR (Fig. 3A). Importantly, rACLP induced a statistically significant increase in Acta2 (SMA) and Col1a2 gene expression in differentiating myofibroblasts. Treatment of primary differentiating fibroblasts with rACLP also induced an increase in SMA protein levels on day 3 post-isolation as compared with untreated control cells (Fig. 3B). These data indicate that exogenous ACLP can enhance myofibroblast formation in differentiating primary lung fibroblasts.
FIGURE 3.
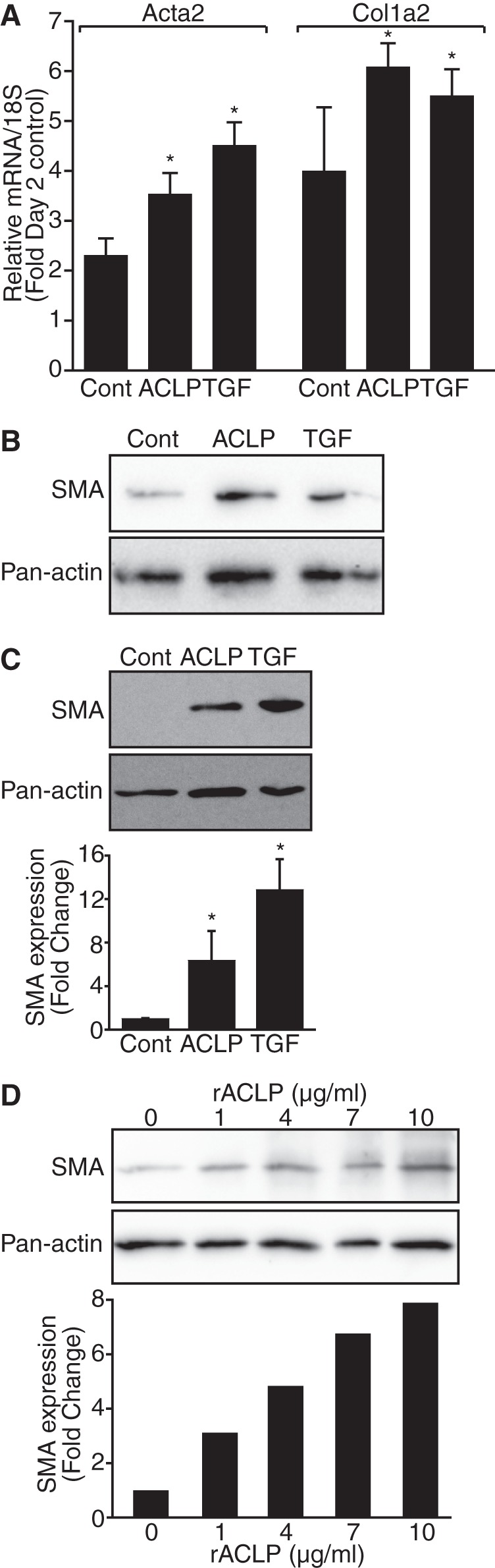
Recombinant ACLP promotes myofibroblast formation. A, mouse primary lung fibroblasts were treated at the time of isolation with 3.75 μg/ml (∼30 nm) rACLP or 1 nm TGFβ. RNA was harvested from treatments in triplicate after 3 days. RT quantitative PCR was used to measure gene expression of Acta2 (SMA) and Col1a2. Gene expression was normalized to 18 S expression of control-treated (Cont.) cells harvested 2 days after isolation. *, p < 0.05 versus control-treated cells. B, mouse primary lung fibroblasts were treated at the time of isolation with 3.75 μg/ml rACLP or 1 nm TGFβ. Protein lysate was harvested on day 3 post-isolation and analyzed by Western blot analysis with antibodies against SMA and pan-actin. C, IMR90 human lung fibroblast cells were treated in low-serum medium with 3.75 μg/ml rACLP or 1 nm TGFβ for 48 h and analyzed by SDS-PAGE and Western blot analysis with antibodies against SMA and pan-actin. SMA protein levels were determined relative to pan-actin levels. *, p < 0.05 versus control-treated cells. D, IMR90 cells were treated in low-serum medium with 0, 1, 4, 7, or 10 μg/ml rACLP for 48 h and analyzed by SDS-PAGE and Western blot analysis with antibodies against SMA and pan-actin. SMA protein levels were determined relative to levels of pan-actin. Data presented are representative of three separate experiments.
To further define the role of ACLP in the fibroblast-to-myofibroblast differentiation process, IMR90 human lung fibroblast cells were used. IMR90 cells do not express endogenous ACLP and are, therefore, a good system to examine the effects of ACLP in gain of function studies. IMR90 cells were grown in medium containing 3.75 μg/ml (30 nm) rACLP or 1 nm TGFβ as a positive control (Fig. 3C). Dose-response experiments revealed that 3.75 μg/ml or ∼30 nm was optimal to induce myofibroblast differentiation (Fig. 3D). Compared with untreated control cells, rACLP induced a 6-fold increase in SMA protein expression in IMR90 cells after a 48-h treatment. Taken together, these data suggest that ACLP promotes the fibroblast-to-myofibroblast differentiation by significantly stimulating expression of SMA and collagen I.
ACLP Stimulates TGFβ Signaling Pathways
Because ACLP is similar to TGFβ in its ability to stimulate SMA and collagen I expression (16), we investigated whether the mechanism of ACLP action might be related to TGFβ signaling pathways. TGFβ signaling is regulated by a complex process involving a number of extracellular factors, including activation of latent TGFβ by thrombospondin (36), integrin signaling (37), and receptor activity (21). Additionally, collagen can interact with the TβR complex independently of the TGFβ ligand (38, 39). TGFβ signaling in fibroblasts induces the phosphorylation of Smad3, which then binds to Smad4 and translocates into the nucleus to promote transcription of myofibroblast markers (22, 40). On the basis of this knowledge, serum-starved IMR90 cells were treated with rACLP for 15, 30, 45, or 60 min or 24 h (Fig. 4A). Acute rACLP treatment stimulated Smad3 phosphorylation, first detected after 15 min, and dramatically increased Smad1 phosphorylation after 30 min. This phosphorylation was transient and was not detectable after 24 h. Interestingly, Smad2/3 protein expression was reduced after long term rACLP treatment. On the basis of these findings, we examined the dose response of rACLP treatment at 30 min. As little as 1 μg/ml rACLP induced Smad3 phosphorylation in IMR90 fibroblasts, whereas higher doses also increased Smad1 phosphorylation (Fig. 4B).
FIGURE 4.
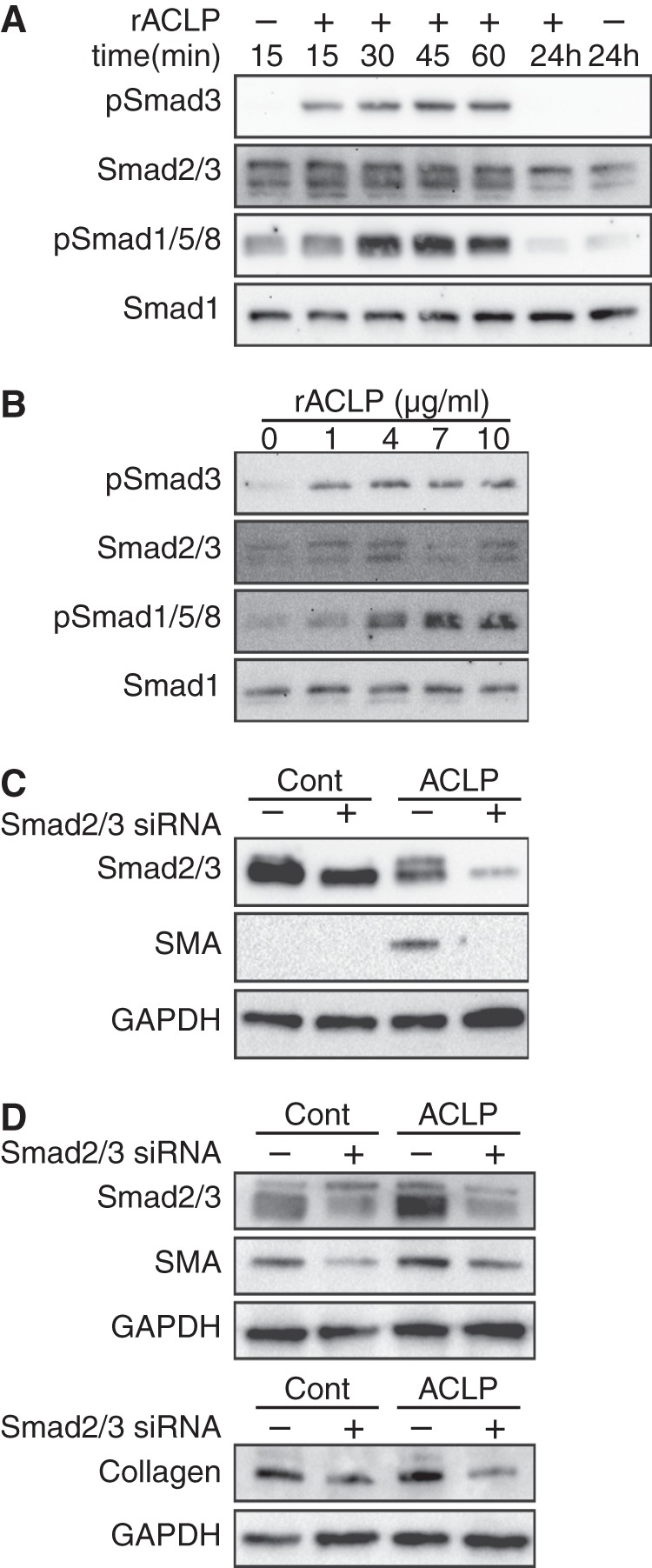
ACLP stimulates Smad3 phosphorylation and enhances SMA protein expression. A, IMR90 human lung fibroblasts were serum-starved overnight and treated with 3.75 μg/ml rACLP for 15, 30, 45, or 60 min or 24 h. Protein lysate was harvested and analyzed by SDS-PAGE and Western blot analysis with antibodies against phospho-Smad3, total Smad2/3, phospho-Smad1/5/8, and total Smad1. B, serum-starved IMR90 cells were treated with 0, 4, 7, or 10 μg/ml rACLP for 30 min. Protein lysate was harvested and analyzed by SDS-PAGE and Western blot analysis with antibodies against phospho-Smad3, total Smad2/3, phospho-Smad1/5/8, and total Smad1. C, IMR90 cells were transfected with siRNA targeting Smad2 and Smad3 and then treated with 3.75 μg/ml rACLP for 48 h. Protein lysates were analyzed by Western blot analysis with antibodies against SMA, total Smad2/3, and GAPDH. Cont., control. D, primary mouse lung fibroblasts were transfected with siRNA targeting Smad2 and Smad3 2 days post-isolation and then treated with 3.75 μg/ml rACLP for 48 h. Protein lysates were analyzed by Western blot analysis with antibodies against SMA, total Smad2/3, and GAPDH. A second gel was run and analyzed by antibodies against collagen and GAPDH. Data presented are representative of three separate experiments.
Smad2/3 Knockdown inhibits ACLP-induced Fibroblast-to-Myofibroblast Transition
Short-term rACLP treatment of IMR90 cells induced Smad3 phosphorylation, whereas long-term rACLP treatment of IMR90 cells induced SMA protein expression. TGFβ is known to mediate SMA protein expression in lung myofibroblasts via several pathways, including Smad3 phosphorylation (41). Therefore, we hypothesized that ACLP increases SMA protein expression by inducing Smad3 phosphorylation. IMR90 cells were transfected with siRNA targeting Smad2 and Smad3 and treated with rACLP for 48 h (Fig. 4C). A knockdown of ∼40–50% of Smad2/3 resulted in a dramatic reduction in SMA protein expression. Although the transfection efficiency of IMR90 cells is relatively low, ACLP-induced SMA protein expression was dependent on Smad2/3. These data suggest that ACLP stimulates Smad3 phosphorylation to promote myofibroblast differentiation. Additionally, primary lung fibroblasts were transfected with siRNA targeting Smad2 and Smad3 on day 2 post-isolation, followed by treatment with rACLP (Fig. 4D). Smad2/3 siRNA reduced Smad2/3 expression in these primary cells by 60–70%. rACLP increased SMA protein expression in non-targeting control transfected cells, and this increase was blunted in the cells with Smad2/3 knocked down. Similarly, rACLP increased collagen protein expression in non-targeting control transfected cells, and this increase was reduced in cells transfected with Smad2/3 siRNA. Together, these findings indicate that Smad2/3 participates in ACLP-mediated myofibroblast differentiation.
ACLP Stimulates Smad2/3 Nuclear Translocation
Because Smad3 phosphorylation is followed by its translocation into the nucleus (22), the effects of ACLP on Smad3 localization were investigated. Serum-starved IMR90 cells plated onto chamber slides were treated with rACLP for 30 min and stained for Smad2/3 (Fig. 5A). rACLP treatment resulted in a clear nuclear localization of Smad2/3 as compared with untreated control cells. 90% of cells treated with rACLP showed Smad2/3 nuclear translocation. Interestingly, although ACLP-induced Smad3 phosphorylation was transient by Western blot analysis (Fig. 4A), there was a persistent increase in nuclear localization of Smad2/3 in cells stained after 48-h treatment with rACLP (Fig. 5B). 96% of IMR90 cells treated with rACLP for 48 h exhibited Smad2/3 nuclear translocation.
FIGURE 5.
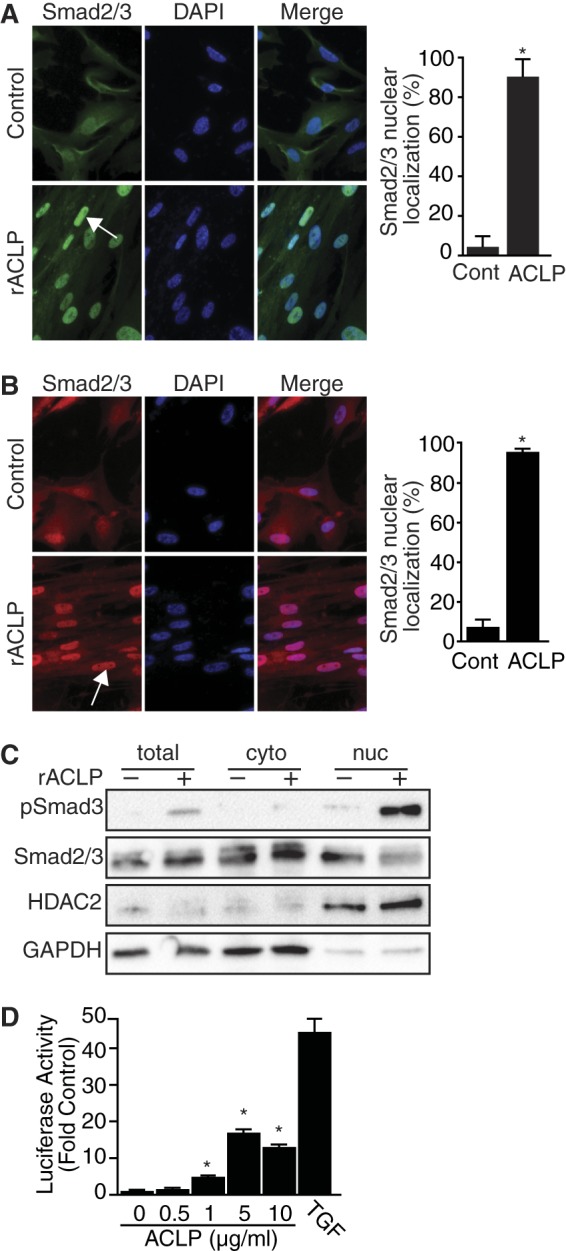
ACLP induces Smad3 nuclear translocation and transcriptional reporter activity. A, IMR90 fibroblasts were treated for 30 min in low-serum medium containing 3.75 μg/ml rACLP. Cells were fixed and stained for Smad2/3 using Alexa Fluor 488 goat anti-rabbit IgG as a secondary antibody. Nuclei were counterstained with DAPI. Images were captured at identical exposures. Cont., control. B, IMR90 fibroblasts were treated for 48 h in low-serum medium containing 3.75 μg/ml rACLP. Cells were fixed and stained for Smad2/3 using Alexa Fluor 568 goat anti-rabbit IgG as a secondary antibody. Nuclei were counterstained with DAPI. Images were captured at identical exposures. C, IMR90 human lung fibroblasts were serum-starved overnight and treated with 3.75 μg/ml rACLP for 30 min. Cytoplasmic (cyto) and nuclear (nuc) extracts were isolated and analyzed analyzed by SDS-PAGE and Western blot analysis with antibodies against phospho-Smad3, total Smad2/3, HDAC2, and GAPDH. D, mink lung epithelial cells stably transfected with the PAI-1 promoter driving luciferase activity (MLEC-TGFβ) were treated for 24 in low-serum medium containing 0, 0.5, 1, 5, or 10 μg/ml rACLP. Cell lysates were analyzed for luciferase activity. Data are presented as relative to untreated control cells. *, p < 0.05) versus control-treated cells. Data presented are representative of three separate experiments.
To further explore rACLP-induced Smad2/3 nuclear translocation, nuclear and cytoplasmic extracts were harvested from IMR90 cells treated with rACLP for 30 min (Fig. 5C). The nuclear fraction of cells treated with rACLP for 30 min had an increase in phospho-Smad3 expression as compared with control-treated cells. There were very low levels of phospho-Smad3 found in the cytoplasm of ACLP-treated cells, confirming nuclear translocation. Taken together, these results indicate that ACLP stimulates Smad signaling pathways important to the fibroblast-to-myofibroblast transition.
ACLP Stimulates TGFβ Reporter Activity
To identify the mechanism of action of ACLP, MLEC-TGFβ cells were treated with increasing amounts of rACLP for 24 h (Fig. 5D) Protein was harvested, and luciferase activity was measured. Cells treated with as little as 1 μg/ml rACLP had a statistically significant increase in luciferase activity as compared with control-treated cells.
ACLP Stimulates the Fibroblast-to-Myofibroblast Transition, in Part, through the TGFβ Receptors
Smad3 phosphorylation is induced upon TβR-II activation (22). Given the induction of Smad3 phosphorylation of ACLP, we examined the relationship between ACLP and the TβRs. Serum-starved IMR90 cells were treated with rACLP in the presence of a TGFβ-receptor kinase inhibitor (SB431542, Sigma) for 30 min (Fig. 6A) or 48 h (B). SB431542 inhibited rACLP-induced Smad3 phosphorylation at 30 min and SMA and collagen protein expression after 48 h. These data suggest that ACLP promotion of the fibroblast-to-myofibroblast transition is, at least in part, dependent on TGFβ receptor kinase activity. To further confirm the role of TGFβ receptor kinase activity on ACLP-induced Smad3 signaling, IMR90 cells were transfected with pCMV TβR-I HA K232R, a kinase-dead (dominant negative) mutant of TβR-I (21), serum-starved, treated with rACLP for 30 min, and stained for the HA tag and Smad2/3 localization (Fig. 6C). Compared with cells negative for HA, cells positive for TβR-I HA K232R exhibited a strong reduction (∼82%) in Smad2/3 nuclear translocation.
FIGURE 6.
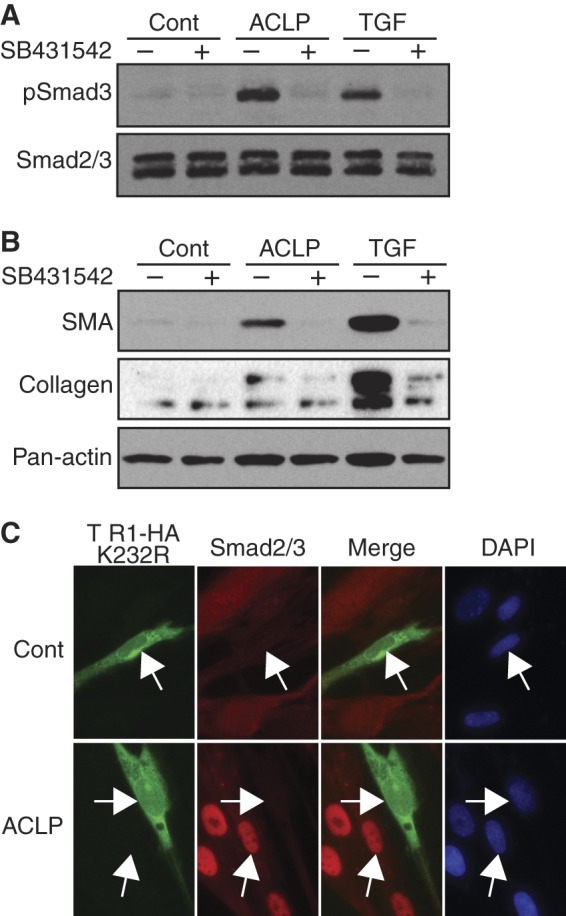
ACLP induction of TGFβ signaling pathways and myofibroblast protein expression is dependent on TGFβ receptor activity. IMR90 lung fibroblasts were treated in low-serum medium with 3.75 μg/ml rACLP or 1 nm TGFβ with either dimethyl sulfoxide as a vehicle control or 5 μm SB431542, a TGFβ receptor inhibitor. A, cell lysates were analyzed by Western blot analysis after 30-min treatment following serum starvation with antibodies against phospho-Smad3 and total Smad2/3. Cont., control. B, cell lysates were analyzed by Western blot analysis after 48 h with antibodies against SMA and pan-actin. C, IMR90 cells plated onto 0.1% gelatin-coated chamber slides were transfected with pCMV TβR-I HA K232R using Lipofectamine 2000, serum-starved overnight, and then treated with 3.75 μg/ml rACLP for 30 min. Cells were fixed and stained for HA and Smad2/3 using Alexa Fluor 488 goat-anti rat IgG and Alexa Fluor 568 goat anti-rabbit IgG as secondary antibodies. Nuclei were counterstained with DAPI. Data presented are representative of three separate experiments.
ACLP Binds to the TGFβ Receptor II
Several proteins, including bone morphogenic proteins, growth and differentiation factors, Müllerian inhibiting substance, nodals, activins, and inhibins, as well as collagen peptides, can modulate TGFβ receptor activity and Smad signaling (38, 42). Because ACLP can stimulate downstream Smad signaling, we examined whether ACLP binds directly to TβR-II. rACLP was biotinylated, immobilized on a streptavidin-coated plate, and incubated with increasing amounts of TβR-II Fc chimera (R&D Systems), and then binding was detected with human IgG-HRP (Fig. 7A). The TβR-II Fc chimera contains the extracellular domain of mouse TβR-II fused to the Fc domain of human IgG and has been used as a global inhibitor of TGFβ (43). In these assays, TβR-II Fc did not bind to streptavidin wells without rACLP. Wells with immobilized rACLP had statistically significantly more binding to the TβR-II Fc chimera as compared with control-coated wells.
FIGURE 7.
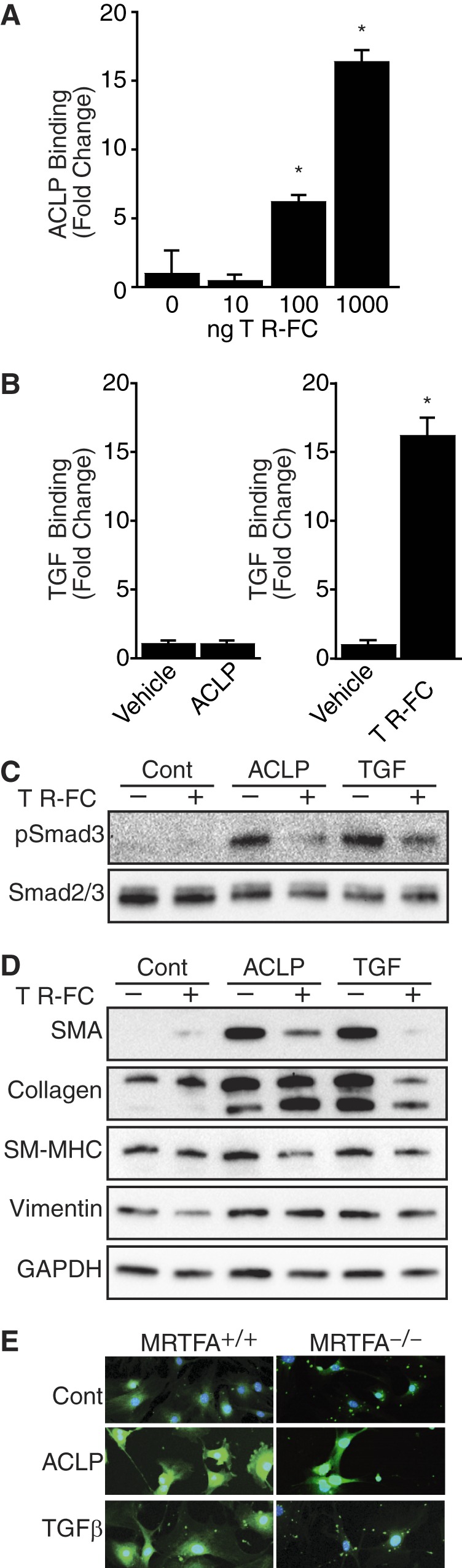
ACLP promotes myofibroblast protein expression via direct interaction with the TGFβ receptor. A, rACLP was biotinylated and immobilized to streptavidin-coated plates. Binding buffer alone was used as a control. The wells were blocked, incubated with increasing amounts of TβR-II Fc chimera (0, 10, 100, 1000 ng), followed by human IgG-HRP and 3,3′,5,5′-tetramethylbenzidine substrate. The reaction was quenched with 2 m H2SO4, and binding was measured by reading the absorbance at 450 nm. Data are presented as binding minus TβR-II Fc background binding. *, p < 0.05) versus control-treated cells. B, TGFβ was biotinylated and immobilized to streptavidin-coated plates overnight. Binding buffer alone was used as a control. The wells were blocked and incubated with either rACLP or TβR-II Fc chimera, followed by anti-ACLP and rabbit IgG-HRP or human IgG-HRP and 3,3′,5,5′-tetramethylbenzidine substrate. The reaction was quenched with 2 m H2SO4, and binding was measured by reading the absorbance at 450 nm. Results are presented as binding minus ACLP (left panel) or TβR-II Fc (right panel) background binding. *, p < 0.05 versus control-treated cells. C, serum-starved IMR90 cells were treated in low-serum medium containing 100 ng/ml of a TβR-Fc chimera (R&D Systems) with 3.75 μg/ml rACLP or 100 pm TGFβ for 30 min. Protein lysates were analyzed by Western blot analysis with antibodies against phospho-Smad3 or total Smad2/3. Cont., control. D, IMR90 cells were treated in low-serum medium containing 100 ng/ml of a TβR-Fc chimera (R&D Systems) with 3.75 μg/ml rACLP or 100 pm TGFβ for 48 h. Protein lysates were analyzed by Western blot analysis with antibodies against SMA, collagen I, smooth myosin heavy chain (SM-MHC), vimentin, and GAPDH. E, primary lung myofibroblasts isolated from MRTFA+/+ and MRTF−/− mice were treated with 3.75 μg/ml rACLP or 1 nm TGFβ for 48 h. Cells were then fixed in 1× PHEM buffer, 3.7% paraformaldehyde and mounted with DAPI counterstain. Pictures were taken on a Zeiss LSM confocal microscope with constant exposure times. Data presented are representative of three separate experiments.
ACLP and TGFβ Do Not Bind Directly
To determine whether ACLP might bind directly to TGFβ, biotinylated TGFβ was immobilized on a streptavidin-coated plate and incubated with either rACLP or the TβR-II Fc chimera, and binding was detected with either anti-ACLP and rabbit IgG-HRP or human IgG-HRP. Wells incubated with ACLP showed no binding above background (Fig. 7B, left panel). In contrast, wells incubated with the TβR-II Fc chimera had binding significantly above background (Fig. 7B, right panel).
ACLP Interacts with the TGFβ Receptor to Promote Fibroblast-to-Myofibroblast Differentiation
Smad3 phosphorylation is known to be induced when the TGFβ ligand binds to TβR-II (22). Because ACLP stimulates Smad3 phosphorylation in fibroblasts and binds to TβR-II in vitro, we hypothesized that the effects of ACLP might be inhibited by the TβR-II Fc chimera. IMR90 fibroblasts were treated with rACLP or TGFβ in the presence of 100 ng/ml of the TβR-II Fc chimera. Cotreatment of fibroblasts with rACLP and TβR-II Fc for 30 min resulted in a reduction of Smad3 phosphorylation as compared with cells treated with rACLP alone (Fig. 7C). Similarly, fibroblasts treated with rACLP and TβR-II for 48 h had lower levels of SMA as compared with cells treated with rACLP alone (Fig. 7D). Similar effects were observed with smooth myosin heavy chain and vimentin. Although the TβR-II Fc treatment blunted TGFβ-induced collagen expression, it did not alter ACLP-dependent collagen changes. These data suggest that ACLP interacts with TβR-II to promote Smad3 phosphorylation and nuclear translocation to promote SMA protein expression and myofibroblast differentiation. These studies also indicated that ACLP regulates collagen expression through a pathway that is distinct from that used by TGFβ. Furthermore, these studies also uncovered a differential regulation of SMA and collagen in lung fibroblasts.
To explore the mechanism by which ACLP stimulates collagen expression (Fig. 3A) and to probe the differences in pathways stimulated by ACLP and TGFβ, we examined the role of MRTFA. Our recent work established an important role for MRTFA in stimulating type I collagen expression through complexes with both serum response factor and Sp1 (18). We examined lung fibroblasts isolated from MRTFA+/+ and MRTF−/− mice harboring the Col1a1 promoter driving GFP expression (col3.6GFPtpz). Unlike TGFβ, ACLP stimulated collagen promoter activity in the absence of MRTFA (Fig. 7E). Together, these results indicate that ACLP stimulation of collagen expression does not absolutely require the TGFβ receptor or the serum response factor coactivator MRTFA.
ACLP Knockdown Slows and Partially Reverts the Fibroblast-to-Myofibroblast Transition
We have shown previously that ACLP knockout mice are protected from bleomycin-induced fibrosis (25) and that ACLP knockdown slows myofibroblast differentiation in primary mouse lung fibroblasts (Fig. 1B). Therefore, we examined whether knockdown of ACLP in primary lung myofibroblasts would cause them to revert back into fibroblasts. Passaged primary lung myofibroblasts were transfected with siRNA targeting ACLP (Fig. 8A). ACLP knockdown resulted in a decrease in SMA protein expression as compared with non-targeting control-transfected cells. As seen in differentiating myofibroblasts, ACLP knockdown alone is not sufficient to significantly decrease collagen expression. These results suggest that ACLP plays an integral role in the differentiation of fibroblasts to myofibroblasts by signaling through TβR-II to promote Smad3 phosphorylation and nuclear translocation to stimulate SMA. Additionally, ACLP signals through an MRTFA-independent pathway to promote collagen expression (Fig. 8B).
FIGURE 8.
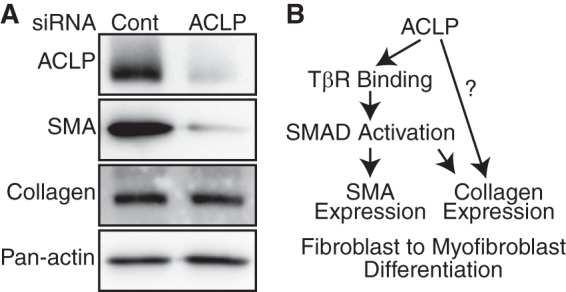
ACLP knockdown partially reverses the myofibroblast SMA expression. A, primary lung myofibroblasts were transfected at low passage number with 2 nm ACLP or non-targeting control siRNA (Cont.) using RNAiMax (Invitrogen). Protein was harvested 48 h later and analyzed with antibodies against ACLP, SMA, collagen I, and pan-actin. Data presented are representative of three separate experiments. B, proposed scheme for ACLP induction of the fibroblast-to-myofibroblast transition.
DISCUSSION
This study identified a novel mechanism by which ACLP controls lung myofibroblast differentiation. Specifically, we discovered that ACLP interacts with and activates TGFβ receptors and stimulates downstream Smad3 signal transduction pathways to enhance SMA expression and signals through an MRTFA-independent pathway to promote collagen expression. Furthermore, we identified two strategies to interfere with ACLP-mediated myofibroblast differentiation. Together with the known roles of TGFβ signaling in fibrotic lung disease, these studies indicate that ACLP is a novel regulator that stimulates and maintains SMA, collagen, and myofibroblast gene expression.
Idiopathic pulmonary fibrosis is a rapidly progressive and fatal disease with no effective therapies (1). It is characterized by an increased deposition of collagen and other ECM proteins as well as increased expression of SMA (2), leading to scarring and thickening of tissue and a progressive decline of lung function (44). One key effector cell of IPF is the myofibroblast. It is responsible for increased ECM protein deposition and increased expression of SMA (2). Our laboratory has previously shown an increase in ACLP in human fibrotic lungs and early up-regulation of ACLP in the bleomycin model of fibrosis in mice (25). Using a rapidly differentiating lung fibroblast model, we have shown that ACLP precedes SMA and collagen expression in differentiating myofibroblasts (Fig. 1A) and that exogenous ACLP is sufficient to replicate this process (Fig. 3).
ACLP knockdown slows the fibroblast-to-myofibroblast transition, as evidenced by a subsequent reduction in SMA protein expression. However, collagen protein expression was not reduced significantly. This could be due to the stability of collagen or to other factors playing a role in regulating its expression. Collagen and SMA can both be regulated by TGFβ signaling through Smad phosphorylation and MRTFA localization (17, 18). However, our results suggest that there is a differential regulation of SMA and collagen by ACLP. Additionally, the promotion of collagen expression of ACLP is not inhibited by the TβR-II Fc chimera, whereas TGFβ-induced collagen expression is blocked by the TβR-II Fc chimera. This indicates that ACLP and TGFβ promote collagen expression via distinct pathways. This is also shown when ACLP knockdown in either differentiating myofibroblasts or fully differentiated myofibroblasts results in only a small reduction in collagen expression. Differential regulation of the myofibroblast differentiation by ACLP and TGFβ was confirmed in our MRTFA knockout system. It will be interesting to study the effects of ACLP on SMA expression and stress fiber formation in the MRTFA-null system. TGFβ requires the presence of MRTFA to promote collagen promoter activity, whereas ACLP promotes collagen promoter activity in an MRTFA-independent manner. Collagen transcription can be positively regulated by several transcription factors, including Sp1, AP1, Ets1, Sp3, and CBF (45). These factors represent several possible targets of ACLP signaling.
Our results suggest that ACLP is a novel regulator of TβR signaling. ACLP binds to TβR-II without binding directly to TGFβ. The activity of ACLP is inhibited by the soluble TβR-II, is dependent on TβR kinase activity, is blocked by a dominant negative form of the TβR-I, and is dependent on the presence of Smad2/3. On the basis of the findings reported here, ACLP may act to stabilize the TGFβ-TβR complex, or ACLP may bind to TβR-II to promote its recruitment of TβR-I in a TGFβ-independent manner. TβR signaling may be activated independently of the TGFβ ligand. For example, collagen fragments activate Smad signaling to promote cross-talk between integrins and the TβR complex (38). Collagen I also stimulates the epithelial-mesenchymal transition by signaling through TβR-I independently of TGFβ ligand (39). Consistent with these findings, ACLP may interact with collagen at the cell surface via its discoidin domain and promote collagen expression as it facilitates the fibroblast-to-myofibroblast transition. Therefore, it is possible that ACLP mediates Smad signaling by facilitating the interaction between collagen and the TβR complex.
Previous studies have identified roles for ACLP in cell differentiation processes. For example, ACLP is down-regulated during adipogenesis (46), and retroviral ACLP overexpression inhibits adipogenesis while promoting a smooth muscle-like phenotype with increased contractile protein expression and enhanced stress fiber formation (47). One significant advance of this work over previous studies is the use of rACLP, which allowed us to examine ACLP function in signaling assays as well as to identify ACLP as playing a role in regulating collagen expression. Sorisky and colleagues (48) demonstrated that the effects of ACLP on adipogenesis may be mediated by its interaction with collagen. This collagen interaction is consistent with our previous results showing that ACLP is expressed in collagen-rich tissues (28) and enhances the contractility of lung fibroblasts via its collagen-binding discoidin domain (25).
Myofibroblasts are mechanically active cells that generate a feed-forward mechanism, promoting their differentiation (49). Work from Hinz and colleagues and others (50–52) has shown that myofibroblast contractility is an independent driver of fibrosis, including activation of latent forms of TGFβ and Rho-Rho kinase pathways. The increased stiffness of fibrotic lungs has been attributed to the presence of myofibroblasts (53). A recent study suggests that targeting the ability of a myofibroblast to contract the matrix could reduce fibrosis by blocking myofibroblast differentiation from precursor cells and by inducing myofibroblast apoptosis (52).
In this study, we have shown that removing ACLP leads to a decrease in SMA expression in differentiated myofibroblasts. Our study and the Zhou study (52) suggest the possibility of targeting different components of the TGFβ signaling pathway as disease progression occurs. TGFβ itself is not considered to be a good IPF drug target because of the chronic nature of the disease (54) and because blocking TGFβ long-term could lead to many detrimental side effects. Our findings that ACLP functions through both TGFβ receptor-dependent and -independent pathways may lead to future therapies for fibrotic conditions.
Acknowledgments
We thank Dr. Eric Olson (UT Southwestern Medical Center) for the MRTF-A heterozygous mice and Dr. David Rowe (University of Connecticut) for col3.6GFPtpz. We also thank Xaralabos Varelas for suggestions and reagents, Larry Luchsinger for creating the original cross of the mouse strains, and Elena Kandror for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grants HL078869 and HL078869-04S1 (to M. D. L.). This work was also supported by Department of Biochemistry start-up funds.
- IPF
- idiopathic pulmonary fibrosis
- ECM
- extracellular matrix
- SMA
- α-smooth muscle actin
- MRTFA
- myocardin-related transcription factor A
- ACLP
- aortic carboxypeptidase-like protein
- rACLP
- recombinant aortic carboxypeptidase-like protein.
REFERENCES
- 1. Ley B., Collard H. R., King T. E., Jr., (2011) Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 183, 431–440 [DOI] [PubMed] [Google Scholar]
- 2. Scotton C. J., Chambers R. C. (2007) Molecular targets in pulmonary fibrosis. The myofibroblast in focus. Chest 132, 1311–1321 [DOI] [PubMed] [Google Scholar]
- 3. Desmoulière A., Chaponnier C., Gabbiani G. (2005) Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 13, 7–12 [DOI] [PubMed] [Google Scholar]
- 4. Hashimoto N., Jin H., Liu T., Chensue S. W., Phan S. H. (2004) Bone marrow-derived progenitor cells in pulmonary fibrosis. J. Clin. Invest. 113, 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dunsmore S. E., Shapiro S. D. (2004) The bone marrow leaves its scar. New concepts in pulmonary fibrosis. J. Clin. Invest. 113, 180–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoyles R. K., Derrett-Smith E. C., Khan K., Shiwen X., Howat S. L., Wells A. U., Abraham D. J., Denton C. P. (2011) An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor β receptor. Am. J. Respir Crit. Care Med. 183, 249–261 [DOI] [PubMed] [Google Scholar]
- 7. Kim K. K., Kugler M. C., Wolters P. J., Robillard L., Galvez M. G., Brumwell A. N., Sheppard D., Chapman H. A. (2006) Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. U.S.A. 103, 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lama V. N., Phan S. H. (2006) The extrapulmonary origin of fibroblasts. Stem/progenitor cells and beyond. Proc. Am. Thorac. Soc. 3, 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., Brown R. A. (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 [DOI] [PubMed] [Google Scholar]
- 10. Werner S., Grose R. (2003) Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 83, 835–870 [DOI] [PubMed] [Google Scholar]
- 11. Hinz B. (2006) Masters and servants of the force. The role of matrix adhesions in myofibroblast force perception and transmission. Eur. J. Cell Biol. 85, 175–181 [DOI] [PubMed] [Google Scholar]
- 12. Broekelmann T. J., Limper A. H., Colby T. V., McDonald J. A. (1991) Transforming growth factor β1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. U.S.A. 88, 6642–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wynn T. A. (2008) Cellular and molecular mechanisms of fibrosis. J. Pathol. 214, 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raghow B., Irish P., Kang A. H. (1989) Coordinate regulation of transforming growth factor β gene expression and cell proliferation in hamster lungs undergoing bleomycin-induced pulmonary fibrosis. J. Clin. Invest. 84, 1836–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sime P. J., Xing Z., Graham F. L., Csaky K. G., Gauldie J. (1997) adenovector-mediated gene transfer of active transforming growth factor-β1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100, 768–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desmoulière A., Geinoz A., Gabbiani F., Gabbiani G. (1993) Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Small E. M., Thatcher J. E., Sutherland L. B., Kinoshita H., Gerard R. D., Richardson J. A., Dimaio J. M., Sadek H., Kuwahara K., Olson E. N. (2010) Myocardin-related transcription factor-a Controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 107, 294–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luchsinger L. L., Patenaude C. A., Smith B. D., Layne M. D. (2011) Myocardin-related transcription factor-a complexes activate type I collagen expression in lung fibroblasts. J. Biol. Chem. 286, 44116–44125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flaumenhaft R., Abe M., Sato Y., Miyazono K., Harpel J., Heldin C. H., Rifkin D. B. (1993) Role of the latent TGF-β binding protein in the activation of latent TGF-β by co-cultures of endothelial and smooth muscle cells. J. Cell Biol. 120, 995–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khalil N., Parekh T. V., O'Connor R. N., Gold L. I. (2002) differential expression of transforming growth factor-β type I and II receptors by pulmonary cells in bleomycin-induced lung injury. Correlation with repair and fibrosis. Exp. Lung Res. 28, 233–250 [DOI] [PubMed] [Google Scholar]
- 21. Wrana J. L., Attisano L., Wieser R., Ventura F., Massagué J. (1994) Mechanism of activation of the TGF-β receptor. Nature 370, 341–347 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y., Feng X., We R., Derynck R. (1996) Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature 383, 168–172 [DOI] [PubMed] [Google Scholar]
- 23. Liu F., Pouponnot C., Massagué J. (1997) Dual role of the Smad4/Dpc4 tumor suppressor in TGFβ-inducible transcriptional complexes. Genes Dev. 11, 3157–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Layne M. D., Endege W. O., Jain M. K., Yet S. F., Hsieh C. M., Chin M. T., Perrella M. A., Blanar M. A., Haber E., Lee M. E. (1998) Aortic carboxypeptidase-like protein, a novel protein with discoidin and carboxypeptidase-like domains, is up-regulated during vascular smooth muscle cell differentiation. J. Biol. Chem. 273, 15654–15660 [DOI] [PubMed] [Google Scholar]
- 25. Schissel S. L., Dunsmore S. E., Liu X., Shine R. W., Perrella M. A., Layne M. D. (2009) Aortic carboxypeptidase-like protein is expressed in fibrotic human lung and its absence protects against bleomycin-induced lung fibrosis. Am. J. Pathol. 174, 818–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reznik S. E., Fricker L. D. (2001) Carboxypeptidases from A to Z. Implications in embryonic development and Wnt binding. Cell Mol. Life Sci. 58, 1790–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tumelty K. E., Layne M. D. (2013) in Handbook of Proteolytic Enzymes (Rawlings N. D., Salvesen G. S., eds) pp. 1348–1353, Academic Press, Oxford [Google Scholar]
- 28. Ith B., Wei J., Yet S. F., Perrella M. A., Layne M. D. (2005) Aortic carboxypeptidase-like protein is expressed in collagen-rich tissues during mouse embryonic development. Gene. Expr. Patterns 5, 533–537 [DOI] [PubMed] [Google Scholar]
- 29. Layne M. D., Yet S. F., Maemura K., Hsieh C. M., Bernfield M., Perrella M. A., Lee M. E. (2001) Impaired abdominal wall development and deficient wound healing in mice lacking aortic carboxypeptidase-like protein. Mol. Cell Biol. 21, 5256–5261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li S., Chang S., Qi X., Richardson J. A., Olson E. N. (2006) Requirement of a myocardin-related transcription factor for development of mammary myoepithelial cells. Mol. Cell Biol. 26, 5797–5808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kalajzic Z., Li H., Wang L. P., Jiang X., Lamothe K., Adams D. J., Aguila H. L., Rowe D. W., Kalajzic I. (2008) Use of an α-smooth muscle actin Gfp reporter to identify an osteoprogenitor population. Bone 43, 501–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abe M., Harpel J. G., Metz C. N., Nunes I., Loskutoff D. J., Rifkin D. B. (1994) An assay for transforming growth factor-β Using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal. Biochem. 216, 276–284 [DOI] [PubMed] [Google Scholar]
- 33. Hochuli E., B. W., Dobeli H., Gentz R., Stuber D. (1988) Genetic approach to facilitate purification of recombinant proteins with a novel metal chelate adsorbent. Nat. Biotech. 6, 1321–1325 [Google Scholar]
- 34. Murphy-Ullrich J. E., Schultz-Cherry S., Höök M. (1992) Transforming growth factor-β complexes with thrombospondin. Mol. Biol. Cell 3, 181–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sobue K., Fujio Y., Kanda K. (1988) Tumor promoter induces reorganization of actin filaments and calspectin (fodrin or nonerythroid spectrin) in 3t3 cells. Proc. Natl. Acad. Sci. U.S.A. 85, 482–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crawford S. E., Stellmach V., Murphy-Ullrich J. E., Ribeiro S. M., Lawler J., Hynes R. O., Boivin G. P., Bouck N. (1998) Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 93, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 37. Munger J. S., Huang X., Kawakatsu H., Griffiths M. J., Dalton S. L., Wu J., Pittet J. F., Kaminski N., Garat C., Matthay M. A., Rifkin D. B., Sheppard D. (1999) The integrin αV β6 binds and activates latent TGF β1. A mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 [DOI] [PubMed] [Google Scholar]
- 38. Garamszegi N., Garamszegi S. P., Samavarchi-Tehrani P., Walford E., Schneiderbauer M. M., Wrana J. L., Scully S. P. (2010) Extracellular matrix-induced transforming growth factor-β receptor signaling dynamics. Oncogene 29, 2368–2380 [DOI] [PubMed] [Google Scholar]
- 39. DeMaio L., Buckley S. T., Krishnaveni M. S., Flodby P., Dubourd M., Banfalvi A., Xing Y., Ehrhardt C., Minoo P., Zhou B., Crandall E. D., Borok Z. (2012) Ligand-independent transforming growth factor-β type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition. J. Pathol. 226, 633–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Macías-Silva M., Abdollah S., Hoodless P. A., Pirone R., Attisano L., Wrana J. L. (1996) Madr2 is a substrate of the TGFβ receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell 87, 1215–1224 [DOI] [PubMed] [Google Scholar]
- 41. Gu L., Zhu Y. J., Yang X., Guo Z. J., Xu W. B., Tian X. L. (2007) Effect of TGF-β/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol. Sin. 28, 382–391 [DOI] [PubMed] [Google Scholar]
- 42. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 43. Horan G. S., Wood S., Ona V., Li D. J., Lukashev M. E., Weinreb P. H., Simon K. J., Hahm K., Allaire N. E., Rinaldi N. J., Goyal J., Feghali-Bostwick C. A., Matteson E. L., O'Hara C., Lafyatis R., Davis G. S., Huang X., Sheppard D., Violette S. M. (2008) Partial inhibition of integrin αVβ6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 177, 56–65 [DOI] [PubMed] [Google Scholar]
- 44. Ask K., Martin G. E., Kolb M., Gauldie J. (2006) Targeting genes for treatment in idiopathic pulmonary fibrosis. Challenges and opportunities, promises and pitfalls. Proc. Am. Thorac. Soc. 3, 389–393 [DOI] [PubMed] [Google Scholar]
- 45. Ramirez F., Tanaka S., Bou-Gharios G. (2006) Transcriptional regulation of the human α 2(I) collagen gene (Col1a2), an informative model system to study fibrotic diseases. Matrix Biol. 25, 365–372 [DOI] [PubMed] [Google Scholar]
- 46. Gagnon A., Landry A., Proulx J., Layne M. D., Sorisky A. (2005) Aortic carboxypeptidase-like protein is regulated by transforming growth factor β in 3t3-L1 preadipocytes. Exp. Cell Res. 308, 265–272 [DOI] [PubMed] [Google Scholar]
- 47. Abderrahim-Ferkoune A., Bezy O., Astri-Roques S., Elabd C., Ailhaud G., Amri E. Z. (2004) Transdifferentiation of preadipose cells into smooth muscle-like cells. Role of aortic carboxypeptidase-like protein. Exp. Cell Res. 293, 219–228 [DOI] [PubMed] [Google Scholar]
- 48. Gusinjac A., Gagnon A., Sorisky A. (2011) Effect of collagen I and aortic carboxypeptidase-like protein on 3t3-L1 adipocyte differentiation. Metabolism 60, 782–788 [DOI] [PubMed] [Google Scholar]
- 49. Hinz B. (2010) The myofibroblast. Paradigm for a mechanically active cell. J. Biomech. 43, 146–155 [DOI] [PubMed] [Google Scholar]
- 50. Wipff P. J., Rifkin D. B., Meister J. J., Hinz B. (2007) Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 179, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu F., Mih J. D., Shea B. S., Kho A. T., Sharif A. S., Tager A. M., Tschumperlin D. J. (2010) Feedback amplification of fibrosis through matrix stiffening and Cox-2 suppression. J. Cell Biol. 190, 693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou Y., Huang X., Hecker L., Kurundkar D., Kurundkar A., Liu H., Jin T. H., Desai L., Bernard K., Thannickal V. J. (2013) Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Invest. 123, 1096–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hinz B. (2009) Tissue Stiffness, Latent Tgf-β1 Activation, and mechanical signal transduction. Implications for the pathogenesis and treatment of fibrosis. Curr. Rheumatol. Rep. 11, 120–126 [DOI] [PubMed] [Google Scholar]
- 54. Leask A., Abraham D. J. (2004) TGF-β signaling and the fibrotic response. FASEB J. 18, 816–827 [DOI] [PubMed] [Google Scholar]