

Background: Phytochrome photoreceptors are activated by light-induced isomerization of the chromophore cofactor.
Results: Photoactivation of Synechococcus OS-B′ phytochrome breaks an unusual chromophore D-ring hydrogen bond, whereas only subtle changes occur at the A-ring linkage to the protein.
Conclusion: Activation arises from a photoflip of a strongly tilted D-ring.
Significance: The hypothesis that the A-ring rotates upon photon absorption is wrong.
Keywords: Biophysics, Cyanobacteria, Molecular Biology, NMR, Photoreceptors, Protein Structure, Structural Biology, Photoisomerization, Phytochrome
Abstract
Phytochrome photoreceptors in plants and microorganisms switch photochromically between two states, controlling numerous important biological processes. Although this phototransformation is generally considered to involve rotation of ring D of the tetrapyrrole chromophore, Ulijasz et al. (Ulijasz, A. T., Cornilescu, G., Cornilescu, C. C., Zhang, J., Rivera, M., Markley, J. L., and Vierstra, R. D. (2010) Nature 463, 250–254) proposed that the A-ring rotates instead. Here, we apply magic angle spinning NMR to the two parent states following studies of the 23-kDa GAF (cGMP phosphodiesterase/adenylyl cyclase/FhlA) domain fragment of phytochrome from Synechococcus OS-B′. Major changes occur at the A-ring covalent linkage to the protein as well as at the protein residue contact of ring D. Conserved contacts associated with the A-ring nitrogen rule out an A-ring photoflip, whereas loss of contact of the D-ring nitrogen to the protein implies movement of ring D. Although none of the methine bridges showed a chemical shift change comparable with those characteristic of the D-ring photoflip in canonical phytochromes, denaturation experiments showed conclusively that the same occurs in Synechococcus OS-B′ phytochrome upon photoconversion. The results are consistent with the D-ring being strongly tilted in both states and the C15=C16 double bond undergoing a Z/E isomerization upon light absorption. More subtle changes are associated with the A-ring linkage to the protein. Our findings thus disprove A-ring rotation and are discussed in relation to the position of the D-ring, photoisomerization, and photochromicity in the phytochrome family.
Introduction
Phytochromes are photochromic biliprotein photoreceptors controlling development in plants but are also represented in diverse microorganisms. In general, red light absorption by the Pr2 dark state leads to formation of the Pfr state that is photoconverted by far-red light back to Pr, physiological signaling being switched by state transition. The structural characteristics of Pr and Pfr, the photoconversion mechanism, and the physiological signals generated are all subjects of intense research (1–3).
The photosensory module of Class I phytochromes comprises a PAS (Period/Arnt/Singleminded)-GAF (cGMP phosphodiesterase/adenylyl cyclase/FhlA)-(PHY) (phytochrome-specific) tridomain. In plants and cyanobacterial phytochrome Cph1, the bilin chromophore (structure with pyrrole ring and atom labels shown in Fig. 3 inset) is covalently bound to a Cys residue in the GAF domain, whereas in bacteriophytochromes (BphPs) the attachment site is near the N terminus. Crystal structures of BphPs and Synechocystis Cph1 (Cph1) in the Pr state show the chromophore in a periplanar ZZZssa geometry associated with an intricate network of conserved hydrogen bonds, with ring D being poised at 20–40° relative to the B/C-ring plane (4–7). Early work (8) had suggested that photoconversion involved a D-ring photoflip, and indeed the Pfr conformation was shown to be ZZEssa in the unusual BphP from Pseudomonas aeruginosa in which Pfr is the lowest energy dark state (9–11). Studies with sterically locked chromophores also supported these conclusions (12). From MAS NMR of Pr and Pfr of Cph1, we showed unambiguously that they are associated with the ZZZssa and ZZEssa geometries, respectively (13). How and even whether the Z→E photoisomerization and associated D-ring photoflip itself leads to red/far-red photochromicity and triggers signaling remains uncertain, however. In any case, it is clear that photoconversion involves changes in the molecule in addition to the D-ring flip, including major salt bridge rearrangements associated with the chromophore and more widespread changes in rigidity (1, 2, 13, 14). Proton transfer is also involved, but although the chromophore probably deprotonates transiently prior to Pfr formation, all four pyrrole nitrogens are protonated in both Pr and Pfr (13, 15–20).
FIGURE 3.
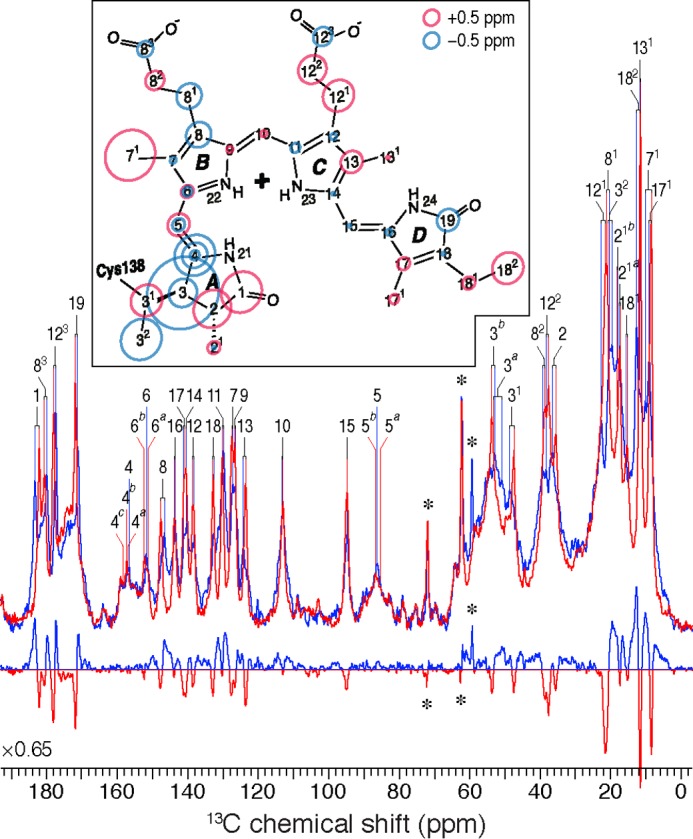
One-dimensional 13C CP/MAS NMR spectra of [U-13C,13N]PCB-SyB.Cph2(GAF) as P630 (red) and P690 (blue) and the P690–P630 difference spectrum (bottom). 13C resonances as P630 and P690 are labeled. Resonances of the natural abundance glycerol carbons are indicated by asterisks. The inset shows the changes in 13C shifts of the chromophore during photoconversion. The P630 state is taken as reference, and the size of the circles is proportional to the difference as P690–P630. Carbons showing splittings are labeled with multiple circles.
Liquid NMR studies of phytochrome have been hampered by both the large size of the photochemically functional PAS-GAF-PHY module and the high mobility of the chromophore within it (19–21). Vierstra and co-workers (22) described a phytochrome from the thermotolerant cyanobacterium Synechococcus OS-B′ (here termed SyB.Cph2 as suggested (3)), which showed orange/far-red photochromicity resembling that of canonical phytochromes despite missing the N-terminal PAS domain. Photochromicity was retained by the isolated 23-kDa GAF-domain fragment (SyB.Cph2(GAF)), a remarkable finding, as phytochromes generally lose their ability to form bona fide Pfr if PHY domain function is compromised. However, the “Pfr” state of SyB.Cph2(GAF) not only showed a ∼30 nm blue shift (λmax at 630 and 690 nm (see Fig. 1G), and thus the parent states are termed P630 and P690, respectively) but also lost the Pfr typical resonance Raman band at 815 cm−1 (22). Despite these unusual properties, the 23-kDa SyB.Cph2(GAF) fragment was a potentially valuable object for liquid NMR structural studies, and indeed from such data model ensembles for both P630 and P690 were published (23, 24). These led to the proposal that a photoflip of ring A rather than ring D occurred in SyB.Cph2(GAF), and indeed it was argued that this might be the case in all phytochromes (24). Although changes in addition to D-ring rotation are well known, the notion of an A-ring photoflip as the primary photochemical event conflicts with a large body of published data. This as well as experimental and theoretical problems associated with the work itself (discussed below) prompted us to apply the MAS NMR methods that we have established for canonical phytochromes (2) to SyB.Cph2(GAF).
FIGURE 1.

Two-dimensional MAS NMR 13C–13C homonuclear dipolar correlation spectra of [U-13C,15N]PCB-SyB.Cph2(GAF). Polarization transfer times of 5 and 50 ms were employed for the P630 (A, red and purple, respectively) and P690 (B, orange and cyan, respectively) states. The blue lines in two full contour plots indicate sequences of nearest-neighbor correlations (for numbering see Fig. 3, inset; for full assignments, including indirect-bonded correlations for P630 and P690 and one-dimensional spectra, see supplemental Figs. S2 and S3, respectively). The observed 13C signal splittings of a subset of carbon resonances are illustrated in C–F for P630 and P690, respectively (expansions are inset). The data were collected with an 8-ms evolution in the indirect dimension; 1434 complex t2 and 128 real t1 points with 2048 scans. A relaxation delay of 1.5 s was applied. G, UV-visible absorbance spectra of the SyB.Cph2(GAF) preparations used in this study. The spectra of P630 and of the P690/P630 photoequilibrium mixture were measured following irradiation with >700 and 525 nm light, respectively, as described.
Although, as in canonical phytochromes, major 13C chemical shift (δC) changes are associated with the A-ring linkage to the protein at C3 and C31 (for atom numbering, see Fig. 3, inset), an A-ring photoflip can be ruled out as the 1H–15N heteronuclear correlations between the B-ring nitrogen (N22) and the proton attached to A-ring nitrogen (HN21) are seen in both states. Although significant δC changes associated with the ring D are seen (for example at C19), these are quite different from and much smaller than those in canonical phytochromes. Denaturation experiments showed conclusively, however, that ring D does undergo a Z→E photoflip in the native holoprotein. The discrepancy can be resolved on the basis of a strongly twisted C15=C16 double bond, however. These findings together with others imply that phytochrome photochromicity involves a complex interplay between ring D and the surrounding protein, changes associated with ring A, including the covalent linkage possibly playing an important role in intramolecular signaling.
EXPERIMENTAL PROCEDURES
Sample Preparation
The SyB.Cph2(GAF) sequence was synthesized following codon optimization for expression in Escherichia coli (residues 1–200 plus Ser-Leu linker and His6 tag, see supplemental Fig. S1). The apoprotein was overproduced from pPROLar in BL21pro for 4 h at 37 °C following induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside and 0.2% arabinose. The cells were lysed into TESβ (50 mm Tris/HCl, pH 7.8, 5 mm EDTA, 300 mm NaCl, with 1 mm β-mercaptoethanol added freshly), and the crude extract was clarified at 45,000 × g prior to in vitro autoassembly with ∼20% excess uniformly 13C- and 15N-labeled phycocyanobilin ([u-13C,15N]PCB) prepared as described (13). The holoprotein was precipitated with ammonium sulfate, redissolved in 50 mm Tris/HCl, pH 7.8, 1 mm iminodiacetic acid, 300 mm NaCl, 10 mm imidazole with 1 mm β-mercaptoethanol freshly added, and affinity-purified over nickel-nitrilotriacetic acid eluting with a similar buffer containing 100 mm imidazole. Peak fractions were pooled, precipitated with ammonium sulfate again, and resuspended at ∼2 mg/ml in TESβ. Working in darkness using infrared visualization, the sample was then photoconverted to P630 by saturating irradiation with far-red light (>700 nm), purified by size-exclusion chromatography over Superdex 200, re-buffered into 50 mm Tris/HCl, pH 7.8, with 5% glycerol, concentrated to ∼4 mm by ultrafiltration, transferred to an MAS rotor, and frozen at −80 °C. A photoequilibrium mixture containing ∼50% P690 was obtained by saturating irradiation of the concentrated sample at 525 nm in an ∼100 μm diameter capillary prior to freezing in the rotor. UV-visible absorbance spectra (Agilent 8453) of the samples (Fig. 1G) were measured by scratching ∼3 μl of frozen material from the rotor followed by dilution into TESβ.
PCB and phytochromobilin (PΦB) adducts of SyB.Cph2(GAF) and Cph1(PAS-GAF-PHY) were produced in vivo by coexpressing the appropriate phytochrome gene with cyanobacterial hemoxygenase and pcyA or Arabidopsis HY2 in E. coli. Working in darkness, the purified holophytochrome at ∼10 μm was photoconverted with an appropriate LED source and diluted 10-fold into 8 m urea/HCl, pH 1.5, the absorbance spectrum thereby indicating the proportion of Z- and E-isomers present (25, 26).
MAS NMR Studies
All solid-state MAS NMR spectra were performed on a Bruker AV-750 WB spectrometer (Karlsruhe, Germany) equipped with a 4-mm triple-resonance CP/MAS probe (Bruker). The 1H channel was tuned to 750.13 MHz for broadband decoupling, and the X channel was tuned to 188.62 MHz for 13C and 76.01 MHz for 15N. In each experiment, a 4-mm diameter zirconia MAS rotor was loaded with ∼8 mg of protein in ∼90 μl. The MAS spinning frequency in all experiments was set to 13 kHz and controlled to ±3 Hz with a Bruker MAS control unit. The temperature over the sample volume of in situ MAS probe at the νR = 13 kHz was calibrated via the T1 of 79Br in KBr (27). In this study, 233 ± 0.5 K was maintained throughout the measurements. Optimized 1H π/2 and 13C π pulse lengths were 3.1 and 8.0 μs, respectively. The 1H power was ramped 100–80% during CP, and 1H r.f. field strength of 72 kHz was employed for two-pulse phase modulation decoupling during acquisition. NMR spectra for P690 were recorded with a photoequilibrium mixture of P630 and P690. Spectra for P690 were obtained by subtraction of the P630 spectra from the P630/P690 mixture with an appropriate weighting constant of 0.5. The raw spectra for P630 and the mixture were processed and phased identically prior to subtraction.
Two-dimensional 13C–13C DARR experiments were used to assign 13C chemical shifts of the PCB chromophore. The DARR spectra shown in Fig. 1 were recorded with two 13C homonuclear recoupling mixing times of 5 and 50 ms, respectively. During the mixing period, the 1H–13C dipolar interaction was recovered by 1H continuous wave irradiation with the intensity satisfying the n = 1 rotary-resonance condition (28). The data were obtained with the following acquisition parameters: an 8-ms evolution in the indirect dimension, a relaxation delay of 1.5 s for each t1 slice, and 128 slices of 2048 scans each, leading to a total experimental time of ∼110 h. The two-dimensional 1H–15N heteronuclear dipolar correlation spectra shown in Fig. 2 were measured with 2152 transients, a relaxation delay of 1.8 s for each t1 slice, and a total of 128 t1 slices. An optimized CP contact time of 2.048 ms was used. The spectrum was recorded over a period of ∼139 h. Prior to Fourier transformation, the data were zero-filled to 4096 points, and an exponential apodization of 15 Hz was applied. Other acquisition parameters are given in the figure legends.
FIGURE 2.
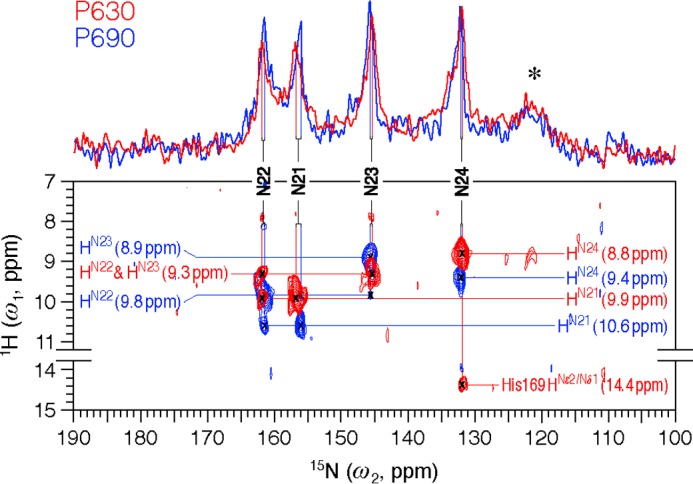
Two-dimensional MAS NMR 1H–15N frequency-switched Lee-Goldburg decoupled dipolar correlation spectra of [u-13C,15N]-PCB-SyB.Cph2(GAF) as P630 (red) and P690 (blue). The regions without resonances are omitted. Two one-dimensional traces onto the 15N dimension are shown. 15N resonances (N21–N24) are indicated by vertical lines. All four NH protons (HN21-N24) are fully resolved and marked by horizontal lines. The asterisk indicates protein backbone signals in natural abundance. The spectra were acquired with 1536 complex t2 and 128 real t1 points and 2152 scans. An LG-CP contact time of 2.048 ms was applied. The relaxation delay was 1.8 s.
For the two-dimensional 1H–15N HETCOR experiment acquired with a Lee-Goldburg-CP contact time of 2.0 ms, the maximum detection limit for heteronuclear transfers can be set to ∼3.5 Å. This limit is not overestimated, as experimentally determined by the poorly resolved P630 correlation signals between the pyrrole nitrogens of rings B–C and the pyrrole water at δH of ∼7.9 ppm (red, Fig. 2). In the Cph1 2VEA structure (5), this pyrrole water is associated with the rings A–C at distances of ∼3.3 Å. Also, the two-dimensional 1H–15N MELODI-HETCOR spectrum of cationic histidine with an 800-μs Lee-Goldburg-CP period revealed two intermolecular hydrogen bonds between the imidazole nitrogens and water protons at distances up to 3.5 Å (extracted from the crystal structure of l-histidine hydrochloride monohydrate) (29).
13C and 15N chemical shifts were externally referenced with respect to backbone CO resonance of solid glycine·HCl at 176.04 ppm and the backbone Nα resonance of solid histidine·HCl at 49.04 ppm, respectively. 1H chemical shifts were calibrated by running a 1H–13C HETCOR experiment on the histidine·HCl through its predominantly down- and up-field peaks centered at 9.21 and 18.42 ppm, respectively. The NMR data were processed with Bruker Topspin 3.0 and further analyzed by using the Sparky 3.114 (41). The 13C and 15N CP/MAS spectra were transferred to WaveMetrics Igor Pro 6.10 (Lake Oswego, OR) for the full-width at half-maximum measurements using the integrated multiple-peak fitting package.
RESULTS
Two-dimensional homo- and heteronuclear MAS NMR dipolar correlation spectroscopy on SyB.Cph2(GAF) holophytochrome assembled in vitro with [u-13C,15N] PCB enabled complete and unambiguous 13C and 15N assignments of the chromophore for both P630 and P690 (Figs. 1 and 2).
Two-dimensional 13C–13C DARR Spectra
Contour plots of 13C–13C DARR spectra are shown in Fig. 1, A and B, for P630 and P690, respectively. The spectra were recorded with two 13C homonuclear recoupling mixing times of 5 and 50 ms (Fig. 1, A and B, red and purple for P630, orange and cyan for P690, respectively) for 13C assignment of the chromophore; initial assignments were obtained from the well defined correlation networks connecting directly bonded carbons and confirmed by the paired peaks originated from weak polarization transfers among indirectly bonded carbons. For example in P630, paired peaks of the D-ring ethyl side chain (C18–C181–C182) are clearly present in the spectrum recoded with a mixing time of 5 ms (red, Fig. 1A), and the assignment of C181 (15.0 ppm) and C182 (11.3 ppm) is verified by the correlations involving neighboring D-ring carbons, like C171 (8.5 ppm)–C181/C182 and C19 (171.7 ppm)–C181/C182, resolved in the spectrum at the 50-ms dipolar mixing time (purple, Fig. 1A). Also, the correlation network in the A-ring region shows a single set of δC for C1 (182.0 ppm), C2 (35.2 ppm), and the ethylidene side chain (C31, 47.4 ppm and C32, 20.6 ppm), while revealing a correlation network split for its methyl side chain (C1-C2–[C21a (17.2 ppm)/C21b (18.1 ppm)]/–(C3b (53.6 ppm)), Fig. 1C) as well as a doubling for the C3 response with a δC change of 0.3 ppm (C3a (53.3 ppm)/C3b–C31, Fig. 1C) and a signal tripling for C4 (C3b–C4c (157.9 ppm)/C4b (157.4 ppm)/C4a (156.9 ppm), Fig. 1D). Similarly, a 1.1-ppm resonance separation is observed for C5 (85.4/86.5 ppm, Fig. 1D) and a 0.7-ppm difference for its B-ring neighbor, C6 (151.4/152.1 ppm, Fig. 1E). Such signal doubling is also observed in P690 for C21 and C3 (17.6/18.0 and 51.0/52.7 ppm, respectively, Fig. 1F), whereas C4-C6 splitting was not apparent. These observations indicate at least two different conformations of the chromophore in both states and loss of order in P690 (for example in the C4–C6 region).
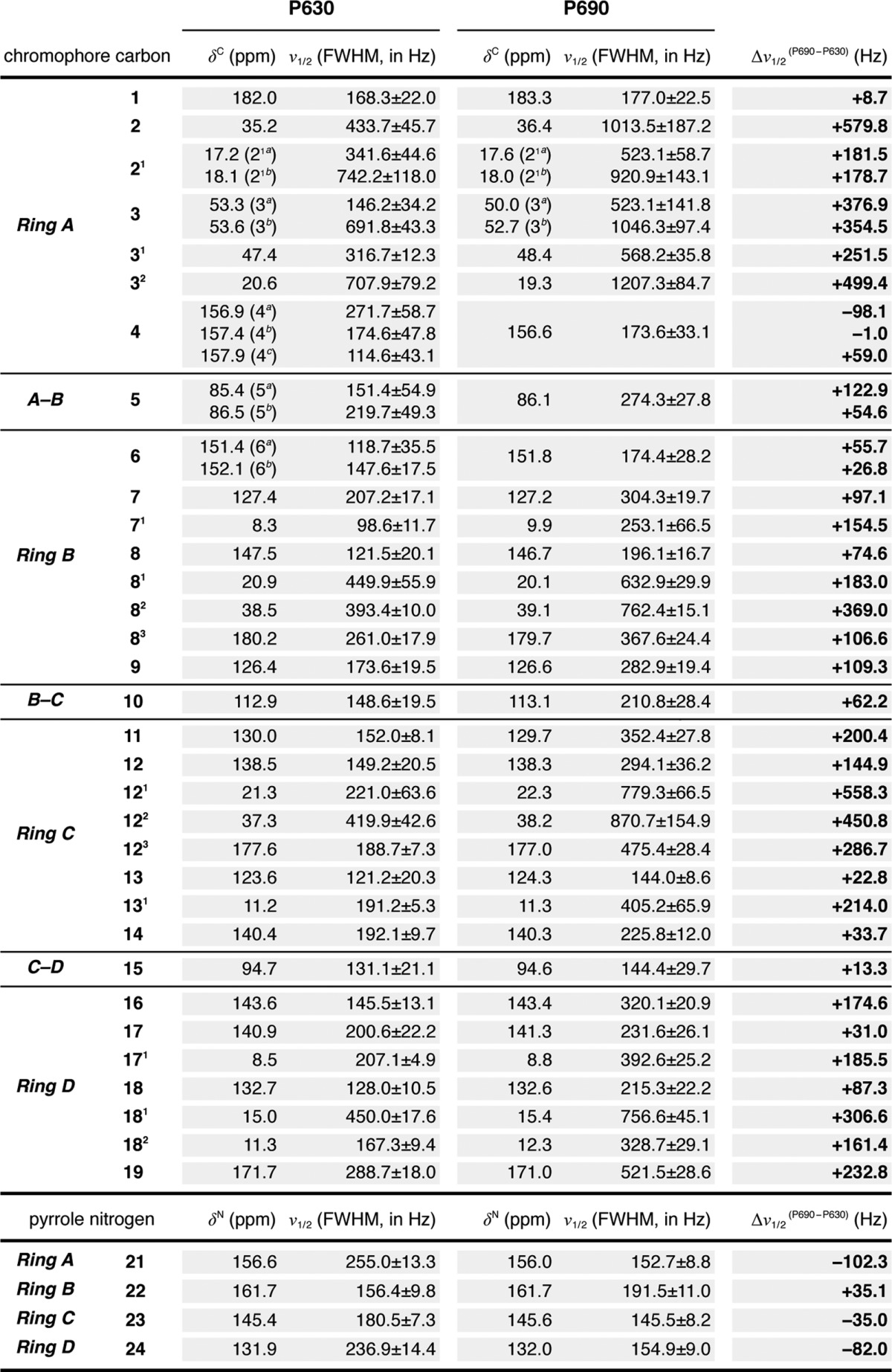
The complete 13C chromophore assignments in both P630 and P690 states are listed in Table 1. The overall similarity between the 13C chemical shifts of the chromophore of SyB.Cph2(GAF) as P630 and of Cph1(PAS-GAF-PHY) as Pr implies that the chromophore adopts a ZZZssa geometry in both proteins. The one-dimensional 13C CP/MAS NMR spectra of SyB.Cph2(GAF) as P630 and P690 are shown in Fig. 3 alongside the associated difference spectrum generated by P630 → P690 photoconversion (bottom). Most 13C resonances of the pyrrolic quaternary carbons show only subtle changes. In particular, those lying between 120 and 160 ppm, including C6–C9 and C11–C14 of rings B and C as well as C4 and C16–C18 of rings A and D, show minimal changes in δC (Fig. 3, inset) and line-widths (expressed as full-width at half-maximum, ν½, Table 2). Consequently, the data provide little support for C15=C16 photoisomerization, calling the expected Z/E photoflip of ring D into question. By way of comparison, the D-ring photoflip in the canonical Cph1 and plant phytochromes is associated with striking δC changes of ∼6 ppm for C14 and most D-ring pyrrolic carbons (see Table 1). Similarly, however, there is also little support for C4=C5 isomerization and the proposed A-ring photoflip (24); the C4 and C6 atoms associated with the A–B methine bridge show a δC change less than 1.3 ppm, far smaller than that from C14 and C16 of the C–D bridge associated with the D-ring photoflip in canonical phytochromes (13, 17, 18). Furthermore, δC changes seen at ring A (C3 (−2.3/−0.9 ppm), its side chain C31 and C32 (+1.0 and −1.3 ppm, respectively) linkage to Cys-138, and pyrrolic carbons C1 (+1.3 ppm) and C2 (+1.2 ppm)) are also too small for an A-ring photoflip. However, the D-ring side chain carbons C171 and C181 (+0.3 and +0.4 ppm, respectively) and C18 (−0.1 ppm) show essentially no change during photoconversion. Thus, we confirm the larger effect on the A-ring C32 relative to that on the D-ring C171 seen in liquid NMR studies of SyB.Cph2(GAF) with the chromophore selectively labeled using 1,2-[13C]α-aminolevulinic acid, a difference that by itself might be construed as evidence for the proposed A-ring photoflip (24).
TABLE 1.
13C and 15N chemical shifts (δC and δN) of the chromophore in Cph1 (PAS-GAF-PHY) and SyB.Cph2(GAF)
Data were obtained from [u-13C, 15N]PCB-Cph1 (PAS-GAF-PHY) as Pr and Pfr (2) and [u-13C, 15N]PCB-SyB.Cph2(GAF) as P630 and P690 (Figs. 1 and 2). ΔδC and ΔδN for Cph1(PAS-GAF-PHY) (Pfr-Pr) as Δδ(Pfr-Pr), SyB.Cph2(GAF) (P690-P630) as Δδ(P690-P630), SyB.Cph2(GAF) P630-Cph1(PAS-GAF-PHY) Pr as Δδ(P630-Pr), and SyB.Cph2(GAF) P690-Cph1(PAS-GAF-PHY) Pfr as Δδ(P690-Pfr) are listed.
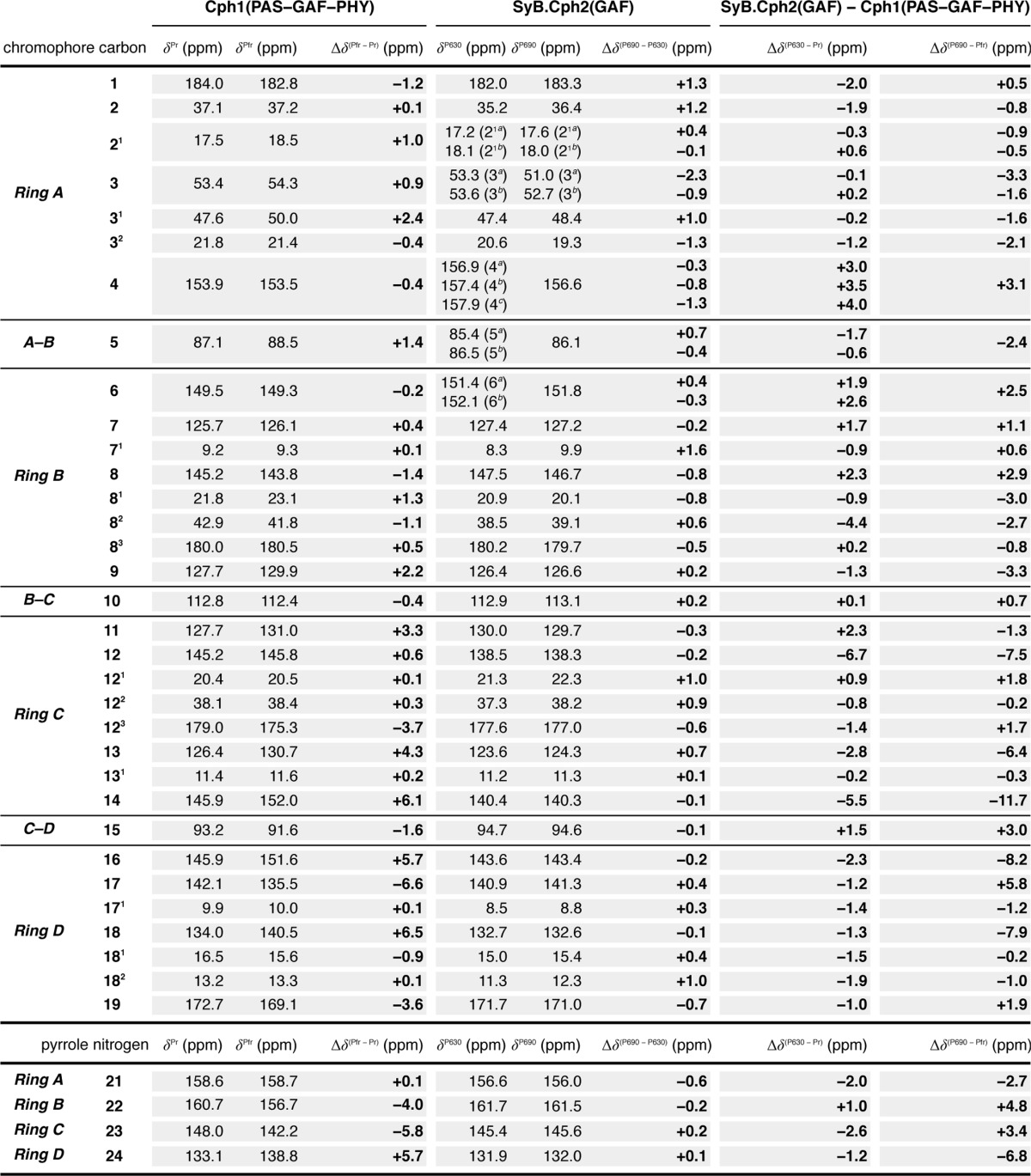
TABLE 2.
Full-width at half-maximum (FWHM) line widths (ν½) of 13C and 15N resonances of the chromophore in SyB.Cph2(GAF)
13C and 15N experimental line shapes were simulated by the Voigt function (convolution of a Lorentzian with a Gaussian at an equal ratio). The v½ values listed (mean ± S.D.) were extracted from the Voigt profile (fitting spectra not shown). Δν½ values are reported as P690-P630 and are listed in the right-most column.
Two-dimensional 1H–15N HETCOR Spectra
Both P630 and P690 spectra presented in Fig. 2 are dominated by correlations between directly bonded NH groups of the chromophore. The four pyrrole nitrogens are fully protonated in both P630 and P690, evidenced by the narrow δN dispersions (∼30 ppm) and all δN values lying below 161 ppm. These findings are in line with those from studies of Cph1 and plant phytochromes (13, 17, 20). In P630 (Fig. 2, red, one-dimensional trace), the areas of the four isolated peaks at 161.7, 156.6, 145.4, and 131.9 ppm correspond to a single nitrogen in each case. All four P630 peaks occur at δN positions almost identical to those observed for Cph1(PAS-GAF-PHY) (Table 1), with the resulting tentative assignments being validated by their correlations with the NH protons. In P630, three different 1H sites at δH = 9.9, 9.3, and 8.8 ppm in the 1H spectral region of 8–11 ppm (red, Fig. 2) are seen. As indicated by the 15N assignment of Cph1 (18), 1H partners of two tetrapyrrole nitrogens of the inner rings B (N22) and C (N23) should resonate at the same frequency of 9.3 ppm, whereas N22 (161.7 ppm) should show a correlation with another HN at ∼9.9 ppm in close proximity (HN21 of ring A). Indeed, an intense correlation signal at 9.9 ppm is seen, and thus the 15N peak at 156.6 ppm can be assigned unambiguously to N21. The 15N assignment is validated by resolved P690 correlations (blue, Fig. 2); HN23 is at 8.9 ppm, and at the same time N23 (145.6 ppm) shows a weak interaction with its neighboring proton at δH = 9.8 ppm, which can be directly assigned to HN22. In the N22 slice centered at 161.5 ppm, a much more intense signal at this 1H frequency is observed. Also seen in this slice, the weak signal at δH = 10.6 ppm is readily attributed to the intramolecular magnetization transfer between N22 and HN21. In the N21 slice (156.0 ppm), two correlations centered at the same 1H frequencies as N22 (9.8 and 10.6 ppm) are seen, but their intensities are reversed. Moreover, in both states, we attribute the remaining 15N resonance locating at the most high field position to the D-ring nitrogen (N24), and its directly bonded 1H partner (HN24) resonates at δH = 9.4 and 8.8 ppm in P630 and P690, respectively.
Intriguingly, the P630 state N24 also correlates with an acidic proton with δH of 14.4 ppm associated with a hydrogen bond to the protein. This unique signal can be only be associated with the ring nitrogens of Trp, His, or perhaps Pro (13, 29). From available phytochrome structures, the only such residue within 6 Å of N24 is the perfectly conserved homolog of His-169SyB.Cph2. In Cph1 and bacteriophytochromes in the Pr state (5), however, this residue is hydrogen-bonded to the D-ring carbonyl oxygen at ∼2.8 Å rather than N24. Such a difference could easily arise by reorientation of the imidazole side chain (30). Indeed, two His-290 isoforms were observed for Cph1 Pr (13). Moreover, the liquid NMR data for SyB.Cph2(GAF) are also compatible with a His-169Nδ1/Nϵ2-H···N24 contact, with 7 of the 20 lowest energy models in the corrected P630 ensemble (2LB9) showing this contact within 3.5 Å. As our detection limit of ∼3.5 Å was calibrated (see under “Experimental Procedures”), this bond is clearly broken in P690.
The 15N chemical shifts of the pyrrole nitrogens in both states are summarized in Table 1. The strongest δN change upon P630 → P690 photoconversion was seen for the A-ring nitrogen N21 but amounted to only −0.6 ppm, whereas the pyrrole nitrogens of rings B–D were essentially unaffected (<0.2 ppm). The small change in δN21 does not support the idea that P630 → P690 photoconversion in SyB.Cph2(GAF) is associated with a Z/E isomerization of C4=C5 and an A-ring photoflip as proposed (24). Crucially, the interaction between the B-ring N22 and A-ring HN21 (10.6 ppm) is retained in P690 (blue, Fig. 2), ruling out an A-ring photoflip. In contrast to the δN change observed for all four nitrogens, their directly bonded protons show prominent δH changes (0.4–0.7 ppm, Fig. 2) associated with photoconversion, because they are all involved in hydrogen bonding with the protein. Thus, a 0.6-ppm δH change seen for HN24 provides additional qualitative support for the putative His-169Nδ1/Nϵ2-H···N24 hydrogen bond and its rupture in P690, implying significant changes at ring D. Also, δH changes correlate well with those of δC for the PCB methyls, like C32, C71, and C182 (Table 1), reflecting the light-driven changes at the periphery of the chromophore.
Denaturation
The findings described above not only rule out an A-ring photoflip, and other than the breakage of the His-169···N24 hydrogen bond, they provide no evidence for D-ring photoflip either. Indeed, the pattern of chemical shift changes contrasts with findings for Cph1(PAS-GAF-PHY) in which Pr/Pfr photoconversion Pfr is clearly associated with a Z/E isomerization of C15=C16 and a D-ring photoflip (13, 17). We obtained independent evidence that Z/E isomerization nevertheless also occurs in the case of SyB.Cph2(GAF) by denaturation under acidic conditions (25, 26). In acidic urea, the bilin Z-isomer shows a photostable absorbance peak at about 665 nm, whereas the E-form absorbs at about 580–610 nm and can be converted to Z in light. The data (Table 3) show conclusively that SyB.Cph2(GAF)-PCB in the P630 state comprises the Z-isomer, whereas the P690/P630 mixture following 630 nm irradiation consists predominantly (∼80%) of the E-isomer. Experiments with Cph1(PAS-GAF-PHY)-PCB gave similar results (as reported earlier (26)). Equivalent measurements were also carried out on PΦB adducts that differ from PCB in carrying a vinyl instead of an ethyl side chain at C18 of ring D. A bathochromic shift was seen in all cases, although the shifts in the case of native SyB.Cph2(GAF) were much smaller (Table 3).
TABLE 3.
UV-visible absorbance maxima (λmax, nm) for the lowest energy ground state (after far-red irradiation, underlined) and the spectral difference maximum after irradiation at the appropriate λmax for the native PCB and PΦB adducts and the λmax values for the same samples after denaturation in 7 m urea at pH 1.5
| PCB adduct |
PΦB adduct |
|||
|---|---|---|---|---|
| Native | Denatured in acidic urea | Native | Denatured in acidic urea | |
| SyB.Cph2(GAF) | 628/686 | 663/603 | 640/693 | 673/610 |
| Cph1(PAS–GAF–PHY) | 655/704 | 663/603 | 675/720 | 673/610 |
DISCUSSION
The state-related changes associated with the D-ring atoms we report for SyB.Cph2(GAF) are very different from those for canonical phytochromes (2). Given the dramatic hypsochromic absorbance shift and the loss of the Pfr-typical 815-cm−1 resonance Raman band associated with deletion of the PHY domain (Fig. 1G and Ref. 22), and the unusual findings reported here might relate to specific properties of the GAF fragment, with a more complete SyB.Cph2 molecule perhaps behaving more similarly to canonical phytochromes. In the latter case, a functional PHY domain seems necessary for Pfr formation or stabilization, with the photoconversion process apparently arresting at a bleached and probably deprotonated meta-R intermediate if the PHY domain is damaged or deleted.
In the apparent absence of the expected D-ring rotation in SyB.Cph2(GAF), Vierstra and co-workers (24) proposed an A-ring photoflip as the basis for photoconversion not only in SyB.Cph2(GAF) but in all phytochromes. Although in an earlier publication (23) they described a structural ensemble (PDB code 2K2N) for SyB.Cph2(GAF), P630 from liquid NMR in which rings A–C were predominantly in roughly coplanar ZZss geometries with almost perpendicularly orientated D-rings, a refined P630 ensemble (PDB code 2KOI) was presented subsequently (24) showing highly contorted chromophores with both rings A and D often almost perpendicular to B and C. This was accompanied by a P690 ensemble (PDB code 2KLI) in which again the rings A–C were approximately coplanar with the D-rings approximately perpendicular. It was thus proposed that photoconversion is associated with an A-ring photoflip, not just in SyB.Cph2(GAF) but in all phytochromes (24). There are several reasons for doubting this as follows.
(i) The NMR data obtained (24) are consistent with a wide variety of model structures, especially in the case of P690, which was necessarily derived by subtraction of the P630 spectra from the P690/P630 mixture. In particular, the limited data for the pyrrolic carbons apparently allowed the algorithm considerable freedom in calculating possible structural models. Indeed, various predicted chromophore geometries show, for example, a C-ring that is flipped such that the propionate points in the opposite direction to that of the B-ring.
(ii) A likely source of error is associated with the P690/P630 mixture itself. As SyB.Cph2(GAF) P690 shows significant dark reversion (22), the sample was continuously irradiated at 620 nm within the magnet, ostensibly to maintain photoequilibrium. With an extinction coefficient of ∼100 mm−1 cm−1 (Fig. 1G), the actinic light would penetrate less than 0.1 mm into the ∼2 mm sample, leading to a significant P690 gradient in the microcell during measurement. (In this study, the sample was passed slowly through a 0.1-mm capillary irradiated omnidirectionally with 525 nm actinic light establishing P630/P690 photoequilibrium prior to freezing.)
(iii) The 2K2N, 2KOI, and 2KLI ensembles (23, 24) all showed significant “puckering” of the chromophore pyrrole rings, although aromatic pyrroles are known to be planar (1, 31). Bilin chemistry also has generally concluded that the B- and C-rings are coplanar, in contrast to the picture presented by the 2K2N, 2KOI, and 2KLI ensembles.
(iv) Corrected/refined model ensembles for P630 and P690 (PDB codes 2LB9 and 2LB5, respectively) were deposited after the planarity problems were pointed out (1), although it would appear that appropriate energy minimization of the methine bridges has not been carried out. These latest ensembles provide little support for an A-ring photoflip as originally proposed on the basis of the same data (24). Although PDB codes 2LB9 and 2LB5 are marked “to be published,” no correction has yet appeared.
In this study, although we found major changes in δC for C3, C31, and C32 associated with the A-ring linkage to the protein, none of the effects that would necessarily accompany an A-ring photoflip is seen. In particular, the fact that the correlation between N22 and HN21 is retained upon photoconversion rules out an A-ring rotation. We suggest that changes in the linkage apparent from C31 together with the problems detailed above might have favored models in which the A-ring was positioned differently in P630 and P690, thereby implying an A-ring photoflip.
Ulijasz et al. (24) attracted particular attention because it contradicted the general consensus that Pr and Pfr geometries are ZZZssa and ZZEssa, respectively, and indeed that the primary photochemical event in phytochrome photoconversion is a D-ring photoflip. The latter conclusion is based on a large body of published data for plant/Cph1-type phytochromes and bacteriophytochromes, culminating in both x-ray crystallographic and MAS NMR-based structural studies (4, 5, 7, 9–11, 13, 17, 18, 32). Although Za→ Ea isomerization at C15 is not in doubt, some reports have nevertheless implied isomerization at the C5 methine. Studies with BphPs assembled with synthetic biliverdin derivatives in which the D-ring was locked by covalent bridges to the C-ring yielded Pr- and Pfr-like spectra associated with Za and Ea geometries, respectively, at C15 methine (12). However, further work with locked 15Ea, in which the A-ring too was locked, implied Zs → Za or even Zs → Ea hula-twist isomerization at C5 (33). Although our data exclude the possibility of the A-ring rotation associated with Zs → Za, we cannot rule out a hula-twist in which the A-ring only shifts slightly. Planar Za geometry at C5 would be associated with clashes between A- and B-rings, however, requiring that the A-ring be strongly tilted. The same applies to the D-ring in both Pr (ZZZssa) and Pfr (ZZEssa), except that ample space around the D-ring for the tilt and photoflip exists, whereas close packing of the protein side chains around the A-ring in Pr makes such a tilt unlikely in Pfr. Of course, the PaBphP Pfr crystal structure shows Zs at C5 anyway (9). We suggest that side effects of the locking modifications to the chromophore might explain the discrepancy, however. There is little room in the pocket for the atoms of the synthetic bridges between the A- and B-rings. Moreover, we would expect that major changes would follow removal of the protons associated with the pyrrole nitrogens. The appropriate controls for this are in any case missing.
However, there is no doubt that changes in the A-ring are associated with photoconversion. In liquid NMR studies of Cph1 from Synechocystis 6803, although a photoflip was only evident for ring D, significant state-induced changes were also seen at the C21 and C32 methyl groups associated with the A-ring and its linkage to the protein (20). We suggest that these effects arise indirectly from post-photochemical relaxation processes involving cofactor and protein. The δC changes seen here for C3, C31, and C32 are quantitatively similar to changes at these atoms in the case of both Cph1 and plant phytochromes during photoconversion, although there the changes associated with ring D predominate (17). X-ray crystallographic data for the PaBphP Q188L mutant also imply major changes at the bilin-protein linkage associated with photoconversion (10). As these effects seem to be widespread in the phytochrome family, it might be that the covalent linkage is an important component of the molecular mechanism of phytochrome action. Nevertheless, at least photochromicity is retained in phytochromes in which the Cys attachment site has been blocked or mutated (34–37). In any case, several other carbon atoms (e.g. C1, C2, C4, C71, C121, and C182) also show ∼1 ppm δC changes, indicating that the structural changes in the protein are not confined to the linkage.
The D-ring changes associated with photoconversion seen in this study are clearly distinct from those in canonical phytochromes; thus, we were surprised to find that the P630 and P690 states indeed represent Z and E isomers, respectively (see Table 3). Although it might be that the photoconversion mechanism in the SyB.Cph2(GAF) fragment is unusual, it is sobering to note that a satisfactory explanation for the ∼50-nm bathochromic shift in λmax characteristic of Pr → Pfr photoconversion in canonical phytochromes has yet to be offered. It is certainly not a direct consequence of the Z→E isomerization of C15=C16 and the associated rotation of ring D, because the absence of protein interactions, the isomerization is associated with an ∼50-nm shift in the opposite direction (as apparent from Table 3). Thus, although photochromicity arises from the bilin, the direction of the spectral shift is reversed by the protein, perhaps in association with changes in the hydrogen-bonding network modifying the charge distribution. Counter ion changes might control orbital energies and conjugation defects. Interactions with the protein leading to strains and torsions of the chromophore methine bridges would also have major effects on the absorbance properties, as extensive studies of free bilins have shown (38). Recent work on PaBphP bacteriophytochrome indicates that the C15 methine bridge between rings C and D is significantly stretched in the Pfr state (11).
The His-169···N24 contact we observe specifically for P630 is intriguing as the equivalent His interaction in the crystal structures of canonical phytochromes is to the D-ring carbonyl (5). A contact to the nitrogen might arise if the ring was strongly tilted, however. Such a situation is indeed seen in several models derived from the liquid NMR studies (PDB code 2LB9, compare with Ref. 24 and see Fig. 4). Such a geometry might derive from hydrophobic interactions with Phe-82 and/or Val-167. It might also reconcile the unexpectedly small δC changes at C14 and C16 with a Z → E photoisomerization as well as the missing 815 cm−1 Raman band in P690 of the SyB.Cph2(GAF) fragment (22). However, substituting the D-ring ethyl side chain with a vinyl group leads to bathochromic shifts in SyB.Cph2(GAF), albeit much smaller than those seen in canonical phytochromes (Table 3), implying that ring D is weakly conjugated with the rest of the chromophore in both states. According to Hückel theory, conjugation is proportional to cos2 of the angle between the π-electron axes, and thus D-ring tilts associated with dihedral angles up to 70° or beyond 110° would still allow significant conjugation. When the D-ring is almost orthogonal, however, cos2 of the dihedral angle is insignificant and conjugation is lost, as is readily apparent in α-phycoerythrocyanin and certain cyanobacteriochromes (39, 40). Hence, we suggest that in SyB.Cph2(GAF), the C15=C16 double bond twisted before and after photoconversion, with the dihedral angle changing from ∼70° over 90° to ∼110° in association with the D-ring photoflip; this would explain all the current data and imply that the missing PHY domain significantly influences the geometry of the D-ring in the case of P690. The PHY domain is in any case essential for physiological signaling.
FIGURE 4.
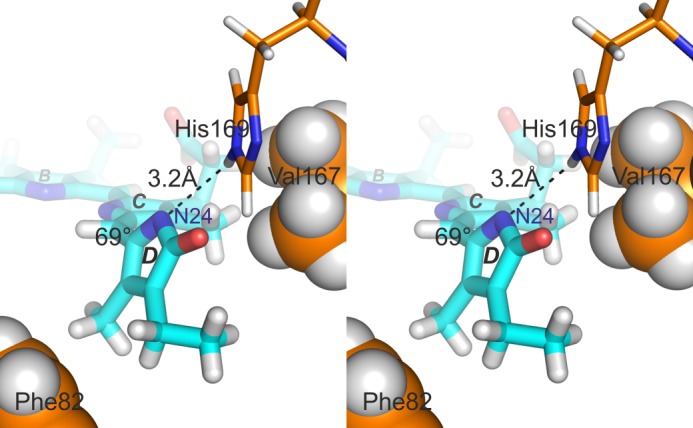
Stereo pair illustrating the proposed His-169···N24 hydrogen bond associated with a strongly tilted D-ring. In this model (model 5 from the corrected 2LB9 P630 ensemble, compare with Ref. 24), the hydrogen bond length is 3.2 Å and the C15=C16 dihedral angle 69°. Hydrophobic interactions with Phe-82 and Val-167 might also be important. (PyMOL image).
Acknowledgments
We thank Peter Schmieder (Leibniz Institute for Molecular Pharmacology, Berlin-Buch, Germany), Hugo Scheer (Ludwig Maximillian University, Munich, Germany), and J. Clark Lagarias (University of California at Davis) for valuable discussions and Fons Lefeber, Kees Erkelens, and K. B. Sai Sankar Gupta (University of Leiden, The Netherlands) for instrumental assistance.
This work was supported by Deutsche Forschungsgemeinschaft Grant Hu702/8 and Nederlandse Organisatie voor Wetenschappelijk Onderzoek Grants DN 89-190 and ALW 822.02.007.

This article contains supplemental Figs. S1–S3.
- Pr
- phytochrome ground states absorbing principally in red region
- Pfr
- phytochrome ground states absorbing principally in far-red region
- PAS
- Period/Arnt/Single
- GAF
- cGMP phosphodiesterase/adenylyl cyclase/FhlA
- PHY
- phytochrome-specific
- PCB
- phycocyanobilin
- DARR
- dipolar-assisted rotational resonance
- BphP
- bacteriophytochrome
- HETCOR
- heteronuclear correlation
- PDB
- Protein Data Bank
- CP
- cross polarization
- MAS
- magic angle spinning.
REFERENCES
- 1. Hughes J. (2010) Phytochrome three-dimensional structures and functions. Biochem. Soc. Trans. 38, 710–716 [DOI] [PubMed] [Google Scholar]
- 2. Song C., Rohmer T., Tiersch M., Zaanen J., Hughes J., Matysik J. (2013) Solid-state NMR spectroscopy to probe photoactivation in canonical phytochromes. Photochem. Photobiol. 89, 259–273 [DOI] [PubMed] [Google Scholar]
- 3. Rockwell N. C., Lagarias J. C. (2010) A brief history of phytochromes. ChemPhysChem 11, 1172–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wagner J. R., Brunzelle J. S., Forest K. T., Vierstra R. D. (2005) A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 438, 325–331 [DOI] [PubMed] [Google Scholar]
- 5. Essen L.-O., Mailliet J., Hughes J. (2008) The structure of a complete phytochrome sensory module in the Pr ground state. Proc. Natl. Acad. Sci. U.S.A. 105, 14709–14714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang X., Stojković E. A., Kuk J., Moffat K. (2007) Crystal structure of the chromophore binding domain of an unusual bacteriophytochrome, RpBphP3, reveals residues that modulate photoconversion. Proc. Natl. Acad. Sci. U.S.A. 104, 12571–12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagner J. R., Zhang J., Brunzelle J. S., Vierstra R. D., Forest K. T. (2007) High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J. Biol. Chem. 282, 12298–12309 [DOI] [PubMed] [Google Scholar]
- 8. Rüdiger W., Thümmler F., Cmiel E., Schneider S. (1983) Chromophore structure of the physiologically active form (Pfr) of phytochrome. Proc. Natl. Acad. Sci. U.S.A. 80, 6244–6248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang X., Kuk J., Moffat K. (2008) Crystal structure of Pseudomonas aeruginosa bacteriophytochrome: photoconversion and signal transduction. Proc. Natl. Acad. Sci. U.S.A. 105, 14715–14720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang X., Kuk J., Moffat K. (2009) Conformational differences between the Pfr and Pr states in Pseudomonas aeruginosa bacteriophytochrome. Proc. Natl. Acad. Sci. U.S.A. 106, 15639–15644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X., Ren Z., Kuk J., Moffat K. (2011) Temperature-scan cryocrystallography reveals reaction intermediates in bacteriophytochrome. Nature 479, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Inomata K., Hammam M. A., Kinoshita H., Murata Y., Khawn H., Noack S., Michael N., Lamparter T. (2005) Sterically locked synthetic bilin derivatives and phytochrome Agp1 from Agrobacterium tumefaciens form photoinsensitive Pr- and Pfr-like adducts. J. Biol. Chem. 280, 24491–24497 [DOI] [PubMed] [Google Scholar]
- 13. Song C., Psakis G., Lang C., Mailliet J., Gärtner W., Hughes J., Matysik J. (2011) Two ground state isoforms and a chromophore D-ring photoflip triggering extensive intramolecular changes in a canonical phytochrome. Proc. Natl. Acad. Sci. U.S.A. 108, 3842–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song C., Psakis G., Lang C., Mailliet J., Zaanen J., Gärtner W., Hughes J., Matysik J. (2011) On the collective nature of phytochrome photoactivation. Biochemistry 50, 10987–10989 [DOI] [PubMed] [Google Scholar]
- 15. van Thor J. J., Borucki B., Crielaard W., Otto H., Lamparter T., Hughes J., Hellingwerf K. J., Heyn M. P. (2001) Light-induced proton release and proton uptake reactions in the cyanobacterial phytochrome Cph1. Biochemistry 40, 11460–11471 [DOI] [PubMed] [Google Scholar]
- 16. Rohmer T., Strauss H., Hughes J., de Groot H., Gärtner W., Schmieder P., Matysik J. (2006) 15N MAS NMR studies of Cph1 phytochrome: Chromophore dynamics and intramolecular signal transduction. J. Phys. Chem. B 110, 20580–20585 [DOI] [PubMed] [Google Scholar]
- 17. Rohmer T., Lang C., Hughes J., Essen L.-O., Gärtner W., Matysik J. (2008) Light-induced chromophore activity and signal transduction in phytochromes observed by 13C and 15N magic-angle spinning NMR. Proc. Natl. Acad. Sci. U.S.A. 105, 15229–15234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rohmer T., Lang C., Bongards C., Gupta K. B., Neugebauer J., Hughes J., Gärtner W., Matysik J. (2010) Phytochrome as molecular machine: Revealing chromophore action during the Pfr → Pr photoconversion by magic-angle spinning NMR spectroscopy. J. Am. Chem. Soc. 132, 4431–4437 [DOI] [PubMed] [Google Scholar]
- 19. Strauss H. M., Hughes J., Schmieder P. (2005) Heteronuclear solution-state NMR studies of the chromophore in cyanobacterial phytochrome Cph1. Biochemistry 44, 8244–8250 [DOI] [PubMed] [Google Scholar]
- 20. Hahn J., Strauss H. M., Schmieder P. (2008) Heteronuclear NMR investigation on the structure and dynamics of the chromophore binding pocket of the cyanobacterial phytochrome Cph1. J. Am. Chem. Soc. 130, 11170–11178 [DOI] [PubMed] [Google Scholar]
- 21. Röben M., Hahn J., Klein E., Lamparter T., Psakis G., Hughes J., Schmieder P. (2010) NMR spectroscopic investigation of mobility and hydrogen bonding of the chromophore in the binding pocket of phytochrome proteins. ChemPhysChem 11, 1248–1257 [DOI] [PubMed] [Google Scholar]
- 22. Ulijasz A. T., Cornilescu G., von Stetten D., Kaminski S., Mroginski M. A., Zhang J., Bhaya D., Hildebrandt P., Vierstra R. D. (2008) Characterization of two thermostable cyanobacterial phytochromes reveals global movements in the chromophore-binding domain during photoconversion. J. Biol. Chem. 283, 21251–21266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cornilescu G., Ulijasz A. T., Cornilescu C. C., Markley J. L., Vierstra R. D. (2008) Solution structure of a cyanobacterial phytochrome GAF domain in the red-light-absorbing ground state. J. Mol. Biol. 383, 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ulijasz A. T., Cornilescu G., Cornilescu C. C., Zhang J., Rivera M., Markley J. L., Vierstra R. D. (2010) Structural basis for the photoconversion of a phytochrome to the activated Pfr form. Nature 463, 250–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao K. H., Scheer H. (1995) Type I and type II reversible photochemistry of phycoerythrocyanin α-subunit from Mastigocladus laminosus both involve Z-E isomerization of phycoviolobilin chromophore and are controlled by sulfhydryls in apoprotein. BBA-Bioenergetics 1228, 244–253 [Google Scholar]
- 26. Ishizuka T., Narikawa R., Kohchi T., Katayama M., Ikeuchi M. (2007) Cyanobacteriochrome TePixJ of Thermosynechococcus elongatus harbors phycoviolobilin as a chromophore. Plant Cell Physiol. 48, 1385–1390 [DOI] [PubMed] [Google Scholar]
- 27. Thurber K. R., Tycko R. (2009) Measurement of sample temperatures under magic-angle spinning from the chemical shift and spin-lattice relaxation rate of 79Br in KBr powder. J. Magn. Reson. 196, 84–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takegoshi K., Nakamura S., Terao T. (2001) 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 344, 631–637 [Google Scholar]
- 29. Li S., Hong M. (2011) Protonation, tautomerization, and rotameric structure of histidine: A comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 133, 1534–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hu F., Luo W., Hong M. (2010) Mechanisms of proton conduction and gating in influenza M2 proton channels from solid-state NMR. Science 330, 505–508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Falk H. (1989) The Chemistry of Linear Oligopyrroles and Bile Pigments, Springer-Verlag, Vienna [Google Scholar]
- 32. Mailliet J., Psakis G., Feilke K., Sineshchekov V., Essen L. O., Hughes J. (2011) Spectroscopy and a high-resolution crystal structure of Tyr263 mutants of cyanobacterial phytochrome Cph1. J. Mol. Biol. 413, 115–127 [DOI] [PubMed] [Google Scholar]
- 33. Inomata K., Khawn H., Chen L. Y., Kinoshita H., Zienicke B., Molina I., Lamparter T. (2009) Assembly of Agrobacterium phytochromes Agp1 and Agp2 with doubly locked bilin chromophores. Biochemistry 48, 2817–2827 [DOI] [PubMed] [Google Scholar]
- 34. Borucki B., Otto H., Rottwinkel G., Hughes J., Heyn M. P., Lamparter T. (2003) Mechanism of Cph1 phytochrome assembly from stopped-flow kinetics and circular dichroism. Biochemistry 42, 13684–13697 [DOI] [PubMed] [Google Scholar]
- 35. Borucki B., Seibeck S., Heyn M. P., Lamparter T. (2009) Characterization of the covalent and noncovalent adducts of Agp1 phytochrome assembled with biliverdin and phycocyanobilin by circular dichroism and flash photolysis. Biochemistry 48, 6305–6317 [DOI] [PubMed] [Google Scholar]
- 36. Hahn J., Strauss H. M., Landgraf F. T., Gimenèz H. F., Lochnit G., Schmieder P., Hughes J. (2006) Probing protein-chromophore interactions in Cph1 phytochrome by mutagenesis. FEBS J. 273, 1415–1429 [DOI] [PubMed] [Google Scholar]
- 37. Lamparter T., Esteban B., Hughes J. (2001) Phytochrome Cph1 from the cyanobacterium Synechocystis PCC6803. Purification, assembly, and quaternary structure. Eur. J. Biochem. 268, 4720–4730 [DOI] [PubMed] [Google Scholar]
- 38. Falk H., Höllbacher G. (1978) Contributions to the chemistry of pyrrole pigments, XXIV: The relation between light absorption and structure of bilatrienes-abc. Monatsh. Chem. 109, 1429–1449 [Google Scholar]
- 39. Rockwell N. C., Martin S. S., Lagarias J. C. (2012) Mechanistic insight into the photosensory versatility of DXCF cyanobacteriochromes. Biochemistry 51, 3576–3585 [DOI] [PubMed] [Google Scholar]
- 40. Schmidt M., Patel A., Zhao Y., Reuter W. (2007) Structural basis for the photochemistry of α-phycoerythrocyanin. Biochemistry 46, 416–423 [DOI] [PubMed] [Google Scholar]
- 41. Goddard T. D., Kneller D. G. (2008) SPARKY, Version 3, University of California, San Francisco [Google Scholar]