

Background: Bacterial IncA/C plasmids distribute antibiotic resistance genes and encode a conserved thioredoxin-fold protein (DsbP).
Results: DsbP shuffles incorrect disulfide bonds in misfolded proteins, and its structure diverges from previously characterized disulfide isomerases.
Conclusion: Plasmid-encoded DsbP is a novel domain-swapped protein-disulfide isomerase.
Significance: IncA/C plasmids may encode this protein proofreading machinery to ensure horizontal gene transfer of antibiotic resistance genes.
Keywords: Antibiotic Resistance, Bacterial Conjugation, Crystal Structure, Disulfide, Enzyme Structure, DsbC/DsbG, Disulfide Isomerase, Domain Swapping, Horizontal Gene Transfer, Multidrug Resistance
Abstract
The multidrug resistance-encoding IncA/C conjugative plasmids disseminate antibiotic resistance genes among clinically relevant enteric bacteria. A plasmid-encoded disulfide isomerase is associated with conjugation. Sequence analysis of several IncA/C plasmids and IncA/C-related integrative and conjugative elements (ICE) from commensal and pathogenic bacteria identified a conserved DsbC/DsbG homolog (DsbP). The crystal structure of DsbP reveals an N-terminal domain, a linker region, and a C-terminal catalytic domain. A DsbP homodimer is formed through domain swapping of two DsbP N-terminal domains. The catalytic domain incorporates a thioredoxin-fold with characteristic CXXC and cis-Pro motifs. Overall, the structure and redox properties of DsbP diverge from the Escherichia coli DsbC and DsbG disulfide isomerases. Specifically, the V-shaped dimer of DsbP is inverted compared with EcDsbC and EcDsbG. In addition, the redox potential of DsbP (−161 mV) is more reducing than EcDsbC (−130 mV) and EcDsbG (−126 mV). Other catalytic properties of DsbP more closely resemble those of EcDsbG than EcDsbC. These catalytic differences are in part a consequence of the unusual active site motif of DsbP (CAVC); substitution to the EcDsbC-like (CGYC) motif converts the catalytic properties to those of EcDsbC. Structural comparison of the 12 independent subunit structures of DsbP that we determined revealed that conformational changes in the linker region contribute to mobility of the catalytic domain, providing mechanistic insight into DsbP function. In summary, our data reveal that the conserved plasmid-encoded DsbP protein is a bona fide disulfide isomerase and suggest that a dedicated oxidative folding enzyme is important for conjugative plasmid transfer.
Introduction
The global prevalence of multidrug-resistant bacteria is increasing at an alarming rate (1). Both the spread of antibiotic-resistant bacteria (2) and the transmission of resistance genes between the same and different species (3), contribute to the increase in antibiotic resistance. For example, since the initial identification of the New Delhi metallo-β-lactamase-1 (NDM-1) gene in Klebsiella pneumoniae, the gene has been identified in a number of other bacterial genera (4). It is now clear that plasmids or integrative and conjugative elements (ICE)5 (a chromosomally integrated mobile genetic element (5, 6)), are important drivers of the dissemination of antibiotic resistance in bacteria (7).
Bacterial cell surface pili, belonging to a subfamily of the type IV conjugation system (known as the type IV secretion system) mediate the transfer of conjugative plasmids and ICE between bacteria (8, 9). The type IV secretion system is a multiple protein component system. Its core functional components comprise: (i) cell surface pili that establish contact with recipient cells; (ii) a multiprotein secretion channel that facilitates transfer of the plasmid across the bacterial cell envelope; and (iii) a type IV coupling protein receptor that recognizes protein-coated conjugative plasmids at the cytoplasmic face and delivers them into the secretion channel (9). The large extrachromosomal conjugative plasmids or ICEs contain a subset of genes located in the transfer (tra) region that encode protein components (Tra proteins) required for building and assembling the complete conjugative machinery (10). In addition to these essential genes and several genes encoding regulatory proteins, the F plasmids and many other drug-resistant conjugative plasmids also contain a gene that encodes a thioredoxin (TRX)-like disulfide oxidoreductase (10–13).
Bacterial disulfide isomerases are periplasmic TRX-like oxidoreductase chaperones that proofread and shuffle incorrect disulfide bonds in secreted proteins harboring multiple cysteines, or protect periplasmic proteins with a single cysteine from oxidative damage (14–18). The best-studied disulfide isomerases are Escherichia coli DsbC and DsbG (EcDsbC and EcDsbG) (18–20). Both have a catalytic domain comprising a TRX-fold with a canonical redox-active CXXC active site motif. Both form a V-shaped protein structure through homodimerization of the N-terminal cystatin-like domains of each subunit. A connecting helix links the N-terminal domain with the C-terminal catalytic domain (16, 17). However, other TRX-like disulfide isomerases are also used by bacteria. For example, a ∼160-residue F-plasmid encoded disulfide reductase and isomerase TrbB, whose sequence remotely resembles that of the extracytoplasmic monomeric thioredoxin ResA (16% identity) (21), is essential for the folding and assembly of components of the conjugative secretion system (13).
In recent years, the incompatibility group IncA/C-conjugative plasmids have received major attention as a consequence of their presence in a wide range of bacterial hosts and their ability to support the spread of multidrug resistance (3, 22). The IncA/C plasmid was first identified in a fish pathogen Aeromonas hydrophila in the 1970s (23, 24). Sequencing analysis of plasmid lineages of blaCMY-2 (extended-spectrum β-lactamase gene) and blaNDM-1 (New Delhi metallo-β-lactamase-1 gene) showed that IncA/C plasmids are highly disseminated across animal and human bacterial pathogens (4, 25–27). Our sequence trawling of several IncA/C plasmids revealed that the tra region encodes a conserved protein, which we named DsbP based on its similarity to other disulfide oxidoreductases and its frequent plasmid location (see Table 1 for a compiled list). The DsbP sequence is remotely related to EcDsbC and EcDsbG (18–20% identity over 214 residues) and is not recognized as homologous with TrbB because it lacks the N-terminal dimerization domain (35% identity, over just 32 residues around the CXXC motif).
TABLE 1.
DsbP gene carrying multidrug-resistant IncA/C plasmids or ICE
The plasmid/ICE size in kilobases (kb) and the number of predicted open reading frames (ORFs), where available, are given in the first column in parentheses. Also provided are names of the organism from which the plasmid/ICE were isolated (second column) and the Uniprot protein ID for DsbP (third column in parentheses).
| Plasmid/ICE | Organism | Uniport ID/anotation |
|---|---|---|
| pSH111_166 (166kb, 201 ORFs) | S. enterica serovar Heidelberg | DsbC thiol:disulfide involved in conjugative transfer (H9TIR9) |
| pSH163_135 (135 kb, 163 ORFs) pSH696_135 (135 kb, 161 ORFs) | ||
| pNDM102337 (166 kb, 191 ORFs) | E.coli | DsbC protein-disulfide isomerase (C4NUX8) |
| pNDM-1_Dok01 (196 kb, 224 ORFs) | ||
| pAR060302 (167 kb, 179 ORFs) | ||
| pNDM10505 (167 kb, 193 ORFs) | ||
| pAPEC1990_61 (161 kb, 173 ORFs) | ||
| peH4H (148 kb, 173 ORFs) | ||
| pUMNK88 (161 kb, 173 ORFs) | ||
| pPG010208 (136 kb, 148 ORFs) | ||
| pCC416 (24 kb) | K. pneumoniae | DsbC thiol:disulfide interchange protein (A6GV51) |
| AOCV01000044.1 (ICE) | K. pneumoniae | DsbC (M7P3V0) |
| pNDM-KN (163 kb) | ||
| pNDM10469 (138 kb, 169 ORFs) | ||
| pR55 (171 kb, 180 ORFs) | ||
| pSN254 (176 kb, 198 ORFs) | Salmonella newport | Uncharacterized protein (A4IVF4) |
| pSD_174 (174 kb, 198 ORFs) | Salmonella dublin | Thiol:disulfide interchange protein DsbC (F5BPS8) |
| pIP1202 (183 kb, 221 ORFs) | Yersinia pestis biovar Orientalis | DsbC (A4IUW6) |
| pTC2 (29 kb) | Providencia stuartii | DsbC (K0HF62) |
| pP99–018 (150 kb, 187 ORFs) | Pasteurella piscicida | DsbC (A0PB07) |
| pP91278 (132 kb, 161 ORFs) | ||
| pRA1 (144 kb, 158 ORFs) | A. hydrophila | DsbC (C6GA31) |
| AB525688.1 (ICE) | Proteus mirabilis | DsbC (D0FZX2) |
| AB571791.1 (ICE) | S. typhimurium | Putative uncharacterized protein (F2Z7X1) |
| AMDF01000024.1 (ICE) | A. baumannii | Disulfide bond isomerase protein (K5QUC4) |
| DQ164214.1 (ICE) | Salmonella newport | DsbC (Q3S5B8) |
IncA/C plasmids also contain mobile elements that facilitate their integration into the bacterial chromosome (22). Consequently, a gene coding for DsbP is also located in ICEs isolated from Proteus mirabilis, K. pneumoniae, Salmonella enterica serovars Typhimurium and Newport, and Acinetobacter baumannii (100% sequence identity to DsbP). Importantly, deletion of a DsbP homolog (47% identity and 71% similarity) in the SXT/R391 family of ICE (closely related to IncA/C plasmids) abolished conjugative transfer (5).
Here, we present three crystal structures of the IncA/C plasmid-encoded DsbP from independent crystal forms. The resulting six structural models of homodimeric DsbP were compared with the structures of chromosomally encoded disulfide isomerases to report on structural diversity, domain motion, and redox state-dependent changes at the active site. We also evaluated the redox activity profiles of DsbP and active site variants in dithiol oxidase, disulfide isomerase, and disulfide reductase assays in comparison with the prototypical EcDsbC and EcDsbG disulfide isomerase enzymes. The results show that DsbP is a novel domain-swapped dimeric disulfide isomerase.
EXPERIMENTAL PROCEDURES
Protein Production
A codon-optimized synthetic gene corresponding to plasmid-encoded DsbP (e.g. H9TIR9 and A6GV51, residues 22–235), lacking the predicted secretion signal sequence was engineered into the bacterial expression vector pMCSG7 (28) by ligation-independent cloning. Residue numbering used here is for the predicted mature DsbP protein (Ser1-Ser2-Lys3-Leu4 correspond to residues 22–235 of the unprocessed gene-encoded protein). Active site variants “CGYC” (DsbP (103CAVC106 → 103CGYC106)) and “CPYC” (DsbP (103CAVC106 → 103CPYC106)) were generated by QuikChangeTM site-directed mutagenesis using the wild-type construct, and the non-catalytic cysteine mutant (Cys144 → Ala, Cys168 → Ala) was made by ligation-independent cloning of a synthetic gene into pMCSG7. Proteins were expressed in BL21(DE3)pLys cells using autoinduction media (29) and purified by immobilized metal affinity chromatography using Talon® resin (Clontech) and standard procedures. Selenomethionine-labeled DsbP was expressed in BL21(DE3)pLys cells grown in M63 minimal media supplemented with 0.05 mg/ml of dl-selenomethionine and induced for 14 h with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 30 °C. The His6 tag was removed by tobacco etch virus protease, leaving three vector-derived residues (Ser−2-Asn−1-Ala0) at the DsbP N terminus. A final size exclusion chromatography step using a Superdex 75 column was performed to yield the purified DsbP enzyme used in all steps described below. Oxidized or reduced DsbP was prepared using a 25-fold molar excess of copper(II)/1,10-phenanthroline or dithiothreitol (DTT), respectively. The oxidizing/reducing agent was then removed and the buffer exchanged to 10 mm HEPES, pH 7.4, using Sephadex 25 resin. E. coli DsbB membrane extracts for the peptide oxidation assay were prepared as described previously (30) and resuspended in phosphate-buffered saline containing 10% glycerol.
Crystallization and Diffraction Data Collection
Oxidized DsbP samples aliquoted and stored at −80 °C were used for crystallization trials. DsbP crystals were grown by the hanging drop vapor diffusion method at 293 K, set up using a Mosquito crystallization robot (TTP Labtech) and incubated and imaged in a RockImager 1000 (Formulatrix). Selenomethionine-labeled DsbP crystals and the native orthorhombic crystals (Ox1) were grown from a protein sample (25 mg/ml) in 15–16% PEG3350 and 0.1 m sodium malonate, pH 6.6. Of the two monoclinic crystal forms, DsbP(Red) crystals were grown using a protein concentration of 25 mg/ml in 10% (w/v) PEG8000, 20% (v/v) ethylene glycol, and 0.1 m MOPS/HEPES, pH 7.5. The other monoclinic DsbP(Ox2) crystals were grown using a protein concentration of 50 mg/ml and precipitant solution containing 10% PEG 4000, 20% (v/v) glycerol, 0.1 m MES/imidazole, pH 6.5, and 20 mm each of 1,6-hexanediol, 1-butanol, 1,2-propanediol (racemic), 2-propanol, 1,4-butanediol, and 1,3-propanediol (Morpheus screen, Molecular Dimensions). DsbP crystals grown in PEG3350 based conditions were cryoprotected with perfluoropolyether oil (Hampton Research) and the crystals grown in glycerol and ethylene glycol were directly frozen in liquid nitrogen without additional cryo-protectant.
Data Collection and Structure Determination
Diffraction data sets were measured at the Australian Synchrotron MX1 and MX2 beamlines using the BLU-ICE data collection interface (31). All data sets were integrated in iMosflm (32) or XDS (33), space group possibilities were analyzed and scaled using Pointless/Scala or Aimless from the CCP4 suite (34). The orthorhombic crystal form (Ox1) was processed from 93° data giving a reflection multiplicity of 3.5. The monoclinic crystal forms were processed from 220° (Ox2) and 180° (Red) data yielding comparable multiplicities (Ox2, 4.5 and Red, 3.7) to that of Ox1. Data for Ox2 were processed to the edge of the detector; data beyond 2.21 Å were not recorded. Red crystals diffracted to 2.07 Å resolution. However, these data exhibited significant anisotropy along the c* direction and the data were therefore truncated to 2.3 Å resolution, following the data significance assessment using the CC1/2 criteria. The initial phases were determined using HYSS, SOLVE, and RESOLVE implemented in the AutoSol routine of the PHENIX software suite by the Se-SAD method (35). Identification of all 32 selenium sites (the four protomers in the asymmetric unit contain eight sites each) resulted in a figure of merit of 0.39 for reflections to 2.2 Å resolution. Density modification and automated model building traced 765 of 868 residues resulting in an R-factor/R-free of 0.25/0.29 and a map model correlation coefficient of 0.83. Electron density was poor for residues ∼160- 175 and the automated model therefore was not built in 3 of the 4 molecules in the asymmetric unit. This initial DsbP model was improved by manual building into the experimental map using Coot (36).
The three DsbP native data sets (Ox1, Ox2, and Red) were solved by molecular replacement with PHASER (37) using as a search model of the DsbP structure solved by SAD-phasing. Iterative manual building and refinement were carried out in PHENIX (35) and COOT (36). Each structure contains four DsbP molecules in the crystallographic asymmetric unit, giving a total of 12 monomer subunit structures of DsbP and 6 independent copies of the biological dimers. The active site cysteine residues were modeled in the oxidized form for each of the three structures initially. However, negative difference density between the sulfur atoms of the catalytic cysteines in one of the three structures suggested that the disulfide had become reduced. In the final refinements, the reduced and oxidized forms of the active site cysteines were modeled with occupancies of 0.7 and 0.3, respectively. This structure is referred to as the reduced structure, Red.
Electron density was absent for the 3 vector-derived residues and the N-terminal 11 residues and C-terminal 2 residues of the DsbP construct in most of the modeled molecules. Judging from the quality of electron density and the relatively high B-factor values, the region comprising residues 165–170 is the most flexible in DsbP. In molecules A and D of the Red structure, the main chain electron density for this region is connected only at low contour levels (0.5–0.8 σ). The same residues could not be modeled in Ox2 molecule A and electron density for residues 171–172 of molecule B is also poorly ordered. Several surface-exposed side chains could not be built into Red (16 residues) and Ox2 (6 residues) structures with confidence and were therefore excluded from the final model. A summary of the data processing and refinement statistics are presented in Table 2. The stereochemical quality of DsbP models were assessed using AutoDepInputTool (38), MolProbity (39), and SF check (40). The final Red structure has a high average atomic B value (53 Å2 for all atoms (of which molecule D alone contributes 73 Å2)) consistent with anisotropic diffraction of the crystal along c* direction. The quality of the electron density is shown in Fig. 2, F–H. The coordinates and structure factors have been deposited with the Protein Data Bank (PDB codes 4ML1 (Ox1), 4ML6 (Red), and 4MLY (Ox2)).
TABLE 2.
Data collection and refinement statistics
Statistics for the highest resolution shell are shown in parentheses.
| Orthorhombic (Ox1) | Monoclinic (Ox2) | Monoclinic (Red) | Se-SAD | |
|---|---|---|---|---|
| Wavelength (Å) | 0.9537 | 0.9537 | 0.9537 | 0.9793 |
| Resolution range (Å) | 54.14–1.98 | 42.79–2.21 | 47.67–2.30 | 19.6–2.20 |
| Higher resolution shell (Å) | 2.09–1.98 | 2.27–2.21 | 2.38–2.30 | 2.27–2.20 |
| Space group | P 21 21 21 | P 21 | P 21 | P 21 |
| Unit cell | ||||
| a | 54.3 | 54.6 | 54.7 | 54.1 |
| b | 108.3 | 110.5 | 110.6 | 111.3 |
| c | 139.0 | 74.1 | 80.7 | 72.7 |
| α | 90 | 90 | 90 | 90 |
| β | 90 | 93.1 | 96.8 | 95.5 |
| γ | 90 | 90 | 90 | 90 |
| Total reflections | 204,119 | 197,792 | 155,050 | 652,058 |
| Unique reflections | 58,014 (8,315) | 43,861 (3,564) | 42,056 (4,162) | 43,382 (3,732) |
| Multiplicity | 3.5 (3.4) | 4.5 (4.0) | 3.7 (3.8) | 15.0 (14.3) |
| Completeness (%) | 99.7 (99.2) | 99.4 (93.4) | 99.2 (100) | 99.8 (99.5) |
| Mean I/σ(I) | 8.9 (2.5) | 16.2 (5.0) | 16.0 (3.4) | 16.5 (1.9) |
| Wilson B-factor (Å2) | 24.5 | 34.6 | 38.4 | 29.7 |
| R-merge | 0.079 (0.465) | 0.049 (0.244) | 0.047 (0.366) | 0.158 (1.7) |
| R-pim | 0.050 (0.293) | 0.028 (0.157) | 0.033 (0.247) | 0.047 (0.475) |
| Refinement statistics | ||||
| R-work (%) | 19.2 (23.4) | 18.6 (26.4) | 19.0 (26.0) | |
| R-free (%) | 24.3 (29.2) | 23.4 (33.1) | 24.4 (32.4) | |
| Unique reflections | 57,718 | 43,816 | 41,826 | |
| No. of non-H atoms | ||||
| Protein | 6,278 | 6,231 | 6,233 | |
| Water | 686 | 354 | 326 | |
| Protein residues | 811 | 800 | 807 | |
| Root mean square bond lengths (Å) | 0.014 | 0.015 | 0.013 | |
| Root mean square bond angles (°) | 1.47 | 1.54 | 1.27 | |
| Ramachandran | ||||
| Favored (%) | 99.4 | 98.5 | 98.3 | |
| Outliers (%) | 0 | 0.25 | 0.4 | |
| Average B-factor (Å2) | 33.9 | 43.7 | 53.2 | |
| Molprobity | ||||
| Clashscore (percentile) | 3.1 (99th (714))a | 3.51 (99th (456)) | 3.13 (100th (355)) | |
| Score (percentile) | 1.10 (100th (12309)) | 1.14 (100th (10167)) | 1.10 (100th (8909)) | |
a 100th Molprobity (39) percentile is the best among the structures of comparable resolution. The percentile and the number of structures included in the comparison (n) are given in parentheses.
FIGURE 2.

DsbP forms a domain-swapped dimerization domain. Comparison of the N-terminal regions of DsbP (A) and EcDsbG (B). Topology and fold of DsbP (C) and EcDsbG (D). E, domain-swapped human cystatin C. Three-dimensional domain swapping of DsbP and human cystatin C (PDB code 1G96) are mediated through an exchange of the strand β1. Both EcDsbG and human cystatin C contain the cystatin C-fold. The helix conserved in the dimerization domain of EcDsbC/G and human cystatin C is absent in DsbP. Inter-subunit interaction primarily involves strand β1 (human cystatin C) or β4 (EcDsbG) or both strands β1 and β4 (DsbP). The 2mFo − DFc map (m, figure of merit; D is σ-A weighting factor) is displayed around an interfacing region between subunits A and B at a 1.2-σ contour. F, DsbP Ox1; G, DsbP Ox2; and H, DsbP Red. I, structure-based sequence alignment. Structurally equivalent positions (uppercase), variable regions (lowercase), and insertions (dash) are shown. Secondary structure assignments for DsbP (top) and EcDsbG (bottom) are presented. Residues contributing to inter-subunit hydrogen bonding interactions are shown in blue. The β4 strand (boxed) mediating subunit interaction is conserved between DsbC/G and DsbP.
Disulfide Reductase Activity
Many TRX-fold proteins including the disulfide-bond isomerase EcDsbC can reduce the intermolecular disulfide bonds formed between insulin chain A and B in the presence of a mild reducing agent such as DTT (41). The rate of insulin disulfide reduction was spectroscopically monitored at 650 nm by an increase in turbidity resulting from the production of insoluble insulin chain B. The reaction was initiated by the addition of 0.131 mm insulin to the sample mixture containing 10 μm enzyme, 0.33 mm DTT, and 2 mm EDTA in 100 mm sodium phosphate, pH 7.0. Measurements were repeated three times and the resulting mean ± S.D. were used in plots.
Scrambled RNaseA Isomerization Assay
In vitro disulfide isomerase activity was monitored using scrambled RNaseA as substrate (42). Inactive scrambled RNaseA was produced as previously described (43). Disulfide isomerization of incorrectly formed disulfides in scrambled RNaseA (40 μm) was carried out in a reaction volume of 750 μl containing 100 mm sodium phosphate buffer, pH 7.0, 1 mm EDTA, 10 mm DTT, and 10 μm enzyme at 298 K. At various time intervals, a 50-μl sample was withdrawn and mixed with 150 μl of 3 mm cytidine 3′,5′-cyclic monophosphate (cCMP4) to measure RNaseA activity. Hydrolysis of cCMP4 was measured using a Synergy H1 multimode plate reader (BioTek) at 296 nm. Samples containing native RNaseA and scrambled RNaseA in the absence of any other enzyme served as positive and negative controls, respectively.
Dithiol Oxidation Assay
The ability of DsbP and variants to catalyze dithiol oxidation was assessed using a model peptide substrate (CQQGFDGTQNSCK) described previously (44). Intramolecular disulfide bond formation was followed fluorometrically using a Synergy H1 multimode plate reader (BioTek) with excitation set to 340 nm and emission to 615 nm. A 150-μs delay before reading and 100-μs reading time was used for time-resolved fluorescence. The reaction mixture (50 μl final volume) contained 80 or 160 nm enzyme (DsbP, EcDsbC, and EcDsbG) and 1.6 μm membrane extract of EcDsbB in 50 mm MES, 50 mm NaCl, and 2 mm EDTA at pH 5.5. Peptide substrate (8 μm) was added to initiate the reaction. The reaction in the absence of enzyme was used as negative control, and in the presence of EcDsbA was used as a positive control. Mean ± S.D. were calculated from three replicates.
Determination of Redox Potential
The redox potential of DsbP was measured with a method described previously (45) and using a DsbP variant in which the non-catalytic cysteines were mutated to alanine (Cys144 → Ala, Cys168 → Ala). Briefly, 2 μm DsbP (or EcDsbA as control) was incubated with various ratios of oxidized and reduced glutathione (GSSG (1 mm)/GSH (4 μm to 15 mm)) for ∼12 h at 24 °C in degassed 100 mm sodium phosphate buffer, pH 7.0, and 1 mm EDTA. Protein samples were precipitated with 10% trichloroacetic acid and washed with ice-cold acetone prior to labeling the free thiols with 2 mm 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid in 50 mm Tris-HCl, pH 8.0, and 1% SDS. Reduced/oxidized forms of the protein were separated on a 12% non-reducing SDS-PAGE (SDS-NuPAGE) and stained with Coomassie Brilliant Blue. The fraction of reduced protein was quantified using ImageJ (46). The equilibrium constant Keq was calculated using the equation: Y = ([GSH]2/[GSSH])/(Keq + ([GSH]2/[GSSH])), where Y is the normalized fraction of reduced protein at equilibrium (47). The redox potential of the protein was then calculated using the Nernst equation: E0′ = E0′GSH/GSSG − (RT/nF)lnKeq, where E0′GSH/GSSG is the standard potential of −240 mV (48), R is the universal gas constant 8.314 JK−1 mol−1, T is the absolute temperature in K, n is the number of electrons transferred, F is the Faraday constant 9.648 × 104 C mol−1, and Keq is the equilibrium constant. The mean redox potential was calculated from four independent measurements.
RESULTS
Overall Structure
The DsbP molecular structure comprises an N-terminal subdomain connected to a C-terminal TRX-fold catalytic domain by a linker region (residues 60–83) (Fig. 1A). It is assembled as a homodimer (Fig. 1B). The appearance of DsbP dimer is that of an “inverted” V-shaped protein, by comparison to the previously characterized disulfide isomerases EcDsbC and EcDsbG (Fig. 1C) (16, 17). However, the molecular size of DsbP is comparable with EcDsbC/G with the inner distance measuring 41 Å between the two opposing distal ends, and 24 Å from the distal end to the V-shaped cleft (by comparison, EcDsbC measures 39 and 25 Å and EcDsbG measures 58 and 31 Å).
FIGURE 1.

DsbP forms an inverted V compared with DsbG. A, each DsbP subunit consists of three structural regions: an N-terminal subdomain (violet), a linker segment (cyan), and a catalytic domain (yellow). Cysteine sulfurs are indicated by green spheres, with the catalytic cysteines identified by an asterisk. Comparison of the crystal structures of the (B) DsbP (yellow) and (C) EcDsbG homodimers (green). The relative orientation of DsbP and EcDsbG are defined by structural alignment of the N-terminal subdomain. EcDsbC has the same V-arrangement as DsbG, with a shorter linker. The inter-subunit interface in the crystal structures of DsbP (D) and EcDsbG (E) are shown. The interface region forming the subunit interaction, as analyzed in PISA (79), is shown in pink. The green spheres show cysteine sulfurs, with the catalytic cysteines indicated by an asterisk.
Strikingly, all three regions contribute to the extensive dimer interface (2076 ± 44 Å2 buried surface area) in the crystal structure (Fig. 1D). In contrast, the subunit interaction in EcDsbG and other characterized bacterial disulfide isomerases (EcDsbC, Haemophilus influenzae DsbC and S. enterica serovar Typhimurium DsbC) are mediated only through the N-terminal domain (Fig. 1E), with the buried surface area (790–1000 Å2, PDB codes 1v57, 1eej, and 1t3b) measuring half that of DsbP.
DsbP Forms a Domain-swapped Dimer
The N-terminal domains of bacterial disulfide isomerases that have been characterized structurally contain a cystatin-like fold comprising a helix packing against a twisted 4-stranded antiparallel β-sheet (1–2-3–4, Fig. 2B) (16, 17). In the dimer, the β4 strands from subunits A and B interact to form an up and down 8-stranded dimerization domain (Fig. 2, B and D). This dimerization mediating structure of EcDsbG is conserved in EcDsbC and other disulfide isomerases (16, 17, 49–51).
DsbP does not follow this pattern. Instead, a domain-swapped dimer is formed through an exchange of β1 strands between subunits A and B (Fig. 2A). Following a low complexity N-terminal region (1–14 residues), the DsbP chain of subunit A forms the β1 strand that augments the β2′ end of the 3-stranded antiparallel β-sheet (2′-3′-4′) of subunit B. Through a linker loop (L1), the β1 strand then enters the 3-stranded antiparallel β-sheet (2–3-4) that augments the β1′ end of subunit B. The main chain hydrogen bonding interactions between β4 strands of subunits A and B completes the 8-stranded dimerization domain (Fig. 2, A and C). Remarkably, the architecture of the dimerization domain between DsbP and EcDsbG is similar with a root mean square deviation of 2.0 Å for 58 Cα equivalent atoms. The key difference being that the three-dimensional domain swapping forms the dimerization domain in DsbP (Fig. 2). Additionally, the conserved helix found at the N termini of EcDsbG and other disulfide isomerases is absent in DsbP (Fig. 2). The intersubunit hydrogen bonding interaction between β4 strands appears to be a highly conserved structural feature among TRX-fold disulfide isomerases (Fig. 2I).
DsbP Has Two Canonical Catalytic Domains
The C-terminal region of each DsbP chain contains a canonical “DsbC/DsbG C-terminal domain-like fold” comprised of a non-contiguous TRX domain encompassing a helical domain (Fig. 3A). The structure of DsbP is architecturally similar, but nonetheless, different from the EcDsbC/DsbG catalytic domains (e.g. root mean square deviation between molA of Ox1 and molA of EcDsbG (PDB code 1v57) is 2.9 Å for 127 Cα atoms). The major differences occur within the helical domain (H3-H5) and the C-terminal helix of the TRX-fold, H6, among these three proteins (Fig. 3A). A TRX-fold CXXC catalytic motif is present at the N terminus of helix H2 (Figs. 1A and 3D), and a second non-catalytic disulfide bond is present at a point similar to that of EcDsbC, between the C-terminal end of helix H3 and the connecting loop between helices H4 and H5 (Fig. 1A).
FIGURE 3.
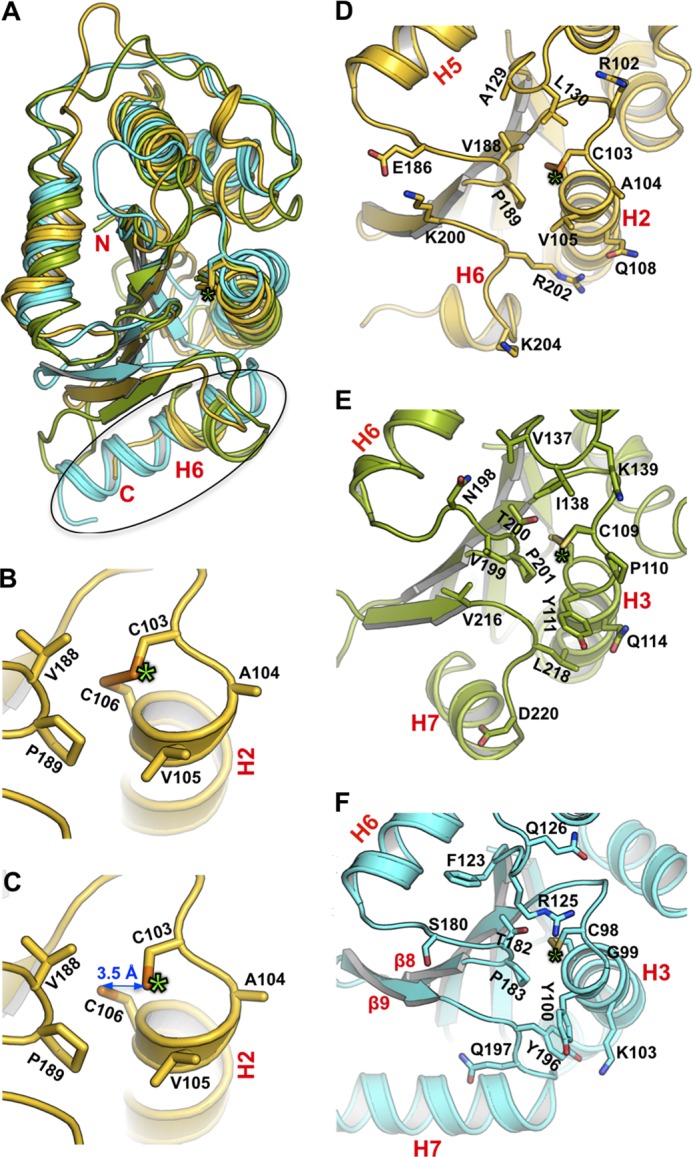
DsbP has a canonical DsbC/DsbG-like catalytic domain. A, structural superposition of the catalytic domains of DsbP (yellow), EcDsbG (green), and EcDsbC (cyan). The structural deviation in helix H6 of DsbP and the equivalent helix H7 of EcDsbC/G is outlined. Active site of oxidized (B) and reduced (C) states of DsbP is shown. The catalytic motif (Cys103-Ala104-Val105-Cys106) and the cis-Pro motif (Val188-Pro189) are labeled. For clarity, only the reduced form of the catalytic cysteines is shown in panel C, although the refined Red structure includes both reduced (70%) and oxidized (30%) forms. The structure of the catalytic face of DsbP (D), EcDsbG (E), and EcDsbC (F) is shown. Residues in the vicinity of catalytic residues are shown. Note DsbP contains four basic residues around the catalytic site (Arg102, Lys200, Arg202, and Lys204), a feature absent in EcDsbG and EcDsbC (cf. panel D with panels E and F). In each panel, the position of the catalytic cysteine is indicated by a green asterisk.
The catalytic motif and the adjacent cis-proline (cis-Pro) motif together form the active site of TRX-fold redox proteins (Fig. 3, D–F). In both EcDsbC and EcDsbG, the catalytic and cis-Pro motifs are Cys-Gly/Pro-Tyr-Cys and Thr-Pro, respectively. However, the residues forming these important regions are different in DsbP: the catalytic motif is Cys103-Ala104-Val105-Cys106 and the cis-Pro motif is Val188-Pro189. Interestingly, a Cys-Ala-Val-Cys motif is present in some structurally characterized FAD/NAD(P)-dependent TRX reductases (52) although there are no NADP or FAD binding sites evident in DsbP. Moreover, the Val-cis-Pro motif is typically found in disulfide oxidases (DsbAs) rather than isomerases of Gram-negative bacteria (53). We determined the standard redox potential of DsbP at pH 7.0 and 25 °C (Fig. 4A) and found that DsbP (−161 mV) is more reducing than EcDsbA (−122 mV), EcDsbC (−130 mV (54)), and EcDsbG (−126 mV, (55)).
FIGURE 4.

Redox characterization of DsbP. A, determination of the redox equilibrium constant (Keq) of DsbP with glutathione at pH 7.0 and 25 °C. A representative plot of four independent experiments showing the normalized fraction of reduced DsbP at various ratios of reduced:oxidized glutathione. The mean Keq (2.0 ± 0.18 mm) was obtained from nonlinear curve fits and the redox potential (−160.6 ± 1.0 mV) calculated as described under “Experimental Procedures.” B, scrambled RNaseA disulfide isomerization activity. 100% activity corresponds to the averaged catalytic rate of native RNaseA. C, enzyme catalyzed insulin precipitation as a result of intermolecular disulfide reduction. D, percent of peptide dithiol oxidase activity relative to control EcDsbA activity. Oxidation rates were calculated over the first 15 min (see “Experimental Procedures”). 100% activity corresponds to the catalytic rate of EcDsbA. For panels B–D, the average and S.D. for each data point were calculated from three replicates; DsbP (square), CGYC (inverted triangle), CPYC (triangle), EcDsbC (diamond), and EcDsbG (circle).
The catalytic disulfide bond of disulfide oxidoreductases is susceptible to synchrotron radiation reduction. The mixed catalytic disulfide bond observed in the Red structure is therefore not completely unexpected and allowed us to capture both redox states of DsbP. Structural comparison of the oxidized and reduced forms of DsbP do not indicate any major conformational changes at the catalytic site (Fig. 3, B and C). Distances of 2.1 Å (oxidized form) and 3.5 Å (reduced form) between the sulfur atoms of the cysteine residues are consistent with those in EcDsbC and EcDsbG structures (16, 17).
Reduced EcDsbC/DsbG is thought to be energetically favored as a consequence of unfavorable contacts in the oxidized form (17). One proposed destabilizing interaction in the oxidized enzyme structures occur between the sulfur of the first Cys in the active site CXXC motif and the main chain nitrogen of the second Cys (3.1 Å). A second unfavorable contact occurs between the sulfur of the first Cys and the side chain oxygen of Thr motif (3.4 Å). Analysis of oxidized DsbP structures yielded a similar distance of 3.1 Å between Cys103 sulfur and the main chain nitrogen of Cys106. However, the natural variation of Val188 in place of Thr in the DsbP cis-Pro motif may relieve the second destabilizing interaction. Intriguingly, mutation of Thr for Val in the EcDsbC cis-Pro motif imparted a more reducing character on the enzyme (56).
DsbP Is a Disulfide Isomerase
We next explored the redox characteristics of DsbP in standard dithiol oxidase, disulfide isomerase, and disulfide reductase assays.
The ability to isomerize disulfide bonds was tested using scrambled RNaseA as described under “Experimental Procedures.” Under the conditions of the assay, DsbP restored 60% of RNase activity within 5 h (Fig. 4B). In comparison, prototypical EcDsbC restored full RNaseA activity in 2 h, whereas EcDsbG restored 45% activity in 5 h. Similarly, EcDsbC exhibited high disulfide reductase activity in an insulin reduction assay, whereas both DsbP (marginally active) and EcDsbG (inactive) were poor reductants in this assay (Fig. 4C). All three enzymes were inactive in the in vitro dithiol oxidation assay (Fig. 4D).
The effect of specific CXXC motifs on redox activity was tested using DsbP active site substitution variants. The DsbP variants were created to mimic the active site of EcDsbC (CGYC, DsbP (CGYC)) or EcDsbG (CPYC, DsbP (CPYC)). The active site variant CGYC almost matched the RNaseA isomerization activity of EcDsbC (Fig. 4B) but only moderately improved the insulin reductase activity. In comparison, the EcDsbG-like CPYC variant exhibited almost equivalent activities to those of the wild-type DsbP (Fig. 4, B-D). Overall, these results show that the plasmid-encoded putative DsbP is a disulfide isomerase, and a less efficient insulin reductant than EcDsbC despite having a Val-cis-Pro motif and a more reducing redox potential.
Orientation of the Linker Helix Alters the Shape of DsbP
To understand the basis for the inverted V-shape of DsbP, we analyzed the 24-residue linker segment (residues 60–83) connecting the dimerization and catalytic domains. Structural alignment suggested that the linker helix (H1) of DsbP is oriented in the opposite direction compared with the equivalent linker helix of EcDsbG (Fig. 5A).
FIGURE 5.

The putative DsbP substrate-binding surface differs from that of EcDsbC/G. A, comparison of the orientation of the linker (arrows) in DsbP (yellow) and EcDsbG (green). B, the linker segment of DsbP forms a tight interaction with the dimerization (DD) and catalytic (CD) domains. C, schematic representation showing how the upper and lower surfaces of the dimerization domain form the cleft in V-shaped EcDsbC/G and inverse V-shaped DsbP, respectively. Structure of DsbP (D), EcDsbG (E), and EcDsbC (F) viewed down the cleft. All three structures are similarly oriented such that strand β4 (blue) points up and the catalytic domains point out of the page. Loops forming the cleft are shown in red and the catalytic cysteine residues are shown as green spheres. Note that strand β4 in DsbP is behind loop L1, which is not the case for EcDsbC/G.
Close examination of the DsbP linker (Fig. 5B) reveals that four hydrophobic and two polar side chains of H1 (Met61, Met64, Val67 and Ala68; Gln63 and Arg65) engage in interactions with the twisted β-sheet closing this end of the dimerization domain. Similarly, helix H1′ seals the other end of the dimerization domain, resembling a closed hemi-cylinder shape. Following helix H1, the segment continues at the interface between the dimerization domain and the catalytic domain before forming a 310-helix and a turn that connects to the catalytic domain. These structural elements contribute six hydrophobic side chains (Ile71, Phe73 Met76, Met78, Val80, and Leu83) that insert and fill the gap between the β-sheet (5–7-6–8-9) and helix H5 of the catalytic domain. Thus, the linker segment mediates tight interdomain interactions unique to DsbP (Fig. 5B).
In both EcDsbC and EcDsbG, the surface formed on the flexible linker helices (H2 and H2′) and the “upper” surface of the cleft created on the dimerization domain (Fig. 5C) are proposed to be involved in substrate recognition (16, 57). By analogy, we hypothesize that the inverted V-shaped DsbP also uses the “bottom” surface of the dimerization domain for substrate recognition. Connecting loops H1-β1 and β2-β3 as well as strand β4 form the V-shaped cleft in EcDsbC/DsbG (Fig. 5, E and F). However, in DsbP, the corresponding cleft is formed by connecting loops β1-β2 (loop L1) and β3-β4 (Fig. 5D). Notably, DsbP strand β4 is located on the opposite face of the putative binding region.
Surface structures of the clefts differ considerably among these three proteins due to variations in length, conformation, and residue composition of loops (Fig. 6, A–C). Despite these differences, DsbP and EcDsbG display an overall negative electrostatic surface potential in their putative substrate binding regions, whereas EcDsbC is largely neutral. Additionally, all three proteins show a positive electrostatic surface potential around the catalytic cysteine, with DsbP displaying the most extended positive potential arising from four basic residues on the catalytic face of the enzyme (Arg102, Lys200, Arg202, and Lys204, see Fig. 3D).
FIGURE 6.

Comparison of electrostatic surface properties and conformational changes. Electrostatic surface comparison of DsbP (A), EcDsbG (B), and EcDsbC (C). Electrostatic surface potential is contoured from −7 (red) to +7 (blue) kT/e. The catalytic domain (CD), dimerization domain (DD), and the catalytic Cys (green asterisk) of each subunit are indicated. The orientations of the molecules in panels A–C show both the catalytic face and the dimerization cleft and are not intended to present the 2-fold axis. D, E, and F, DsbP catalytic domain undergoes conformational changes and wobbly domain motion. Structural superposition of 12 monomers found in the DsbP crystal structures (Ox1, Ox2, and Red1): D, the linker segment, the catalytic domain (E), and the complete DsbP subunit (F) which is aligned to the N-terminal subdomain. The conformational change in the hinge region (around the 310-helix of the linker segment, marked with a circle and black arrow in panels D and F) results in movement of the catalytic domain. The two distinct conformers that showed helix H5 extension are shown in blue (E and F).
DsbP Catalytic Domain Exhibits Domain Motion
The flexibility of linker helix (H2) has been proposed to allow EcDsbC and EcDsbG to accommodate various sizes and shapes of substrate proteins in the cleft (16, 51). However, structural superposition of the linker segments derived from 12 DsbP molecules (independently refined from three high-resolution x-ray diffraction data sets (Ox1, Ox2, and Red, see “Experimental Procedures” for details)) revealed that the equivalent linker is largely unperturbed, albeit a small progressive conformational change was apparent, starting near the 310-helix and running toward the catalytic domain (Fig. 6D). In a similar analysis of the catalytic domain, the TRX domain was found to retain the same overall conformation in each monomer, whereas the embedded helical domain can undergo significant conformational changes, including extension of helix H5 (Fig. 6E). Despite the presence of a (non-catalytic) disulfide bond between Cys143 and Cys168 bridging helix H3 and the loop preceding helix H5 in the helical domain, the residues in this region are highly flexible (see “Experimental Procedures”). The corresponding region in EcDsbC, which also has a disulfide, is also flexible as indicated by high B-factors. Structural superposition of the N-terminal domains of the 12 DsbP molecules revealed that conformational changes in the 310-helix are translated to the catalytic domain causing a “wobbly” motion of the catalytic domain (Fig. 6F) that we propose may contribute to enzymatic activity of DsbP.
DISCUSSION
Genetic plasticity through horizontal gene transfer is a key contributor in the rapid adaptation of bacteria to different environmental challenges (58, 59). The acceleration of reported multidrug resistance strains appears to be one such bacterial response to inappropriate use of antibiotics (60–62). Bacteria disseminate antibiotic resistance genes from one bacterium to another by multiple mechanisms, including a conjugation secretion system that is encoded by a conjugative plasmid or ICE (6).
Plasmid/ICE-encoded disulfide isomerases appear to play an important role in folding and assembly of the conjugative machinery (10, 13). The current body of knowledge of bacterial TRX-fold redox proteins is derived from chromosomally encoded proteins. Accordingly, disulfide oxidase (DsbA in conjunction with its redox partner DsbB) functions as the primary disulfide forming enzyme and is required for oxidative folding of cell-surface and exported proteins including toxins and virulence factors (15). On the other hand, disulfide isomerases (EcDsbC/DsbG) proofread and shuffle incorrect disulfides or protect single cysteine containing proteins from oxidative damage (14–18).
The data presented here reveal the structural and functional details of a conserved plasmid-encoded disulfide isomerase DsbP from a group of multidrug resistance conjugative IncA/C plasmids. We showed that DsbP is atypical due to a domain-swapped dimerization domain, a linker that mediates interdomain interactions, an inverse putative binding site, and a rare CXXC active site motif in the canonical EcDsbC/G-like catalytic domain.
Three-dimensional domain swapping is generally regarded as being independent of protein sequence or secondary structure (63). Interestingly, there are common features between DsbP and human cystatin C, an amyloidogenic cystatin-fold protein that also participates in domain swapping (64). In both proteins it is the β1 strand that is exchanged between the two subunits (Fig. 2E). However, DsbP lacks the N-terminal helix that is otherwise conserved in cystatin and the dimerization interface differs between the two proteins. A structural consequence of three-dimensional domain swapping in DsbP is a much larger subunit interface compared with EcDsbC/DsbG. This may have important implications for function because dimerization is essential for both chaperone and isomerase activities of bacterial disulfide isomerases (65, 66).
A hallmark of disulfide isomerases, including eukaryotic protein-disulfide isomerase, are two catalytic domains that face each other across the putative substrate-binding cleft. Motion of the catalytic domains was shown to be essential for function (16, 51, 67). Our crystal structures of DsbP are consistent with the interpretation that the catalytic domains move relative to the dimerization domain, despite the linker segment being uniquely engaged in inter-domain interactions. Moreover, we speculate that the flexibility/disorder in the region around the non-catalytic disulfide bond may point to this being an important site for substrate binding or regulation of DsbP.
DsbP has in vitro disulfide isomerase activity that is moderately better than that of EcDsbG (60 versus 45% in RNaseA refolding activity). Substitution of the active site motif to that of EcDsbC (CGYC) improved isomerase activity almost to that of EcDsbC. The XX dipeptide in the CXXC active site motif and the residue preceding the cis-proline have been shown previously to affect redox properties and catalytic activities of TRX-fold oxidoreductases (68–73). However, the difference in activity between DsbP (CGYC) and EcDsbC in the insulin reductase assay suggests that other elements also contribute to the catalytic activities of these enzymes.
The relatively acidic surface on the inverted dimerization domain and an extended positive potential surrounding the catalytic site of DsbP compared with EcDsbC/DsbG may suggest different substrate binding specificities for these enzymes. In support of this, a virulence plasmid-encoded DsbA-like enzyme (SrgA from plasmid pStSR100 in Salmonella typhimurium), essential for the oxidative folding of the plasmid-encoded fimbriae major subunit protein (PefA), also exhibited overlapping in vitro activities but different substrate specificity compared with DsbA (74, 75).
Several Tra proteins required for the structure and assembly of conjugative pili contain multiple cysteine residues (10), and are therefore candidate substrates for DsbP. Analysis of functional links for DsbP in the STRING database (76) supports these possibilities (data not shown). Previous attempts to detect substrates for EcDsbC and EcDsbG using two-dimensional gel electrophoresis identified two substrates for EcDsbC (RNase I and MepA) but none for DsbG (77). Another successful strategy employed to trap substrate proteins was through mutagenesis of cis-Pro loop residues (78). Similar approaches may now be used to identify substrates of DsbP. Taken together, our structural and functional studies of DsbP provide a framework to understand the roles of plasmid-mediated disulfide oxidoreductases in the assembly processes of type IV secretion systems and may lead to novel approaches aimed at preventing plasmid- and ICE-mediated transfer of drug resistance genes.
Acknowledgments
We thank the beam-line support team at the Australian Synchrotron for assistance. We also acknowledge use of the University of Queensland Remote Operation Crystallization and X-ray (UQROCX) Diffraction Facility and the assistance of Karl Byriel and Dr. Gordon King.
This work was supported in part by Australian Research Council (ARC) Australian Laureate Fellowship FL0992138 (to J. L. M.).
The atomic coordinates and structure factors (codes 4ML1, 4ML6, and 4MLY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ICE
- integrative and conjugative elements
- TRX
- thioredoxin
- PDB
- Protein Data Bank
- Red
- reduced structure.
REFERENCES
- 1. Carlet J., Jarlier V., Harbarth S., Voss A., Goossens H., Pittet D. (2012) Ready for a world without antibiotics? The Pensieres antibiotic resistance call to action. Antimicrob. Resist. Infect. Control 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Won S. Y., Munoz-Price L. S., Lolans K., Hota B., Weinstein R. A., Hayden M. K. (2011) Emergence and rapid regional spread of Klebsiella pneumoniae carbapenemase-producing Enterobacteriaceae. Clin. Infect. Dis. 53, 532–540 [DOI] [PubMed] [Google Scholar]
- 3. Fernández-Alarcón C., Singer R. S., Johnson T. J. (2011) Comparative genomics of multidrug resistance-encoding IncA/C plasmids from commensal and pathogenic Escherichia coli from multiple animal sources. PLoS ONE 6, e23415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Johnson A. P., Woodford N. (2013) Global spread of antibiotic resistance. The example of New Delhi metallo-β-lactamase (NDM)-mediated carbapenem resistance. J. Med. Microbiol. 62, 499–513 [DOI] [PubMed] [Google Scholar]
- 5. Wozniak R. A., Fouts D. E., Spagnoletti M., Colombo M. M., Ceccarelli D., Garriss G., Déry C., Burrus V., Waldor M. K. (2009) Comparative ICE genomics. Insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 5, e1000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garriss G., Waldor M. K., Burrus V. (2009) Mobile antibiotic resistance encoding elements promote their own diversity. PLoS Genet. 5, e1000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sykes R. (2010) The 2009 Garrod lecture. The evolution of antimicrobial resistance, a Darwinian perspective. J. Antimicrob. Chemother. 65, 1842–1852 [DOI] [PubMed] [Google Scholar]
- 8. Fronzes R., Christie P. J., Waksman G. (2009) The structural biology of type IV secretion systems. Nat. Rev. Microbiol. 7, 703–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zechner E. L., Lang S., Schildbach J. F. (2012) Assembly and mechanisms of bacterial type IV secretion machines. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 1073–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hemmis C. W., Schildbach J. F. (2013) Thioredoxin-like proteins in F and other plasmid systems. Plasmid 70, 168–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu J. H., Moore D., Lee T., Ippen-Ihler K. (1987) Analysis of Escherichia coli K12 F factor transfer genes, traQ, trbA, and trbB. Plasmid 18, 54–69 [DOI] [PubMed] [Google Scholar]
- 12. Elton T. C., Holland S. J., Frost L. S., Hazes B. (2005) F-like type IV secretion systems encode proteins with thioredoxin folds that are putative DsbC homologues. J. Bacteriol. 187, 8267–8277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hemmis C. W., Berkmen M., Eser M., Schildbach J. F. (2011) TrbB from conjugative plasmid F is a structurally distinct disulfide isomerase that requires DsbD for redox state maintenance. J. Bacteriol. 193, 4588–4597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Depuydt M., Leonard S. E., Vertommen D., Denoncin K., Morsomme P., Wahni K., Messens J., Carroll K. S., Collet J. F. (2009) A periplasmic reducing system protects single cysteine residues from oxidation. Science 326, 1109–1111 [DOI] [PubMed] [Google Scholar]
- 15. Heras B., Shouldice S. R., Totsika M., Scanlon M. J., Schembri M. A., Martin J. L. (2009) DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7, 215–225 [DOI] [PubMed] [Google Scholar]
- 16. McCarthy A. A., Haebel P. W., Törrönen A., Rybin V., Baker E. N., Metcalf P. (2000) Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat. Struct. Biol. 7, 196–199 [DOI] [PubMed] [Google Scholar]
- 17. Heras B., Edeling M. A., Schirra H. J., Raina S., Martin J. L. (2004) Crystal structures of the DsbG disulfide isomerase reveal an unstable disulfide. Proc. Natl. Acad. Sci. U.S.A. 101, 8876–8881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Depuydt M., Messens J., Collet J. F. (2011) How proteins form disulfide bonds. Antioxid. Redox Signal. 15, 49–66 [DOI] [PubMed] [Google Scholar]
- 19. Kadokura H., Katzen F., Beckwith J. (2003) Protein disulfide bond formation in prokaryotes. Annu. Rev. Biochem. 72, 111–135 [DOI] [PubMed] [Google Scholar]
- 20. Inaba K. (2009) Disulfide bond formation system in Escherichia coli. J. Biochem. 146, 591–597 [DOI] [PubMed] [Google Scholar]
- 21. Lewin A., Crow A., Hodson C. T., Hederstedt L., Le Brun N. E. (2008) Effects of substitutions in the CXXC active-site motif of the extracytoplasmic thioredoxin ResA. Biochem. J. 414, 81–91 [DOI] [PubMed] [Google Scholar]
- 22. Johnson T. J., Lang K. S. (2012) IncA/C plasmids. An emerging threat to human and animal health? Mob. Genet. Elements 2, 55–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aoki T., Egusa S., Ogata Y., Watanabe T. (1971) Detection of resistance factors in fish pathogen Aeromonas liquefaciens. J. Gen. Microbiol. 65, 343–349 [DOI] [PubMed] [Google Scholar]
- 24. Del Castillo C. S., Hikima J., Jang H. B., Nho S. W., Jung T. S., Wongtavatchai J., Kondo H., Hirono I., Takeyama H., Aoki T. (2013) Comparative sequence analysis of a multidrug-resistant plasmid from Aeromonas hydrophila. Antimicrob. Agents Chemother. 57, 120–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carattoli A., Villa L., Poirel L., Bonnin R. A., Nordmann P. (2012) Evolution of IncA/C blaCMY-(2)-carrying plasmids by acquisition of the blaNDM-(1) carbapenemase gene. Antimicrob. Agents Chemother. 56, 783–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sekizuka T., Matsui M., Yamane K., Takeuchi F., Ohnishi M., Hishinuma A., Arakawa Y., Kuroda M. (2011) Complete sequencing of the bla(NDM-1)-positive IncA/C plasmid from Escherichia coli ST38 isolate suggests a possible origin from plant pathogens. PLoS ONE 6, e25334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drieux L., Decré D., Frangeul L., Arlet G., Jarlier V., Sougakoff W. (2013) Complete nucleotide sequence of the large conjugative pTC2 multireplicon plasmid encoding the VIM-1 metallo-β-lactamase. J. Antimicrob. Chemother. 68, 97–100 [DOI] [PubMed] [Google Scholar]
- 28. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 29. Studier F. W. (2005) Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 30. Bader M., Muse W., Zander T., Bardwell J. (1998) Reconstitution of a protein disulfide catalytic system. J. Biol. Chem. 273, 10302–10307 [DOI] [PubMed] [Google Scholar]
- 31. McPhillips T. M., McPhillips S. E., Chiu H. J., Cohen A. E., Deacon A. M., Ellis P. J., Garman E., Gonzalez A., Sauter N. K., Phizackerley R. P., Soltis S. M., Kuhn P. (2002) Blu-Ice and the distributed control system. Software for data acquisition and instrument control at macromolecular crystallography beamlines. J. Synchrotron Radiat. 9, 401–406 [DOI] [PubMed] [Google Scholar]
- 32. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM. A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 37. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang H., Guranovic V., Dutta S., Feng Z., Berman H. M., Westbrook J. D. (2004) Automated and accurate deposition of structures solved by x-ray diffraction to the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 60, 1833–1839 [DOI] [PubMed] [Google Scholar]
- 39. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vaguine A. A., Richelle J., Wodak S. J. (1999) SFCHECK. A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. D Biol. Crystallogr. 55, 191–205 [DOI] [PubMed] [Google Scholar]
- 41. Bardwell J. C., McGovern K., Beckwith J. (1991) Identification of a protein required for disulfide bond formation in vivo. Cell 67, 581–589 [DOI] [PubMed] [Google Scholar]
- 42. Hillson D. A., Lambert N., Freedman R. B. (1984) Formation and isomerization of disulfide bonds in proteins. Protein disulfide-isomerase. Methods Enzymol. 107, 281–294 [DOI] [PubMed] [Google Scholar]
- 43. Heras B., Kurz M., Jarrott R., Shouldice S. R., Frei P., Robin G., Cemazar M., Thöny-Meyer L., Glockshuber R., Martin J. L. (2008) Staphylococcus aureus DsbA does not have a destabilizing disulfide. A new paradigm for bacterial oxidative folding. J. Biol. Chem. 283, 4261–4271 [DOI] [PubMed] [Google Scholar]
- 44. Vivian J. P., Scoullar J., Rimmer K., Bushell S. R., Beddoe T., Wilce M. C., Byres E., Boyle T. P., Doak B., Simpson J. S., Graham B., Heras B., Kahler C. M., Rossjohn J., Scanlon M. J. (2009) Structure and function of the oxidoreductase DsbA1 from Neisseria meningitidis. J. Mol. Biol. 394, 931–943 [DOI] [PubMed] [Google Scholar]
- 45. Inaba K., Ito K. (2002) Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. EMBO J. 21, 2646–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ. 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wunderlich M., Glockshuber R. (1993) In vivo control of redox potential during protein folding catalyzed by bacterial protein disulfide-isomerase (DsbA). J. Biol. Chem. 268, 24547–24550 [PubMed] [Google Scholar]
- 48. Gilbert H. F. (1995) Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 251, 8–28 [DOI] [PubMed] [Google Scholar]
- 49. Yeh S. M., Koon N., Squire C., Metcalf P. (2007) Structures of the dimerization domains of the Escherichia coli disulfide-bond isomerase enzymes DsbC and DsbG. Acta Crystallogr. D Biol. Crystallogr. 63, 465–471 [DOI] [PubMed] [Google Scholar]
- 50. Zhang M., Monzingo A. F., Segatori L., Georgiou G., Robertus J. D. (2004) Structure of DsbC from Haemophilus influenzae. Acta Crystallogr. D Biol. Crystallogr. 60, 1512–1518 [DOI] [PubMed] [Google Scholar]
- 51. Jiao L., Kim J. S., Song W. S., Yoon B. Y., Lee K., Ha N. C. (2013) Crystal structure of the periplasmic disulfide-bond isomerase DsbC from Salmonella enterica serovar Typhimurium and the mechanistic implications. J. Struct. Biol. 183, 1–10 [DOI] [PubMed] [Google Scholar]
- 52. Dai S., Saarinen M., Ramaswamy S., Meyer Y., Jacquot J. P., Eklund H. (1996) Crystal structure of Arabidopsis thaliana NADPH-dependent thioredoxin reductase at 2.5-Å resolution. J. Mol. Biol. 264, 1044–1057 [DOI] [PubMed] [Google Scholar]
- 53. Shouldice S. R., Heras B., Walden P. M., Totsika M., Schembri M. A., Martin J. L. (2011) Structure and function of DsbA, a key bacterial oxidative folding catalyst. Antioxid. Redox. Signal. 14, 1729–1760 [DOI] [PubMed] [Google Scholar]
- 54. Zapun A., Missiakas D., Raina S., Creighton T. E. (1995) Structural and functional characterization of DsbC, a protein involved in disulfide bond formation in Escherichia coli. Biochemistry 34, 5075–5089 [DOI] [PubMed] [Google Scholar]
- 55. Bessette P. H., Cotto J. J., Gilbert H. F., Georgiou G. (1999) In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J. Biol. Chem. 274, 7784–7792 [DOI] [PubMed] [Google Scholar]
- 56. Ren G., Stephan D., Xu Z., Zheng Y., Tang D., Harrison R. S., Kurz M., Jarrott R., Shouldice S. R., Hiniker A., Martin J. L., Heras B., Bardwell J. C. (2009) Properties of the thioredoxin fold superfamily are modulated by a single amino acid residue. J. Biol. Chem. 284, 10150–10159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Arredondo S. A., Chen T. F., Riggs A. F., Gilbert H. F., Georgiou G. (2009) Role of dimerization in the catalytic properties of the Escherichia coli disulfide isomerase DsbC. J. Biol. Chem. 284, 23972–23979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Levin B. R., Bergstrom C. T. (2000) Bacteria are different. Observations, interpretations, speculations, and opinions about the mechanisms of adaptive evolution in prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 97, 6981–6985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arber W. (2000) Genetic variation. Molecular mechanisms and impact on microbial evolution. FEMS Microbiol. Rev. 24, 1–7 [DOI] [PubMed] [Google Scholar]
- 60. Neu H. C. (1992) The crisis in antibiotic resistance. Science 257, 1064–1073 [DOI] [PubMed] [Google Scholar]
- 61. Austin D. J., Kristinsson K. G., Anderson R. M. (1999) The relationship between the volume of antimicrobial consumption in human communities and the frequency of resistance. Proc. Natl. Acad. Sci. U.S.A. 96, 1152–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kardas P., Devine S., Golembesky A., Roberts C. (2005) A systematic review and meta-analysis of misuse of antibiotic therapies in the community. Int. J. Antimicrob. Agents 26, 106–113 [DOI] [PubMed] [Google Scholar]
- 63. Liu Y., Eisenberg D. (2002) 3D domain swapping. As domains continue to swap. Protein Sci. 11, 1285–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Janowski R., Kozak M., Jankowska E., Grzonka Z., Grubb A., Abrahamson M., Jaskolski M. (2001) Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 8, 316–320 [DOI] [PubMed] [Google Scholar]
- 65. Zhao Z., Peng Y., Hao S. F., Zeng Z. H., Wang C. C. (2003) Dimerization by domain hybridization bestows chaperone and isomerase activities. J. Biol. Chem. 278, 43292–43298 [DOI] [PubMed] [Google Scholar]
- 66. Segatori L., Murphy L., Arredondo S., Kadokura H., Gilbert H., Beckwith J., Georgiou G. (2006) Conserved role of the linker α-helix of the bacterial disulfide isomerase DsbC in the avoidance of misoxidation by DsbB. J. Biol. Chem. 281, 4911–4919 [DOI] [PubMed] [Google Scholar]
- 67. Wang C., Li W., Ren J., Fang J., Ke H., Gong W., Feng W., Wang C. C. (2013) Structural insights into the redox-regulated dynamic conformations of human protein disulfide isomerase. Antioxid. Redox Signal. 19, 36–45 [DOI] [PubMed] [Google Scholar]
- 68. Bessette P. H., Qiu J., Bardwell J. C., Swartz J. R., Georgiou G. (2001) Effect of sequences of the active-site dipeptides of DsbA and DsbC on in vivo folding of multidisulfide proteins in Escherichia coli. J. Bacteriol. 183, 980–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Charbonnier J. B., Belin P., Moutiez M., Stura E. A., Quéméneur E. (1999) On the role of the cis-proline residue in the active site of DsbA. Protein Sci. 8, 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Su D., Berndt C., Fomenko D. E., Holmgren A., Gladyshev V. N. (2007) A conserved cis-proline precludes metal binding by the active site thiolates in members of the thioredoxin family of proteins. Biochemistry 46, 6903–6910 [DOI] [PubMed] [Google Scholar]
- 71. Guddat L. W., Bardwell J. C., Glockshuber R., Huber-Wunderlich M., Zander T., Martin J. L. (1997) Structural analysis of three His32 mutants of DsbA. Support for an electrostatic role of His32 in DsbA stability. Protein Sci. 6, 1893–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mössner E., Huber-Wunderlich M., Glockshuber R. (1998) Characterization of Escherichia coli thioredoxin variants mimicking the active-sites of other thiol/disulfide oxidoreductases. Protein Sci. 7, 1233–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Quan S., Schneider I., Pan J., Von Hacht A., Bardwell J. C. (2007) The CXXC motif is more than a redox rheostat. J. Biol. Chem. 282, 28823–28833 [DOI] [PubMed] [Google Scholar]
- 74. Bouwman C. W., Kohli M., Killoran A., Touchie G. A., Kadner R. J., Martin N. L. (2003) Characterization of SrgA, a Salmonella enterica serovar Typhimurium virulence plasmid-encoded paralogue of the disulfide oxidoreductase DsbA, essential for biogenesis of plasmid-encoded fimbriae. J. Bacteriol. 185, 991–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Heras B., Totsika M., Jarrott R., Shouldice S. R., Guncar G., Achard M. E., Wells T. J., Argente M. P., McEwan A. G., Schembri M. A. (2010) Structural and functional characterization of three DsbA paralogues from Salmonella enterica serovar Typhimurium. J. Biol. Chem. 285, 18423–18432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. von Mering C., Huynen M., Jaeggi D., Schmidt S., Bork P., Snel B. (2003) STRING. A database of predicted functional associations between proteins. Nucleic Acids Res. 31, 258–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hiniker A., Bardwell J. C. (2004) In vivo substrate specificity of periplasmic disulfide oxidoreductases. J. Biol. Chem. 279, 12967–12973 [DOI] [PubMed] [Google Scholar]
- 78. Kadokura H., Tian H., Zander T., Bardwell J. C., Beckwith J. (2004) Snapshots of DsbA in action. Detection of proteins in the process of oxidative folding. Science 303, 534–537 [DOI] [PubMed] [Google Scholar]
- 79. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]