

Background: The HIV-1 accessory protein Vpr interacts with the human nucleotide excision repair protein hHR23A.
Results: NMR and other biophysical/biochemical techniques were used to identify Vpr binding sites on hHR23A.
Conclusion: hHR23A interacts with Vpr via two domains, XPCB and UBA2.
Significance: The involvement of the hHR23A XPCB and UBA2 domains in Vpr binding suggests that cellular effects can be modulated by diverse mechanisms.
Keywords: Base Excision Repair, HIV-1, NMR, Protein-Protein Interactions, Ubiquitination, Binding, UBA2, Vpr, XPCB, hHR23A
Abstract
The human homolog of the yeast DNA repair protein RAD23, hHR23A, has been found previously to interact with the human immunodeficiency virus, type 1 accessory protein Vpr. hHR23A is a modular protein containing an N-terminal ubiquitin-like (UBL) domain and two ubiquitin-associated domains (UBA1 and UBA2) separated by a xeroderma pigmentosum complementation group C binding (XPCB) domain. All domains are connected by flexible linkers. hHR23A binds ubiquitinated proteins and acts as a shuttling factor to the proteasome. Here, we show that hHR23A utilizes both the UBA2 and XPCB domains to form a stable complex with Vpr, linking Vpr directly to cellular DNA repair pathways and their probable exploitation by the virus. Detailed structural mapping of the Vpr contacts on hHR23A, by NMR, revealed substantial contact surfaces on the UBA2 and XPCB domains. In addition, Vpr binding disrupts an intramolecular UBL-UBA2 interaction. We also show that Lys-48-linked di-ubiquitin, when binding to UBA1, does not release the bound Vpr from the hHR23A-Vpr complex. Instead, a ternary hHR23A·Vpr·di-UbK48 complex is formed, indicating that Vpr does not necessarily abolish hHR23A-mediated shuttling to the proteasome.
Introduction
Although more than 30 years have passed since the discovery of the human immunodeficiency virus that causes AIDS, there is still no cure for this devastating disease. Highly active antiretroviral therapy is an effective treatment but fails to eradicate the virus. Several strategies are employed by HIV4 to avoid or derail otherwise effective defense mechanisms of the host. Of particular note in the race between host and virus are the four small accessory proteins of HIV, Vpr, Vif, Vpu, and Nef, which play important roles in viral escape from the innate immune system (1). Removal of any one of these proteins from the genome of HIV impairs viral infectivity in certain cell types and/or pathogenesis in animal models (2–9). This observation is important because it underlies the need to understand the molecular mechanisms of these proteins in the context of the host.
HIV-1 Vpr is required for the efficient infection of non-dividing cells, including macrophages (10, 11), and numerous cellular processes (12, 13) are influenced by the presence of Vpr. In particular, Vpr activates the DNA damage checkpoint, controlled by ataxia-telangiectasia and RAD3-related kinase, promoting G2/M arrest and inducing the expression of natural killer cell-activating ligands, which, in turn, trigger natural killer cell-mediated lysis of infected cells (14–19). These activities involve Vpr binding to a component of the Cullin4-RING (CRL4) E3 ubiquitin ligase complex, suggesting the Vpr-dependent removal of host proteins that interfere with HIV replication in the infected cells (20–30).
hHR23A and hHR23B, human orthologs of the Saccharomyces cerevisiae yeast DNA repair protein RAD23, act together with the xeroderma pigmentosum complementation group C (XPC) protein in recognizing UV-damaged DNA (31–37). hHR23A was found to interact with Vpr, and the hHR23A-Vpr interaction has been suggested as a mechanism by which HIV might interfere with other cellular regulators by targeting them for proteasomal degradation (12, 38–41). Overexpression of full-length hHR23A or a C-terminal hHR23A segment partially abrogated Vpr-induced G2 cell cycle arrest (38–40), although results using Vpr mutants that were deficient in hHR23A binding did not correlate with the involvement of Vpr in cell cycle arrest (42). These conflicting results render the hHR23A-Vpr connection to the HIV-1 life cycle ambiguous.
hHR23A comprises four domains: a ubiquitin-like (UBL) domain, two ubiquitin-associated domains (UBA1 and UBA2) and an XPC binding domain (XPCB). Although hHR23A has been shown previously to bind Vpr via the UBA2 domain (38–40), little detailed structural information about the binding sites has been reported. Using solution NMR and other biochemical and biophysical techniques, we show that both the UBA2 and XPCB domains of hHR23A are involved in complex formation with Vpr. NMR chemical shift perturbation mapping elucidates details of the Vpr-binding site on hHR23A. In addition, we show that Lys-48-linked di-ubiquitin (di-UbK48) is unable to release the bound Vpr from the hHR23A-Vpr complex but, instead, binds to UBA1, resulting in a ternary hHR23A·Vpr·di-UbK48 complex.
EXPERIMENTAL PROCEDURES
Cloning and Construction of Plasmids
Constructs encoding full-length (residues 1–363) and truncated (residues 223–363) hHR23A with C-terminal His6 tags were cloned into pET21 vectors (EMD Chemicals). Full-length Vpr (residues 1–96), Vpr(1–79), Vpr(6–96), Vpr(11–96), Vpr(11–79), and Vpr(6–79) constructs, encoding an N-terminal NusA fusion protein sequence followed by a tobacco etch virus (TEV) protease cleavage site, were cloned into pET43 vectors (EMD Chemicals). For coexpression of the proteins, full-length hHR23A and Vpr(1–79) or hHR23A(223–363) and Vpr(1–79) were cloned into pET-DUET vectors (EMD Chemicals). Plasmids encoding ubiquitin K48R and Asp-77 (an addition of one extra amino acid at the C terminus) mutants were generated by site-directed mutagenesis (Agilent Technologies) using a pET26b wild-type ubiquitin plasmid as a template.
Protein Expression and Purification
All hHR23A, Vpr, and hHR23A/Vpr coexpressed proteins were produced in Escherichia coli Rosetta 2 (DE3) cells (EMD Chemical) using 0.5 mm isopropyl 1-thio-β-d-galactopyranoside for induction over 16 h at 18 °C. Uniform 2H, 15N, and 13C labeling of the proteins was achieved using modified minimal media containing 2H2O, 15NH4Cl, and 13C6-glucose as deuterium, nitrogen, and carbon sources, whereas unlabeled proteins were prepared using Luria-Bertani medium. Cells were harvested by centrifugation at 6000 × g and resuspended in lysis buffer containing 50 mm sodium phosphate (pH 7.5), 10 mm imidazole, and 200 mm NaCl. The cells were lysed using a microfluidizer (Microfluidics, MA), centrifuged at 40, 000 × g, and then the supernatant was applied to a 5-ml HisTrap (GE Healthcare) His6 tag affinity column equilibrated in the lysis buffer. Bound proteins were eluted using a linear gradient of 0–0.5 M imidazole. Protein-containing fractions were further purified over a HiPrep Superdex 200 (2.6 × 60 cm, GE Healthcare) gel-filtration column equilibrated in 25 mm sodium phosphate buffer (pH 7.5), 150 mm NaCl, 1 mm DTT, 10% glycerol, and 0.02% sodium azide. A synthetic Vpr peptide containing residues 1–52 was obtained from California Peptide Research, Inc. (Napa, CA).
The hHR23A-Vpr complex was prepared by mixing purified hHR23A and NusA-Vpr at a molar ratio of 1:3. The NusA tag was removed by TEV protease cleavage (1:20 (w/w) of a protease-to-protein ratio and incubation for 16 h at 4 °C). Proteins were further purified over a Mono-Q column (GE Healthcare) equilibrated in 50 mm Tris-HCl buffer (pH 7.5), 1 mm DTT, and 5% glycerol using a linear 0–1 m NaCl gradient. Protein fractions were pooled, concentrated, and buffer-exchanged against an NMR buffer (10 mm MES (pH 6.5), 50 mm NaCl, 2 mm DTT, and 5% (v/v) D2O, 0.02% sodium azide) in an Amicon Ultra concentrator (Millipore). The molecular masses of proteins were confirmed by LC-electrospray ionization-TOF mass spectrometry (Bruker Daltonics, Billerica, MA). Final protein concentrations for NMR experiments were 0.1–0.6 mm.
The K48R and Asp-77 ubiquitin mutants were produced by growing E. coli Rosetta 2 (DE3) cells (EMD Chemical) in Luria-Bertani medium and inducing them with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside over 16 h at 18 °C. Cells were harvested by centrifugation at 6000 × g and resuspended in 20 mm Tris-HCl buffer (pH 8.0) containing 150 mm NaCl and 3 mm NaN3 for lysis by sonication. After removing cell debris by centrifugation, 70% perchloric acid was slowly added to the supernatant (0.35 ml of acid per 50 ml of supernatant) with vigorous stirring. The milky suspension was recentrifuged, and the soluble fraction was then dialyzed twice against 4 liters of fresh 50 mm sodium acetate buffer (pH 5.0) overnight. The dialyzed proteins were loaded onto an SP column (GE Healthcare) in 50 mm sodium acetate buffer (pH 5.0) and eluted with a linear gradient of NaCl (20–1000 mm). Further purification was obtained by gel-filtration chromatography on Superdex 75 (GE Healthcare) in 20 mm Tris-HCl buffer, 40 mm NaCl, and 3 mm NaN3 (pH 8.0). Protein-containing fractions were collected and concentrated (∼10 mg/ml) using Centriprep devices (Millipore). Protein concentrations were determined by A280, assuming an absorbance of 0.167 for a 1 mg/ml solution.
Nitroxide Spin Labeling of hHR23A
Labeling of the only cysteine residue in full-length hHR23A with (S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanethiosulfonate (MTSL, Toronto Research Chemicals, Inc.) was carried out as described previously (43). Briefly, the reaction was initiated by adding MTSL from a stock solution (28 mm dissolved in 100% acetonitrile) to the hHR23A protein in 20 mm HEPES buffer and 50 mm NaCl (pH 8.0) to a final concentration of [MTSL]:[hHR23A] = 10:1. The reaction mixture was kept at 4 °C overnight, passed over a PD-10 column, equilibrated with 10 mm MES (pH 6.5), 50 mm NaCl, and 0.02% NaN3 (NMR buffer without DTT) to remove excess, unreacted MTSL. The MTSL-labeled hHR23A protein was further buffer-exchanged against the same buffer and concentrated using a Centriprep device (Millipore).
Synthesis of Lys-48-linked Di-ubiquitin
Lys-48-linked di-ubiquitin (di-UbK48) was synthesized using a protocol described previously (44). Briefly, K48R and Asp-77 ubiquitin mutant protein solutions (4 mg of protein each) were added to 5-fold diluted PBDM8 buffer (250 mm Tris-HCl (pH 8.0), 25 mm MgCl2, 50 mm creatine phosphate, 3 units/ml of inorganic pyrophosphatase, and 3 units/ml of creatine phosphokinase), 2.5 mm ATP, 0.5 mm DTT, and 20 μm E2–25K. The conjugation reaction was initiated by adding ∼0.1 μm mammalian E1 (Boston Biochem, MA) and continued for 4–5 h at 37 °C. The reaction was stopped by 50-fold dilution into 50 mm sodium acetate buffer (pH 5.0) at room temperature, and then di-UbK48 was separated from non-reacted ubiquitin mutant monomers by SP cation exchange chromatography (GE Healthcare) in 50 mm sodium acetate buffer (pH 5.0) using a linear gradient of NaCl (20–1000 mm). The fractions containing di-UbK48 were collected, concentrated, and then the buffer was exchanged against NMR buffer using Centriprep devices (Millipore). The final di-UbK48 concentration was determined, assuming an A280 of 0.174 for a 1 mg/ml solution.
Analytical Gel Filtration Column Chromatography
Typically, hHR23A proteins (90 μm) or their mixture with NusA-Vpr (23 μm) in 25 mm sodium phosphate (pH 7.5), 150 mm NaCl, 5% glycerol, 1 mm DTT, and 0.02% sodium azide were applied to a 24-ml analytical Superdex200 column (10 × 300 mm, GE Healthcare) at a flow rate of 0.8 ml/min and equilibrated with the same buffer but lacking the reducing agent. The peak fractions were collected (0.5 ml), concentrated, and analyzed by SDS-PAGE with Coomassie Blue staining.
Multiangle Light Scattering
Size exclusion chromatography/multiangle light scattering (SEC-MALS) data were obtained at room temperature using an analytical Superdex200 column (10 × 300 mm, GE Healthcare) with in-line multiangle light scattering (HELEOS, Wyatt Technology), variable wavelength UV (Agilent 1100, Agilent Technologies), and refractive index (Optilab rEX, Wyatt Technology) detectors. About 100 μl of a protein solution, at ∼2 mg/ml, was loaded onto the column equilibrated with a buffer containing 25 mm sodium phosphate (pH 7.5), 150 mm NaCl, and 0.02% sodium azide at a flow rate of 0.5 ml/min. The molecular masses of eluted protein species were determined using the ASTRA V.5.3.4 program (Wyatt Technologies).
NMR Spectroscopy
NMR data were collected on Bruker 600-, 700-, 800-, and 900-MHz Avance spectrometers. All spectrometers were equipped with z axis gradient, triple-resonance cryoprobes. Experiments were performed at 298 K or 291 K. Heteronuclear two-dimensional 1H-15N HSQC and three-dimensional HNCACB, CBCA(CO)NH, HNCA, HN(CO)CA, HNCO, and HN(CA)CO experiments were performed for the backbone assignment of full-length 13C,15N-hHR23A. Two-dimensional 1H-15N HSQC and three-dimensional HNCACB, CBCA(CO)NH, HNCA, and 13C/15N-edited NOESY data were collected for the backbone assignment of 13C,15N-hHR23A(223–363). HNCA data were collected with a non-linear sampling scheme (45) to achieve higher resolution. Two-dimensional 1H-15N HSQC and three-dimensional HNCA and 13C/15N-edited NOESY spectra (mixing time 120 ms) were collected for the assignment of the coexpressed 13C,15N-hHR23A(223–363)·Vpr(1–79) complex. All data were processed with NMRPipe (46) and analyzed with CARA (47), SPARKY3 (T. D. Goddard and D. G. Kneller), or CCPN (48). Non-linearly sampled data were processed with both NMRPipe (46) and RNMRTK (49).
RESULTS
Vpr Forms a Stable Complex with Full-length hHR23A
No structural or biochemical studies of isolated Vpr under physiological conditions (pH and buffer) have been reported, suggesting that the protein may not be soluble or stable under such conditions. Indeed, numerous initial efforts in our laboratory to express and produce Vpr (containing various regions, namely residues 1–79, 6–79, 11–79, 6–96, 11–96, and 1–96 (full-length)), using a variety of expression systems, failed to produce sufficient amounts of the protein. Therefore, we expressed Vpr as a fusion protein with a large soluble tag (NusA, 61 kDa) at its N terminus. Inclusion of the NusA tag greatly improved the solubility of all Vpr variants (to ∼5 mg/ml in phosphate buffer), with one exception. Full-length Vpr(1–96) remained poorly soluble despite the presence of the NusA tag. To identify the region of Vpr that is crucial for hHR23A binding, several NusA-Vpr constructs were tested for interaction using analytical gel-filtration column chromatography. Vpr variants that lacked several N-terminal residues, (Vpr(6–79), (11–79), (6–96), or (11–96)) did not exhibit binding (data not shown). In contrast, NusA-Vpr(1–79) strongly interacted with hHR23A, as evidenced by coelution of both proteins in a single peak on the gel-filtration column at an elution volume (Fig. 1D, 12 ml, peak 5) lower than that of either hHR23A alone (A, 13.4 ml, peak 1) or NusA-Vpr(1–79) alone (C, 12.6 ml, peak 3). The presence of both proteins in the coeluted peak was confirmed by SDS-PAGE analysis (Fig. 1F, lane 5). These data clearly show that the N-terminal (residues 1–5), but not the C-terminal (residues 80–96), amino acids are required for hHR23A binding. A shorter, synthetic Vpr peptide containing residues 1–52, however, did not interact with hHR23A when tested by NMR chemical shift perturbation studies (data not shown), indicating that Vpr residues 1–79 most likely constitute the minimum region required for hHR23A binding. Here, we refer to Vpr(1–79) simply as Vpr.
FIGURE 1.
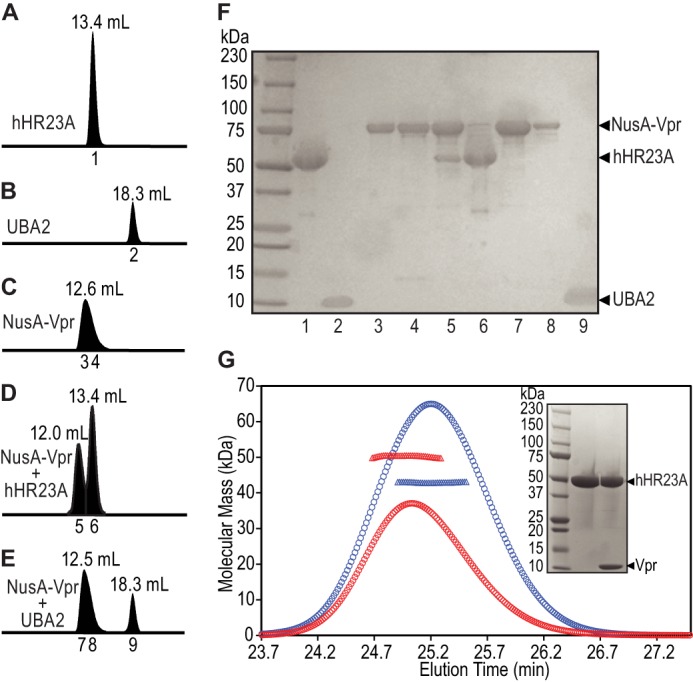
The UBA2 domain of hHR23A is not sufficient for stable Vpr binding. Analytical gel-filtration profiles of hHR23A (A), UBA2 (hHR23A residues 311–363, B), NusA-Vpr (C), NusA-Vpr mixed with hHR23A (D), and NusA-Vpr mixed with UBA2 (E). Elution volumes are indicated above and fraction numbers below the peaks. F, SDS-PAGE analysis of the fractions indicated in A–E, with lane numbers indicating the fraction numbers. G, SEC-MALS analysis of purified hHR23A (blue) and the hHR23A-Vpr complex (after cleavage of the NusA tag, red). Elution profiles are shown by ○, and the weight-averaged molecular masses obtained from the SEC-MALS measurements are shown by ▵ across the peaks. Inset, SDS-PAGE of free (center lane) and the Vpr-complexed (right lane) hHR23A NMR samples used to obtain the 1H-15N HSQC spectra shown in Fig. 2A.
For NMR studies, the large NusA tag in the NusA-Vpr·hHR23A complex was removed by TEV protease digestion (see “Experimental Procedures”). SDS-PAGE analysis after digestion shows that both components, Vpr and hHR23A, are present in the NMR sample (Fig. 1G, inset, right lane). The NMR sample containing purified Vpr-hHR23A complex (comprising 13C,15N-labeled hHR23A and unlabeled Vpr) was also assessed by SEC-MALS (Fig. 1G), revealing a weight-averaged molecular mass of 50.3 kDa (red ▵). This mass (50.3 kDa) is larger than the one seen for free hHR23A (42.9 kDa, Fig. 1G, blue ▵) but slightly smaller than the calculated, expected mass (52.7 kDa) for the binary complex. This suggests that the complex sample contained excess hHR23A, most likely because of incomplete separation on the final Mono-Q column. Indeed, the 1H-15N HSQC spectrum of the complex sample confirms this because resonances from both the Vpr-bound and free hHR23A species are observed (Fig. 2A, red spectrum).
FIGURE 2.

Analysis of resonance intensities permits the identification of Vpr-binding regions in hHR23A. A, superposition of the 800-MHz 1H-15N HSQC spectra of hHR23A alone (blue) and the hHR23A-Vpr complex (red) at 298 K. Selected hHR23A resonances are labeled with residue names and numbers. Vpr-bound hHR23A resonances are encircled. B, ratio of 1H-15N HSQC resonance intensities (Ifree/Icomplex) for free hHR23A resonances in the sample of free hHR23A (Ifree) and the hHR23A-Vpr complex (Icomplex) sample. The data are color-coded according to domains in hHR23A: UBL (orange); LNK1, LNK2, and LNK3 (gray); UBA1 (light blue); XPCB (blue); and UBA2 (green). Ifree/Icomplex values for prolines and unassigned residues were arbitrarily set to 1 and colored according to domains using the same color scheme but in a lighter shade.
The UBA2 Domain of hHR23A Alone Is Insufficient to Form a Stable Complex with Vpr
UBA2 of hHR23A has been identified previously as the Vpr-binding domain on the basis of yeast two-hybrid and pull-down assays (38, 39, 50). However, a direct biophysical measure of the UBA2-Vpr complex, using purified components, has not yet been reported. We initially attempted to prepare the UBA2-Vpr complex by mixing individually purified UBA2 (residues 311–363) and NusA-Vpr. However, analytical gel-filtration chromatography of the mixture revealed an elution profile (Fig. 1E) very similar to the sum of the profiles obtained with the individual components (B and C). This is clearly borne out by SDS-PAGE analysis (Fig. 1F) of fractions 7 and 8 of the mixed sample (E), confirming that NusA-Vpr did not coelute with UBA2. These data demonstrate that the interaction between UBA2 and Vpr is weak and that the complex separates during gel-filtration column chromatography. Thus, additional regions of hHR23A may be required for stable complex formation.
We proceeded to identify all Vpr interaction sites by comparing 1H-15N HSQC spectra of full-length hHR23A alone (Fig. 2A, blue spectrum) and the hHR23A-Vpr complex (red spectrum) using samples in which only hHR23A was uniformly isotope-labeled with 13C and 15N. Backbone resonance assignments for the free hHR23A protein, from multidimensional heteronuclear NMR experiments, yielded >90% assignments for the four structured domains of hHR23A and ∼60% for the linker regions (severe resonance overlap impaired complete assignment of the linker residues). The hHR23A-Vpr complex sample exhibited many new resonances arising from the Vpr-complexed hHR23A protein. Free hHR23A resonances were also present in the complexed sample because of excess hHR23A protein in the sample (Fig. 2A, red spectrum). Resonances of the free and complexed species are in slow exchange on the chemical shift scale, indicating a strong interaction between Vpr and hHR23A, in agreement with the data from gel-filtration and SEC-MALS experiments (Fig. 1). Preliminary binding site mapping was performed by measuring the free resonance intensities in the hHR23A-Vpr complex sample (Icomplex) relative to those of the hHR23A-alone sample (Ifree) (Fig. 2B). On the basis of SDS-PAGE (Fig. 1G, inset), we estimated that the complex sample contained slightly less hHR23A (90%) than the free sample, permitting normalization of the total hHR23A concentration across samples. Thus, resonances associated with non-binding residues are expected to yield ∼1 for Ifree/Icomplex (after applying the normalization factor of 0.9). In contrast, resonances of residues that are involved in Vpr binding will exhibit Ifree/Icomplex > 1 because the chemical shifts for the bound resonances will be different, reducing the free resonance intensities in the complex sample (Icomplex).
As clearly seen from the data presented in Fig. 2B, the largest attenuation of signal intensities is observed for resonances of the UBA2 and XPCB domains, strongly suggesting involvement of these domains in Vpr binding. Tentative assignments of bound resonances, by simply assuming that the free and bound cross-peaks are located close to each other in the spectrum (e.g. Gly-331, Ser-335, Leu-336, Lys-346, and Asn-347 in UBA2 and Glu-233, Asp-237, Asn-266, and Glu286 in XPCB), were later confirmed using heteronuclear three-dimensional experiments (see below). Therefore, both the UBA2 and XPCB domains in full-length hHR23A are required for tight Vpr binding, in accord with the gel-filtration chromatography data (Fig. 1), which showed that the isolated UBA2 domain (residues 311–363) did not coelute with Vpr. Note that a few isolated resonances from the UBL domain and the linker between XPCB and UBA2 domains (LNK3) also exhibited some reduction in intensity because of a Vpr-binding induced conformational change in hHR23A rather than a direct interaction with Vpr (see below).
Direct confirmation that both XPCB and UBA2 participate in the Vpr interaction was obtained from a competition experiment in which truncated protein, comprising only the XPCB, LNK3, and UBA2 domains (residues 223–363), was added to the full-length hHR23A-Vpr complex sample. Upon addition of excess unlabeled hHR23A(223–363) to the 13C/15N-labeled hHR23A(1–363)-Vpr sample, the bound resonances of hHR23A disappeared, whereas the free resonances were unaffected (data not shown), confirming that unlabeled hHR23A(223–363) can bind Vpr, replacing full-length hHR23A in the complex.
Interestingly, although most UBL resonances exhibited little change in their peak intensities, the 1H-15N HSQC resonances of Leu-10 and Gly-52 and their immediate neighbors were attenuated upon Vpr binding (Fig. 2). Previous NMR chemical shift perturbation studies on isolated UBL and UBA2 domains of hHR23A indicated that both Leu-10 and Gly-52 in UBL interact with UBA2 (51). Therefore, we believe that the intensity change in these UBL residues is caused by the loss of the intramolecular interaction, albeit weak, between UBL and UBA2, when intermolecular binding of UBA2 to Vpr occurs.
Interaction between Vpr and XPCB-LNK3-UBA2
Because only the C-terminal region of hHR23A, containing the XPCB and UBA2 domains, was identified as important for Vpr binding, N-terminal deleted hHR23A protein, hHR23A(223–363), was coexpressed with Vpr for detailed binding site mapping. Coexpression was advantageous because significantly improved Vpr yields were observed (1 liter of modified minimal medium was sufficient for preparing an NMR sample with an acceptable concentration (>0.1 mm)), although the expression level of Vpr was consistently lower than that of hHR23A(223–363). The hHR23A(223–363)-Vpr complex was purified using the same protocol used to purify the full-length hHR23A-Vpr complex, indicating that the affinity of hHR23A(223–363) for Vpr is similar to that of full-length hHR23A. The superimposed 1H-15N HSQC spectra of the free and Vpr-complexed hHR23A(223–363) are shown in Fig. 3A. The overall line widths of the Vpr-complexed hHR23A(223–363) resonances are broader than the free ones, reflecting the total mass increase upon complexation. Almost complete backbone assignments for the hHR23A(223–363)-Vpr complex were obtained using heteronuclear, multidimensional NMR experiments, aided by the assignments for free hHR23A(223–363) that were also completed. Combined 1H-15N chemical shift differences between the free and Vpr-complexed hHR23A(223–363) are plotted for each residue in Fig. 3B. Both XPCB and UBA2 exhibit significant resonance frequency perturbations. Frequency shift changes of the resonances associated with the UBA2 domain are generally larger (average value of 0.09 ppm) than those belonging to the XPCB domain (average value = 0.04 ppm), whereas the linker (LNK3) residues exhibit only small changes (average value = 0.02 ppm).
FIGURE 3.

NMR chemical shift changes of hHR23A(223–363) amide resonances induced by Vpr binding. A, superposition of the 900-MHz 1H-15N HSQC spectra of hHR23A(223–363) alone (blue) and the hHR23A(223–363)-Vpr complex (red) at 291 K. Representative hHR23A(223–363) resonances are labeled with residue names and numbers. Note that the hHR23A(223–363)-Vpr complex spectrum (red) also contains Vpr resonances (enclosed by rectangles) because the proteins were coexpressed and, therefore, both labeled with 15N and 13C. B, 1H,15N-combined chemical shift differences, plotted along the linear amino acid sequence, calculated using the equation (ΔδHN2 + (0.14*ΔδN)2)1/2, with ΔδHN and ΔδN representing the 1HN and 15N chemical shift differences between resonances of the free and the Vpr-bound hHR23A(223–363).
Structural mapping of residues affected by Vpr binding onto the NMR structure of the hHR23A XPCB domain (52) clearly positions the binding site predominantly to one face of the protein (Fig. 4A). The interaction surface forms a saddle-like groove delineated by Gln-249 (α2), Leu-255, Ala-257 (the loop between α2 and α3), Leu-259, Gln-260 (α3), Asn-266, Gln-268 (the loop between α3 and α4), Ser-274 (α4), Gln-277, Glu-278, Ile-281, Met-283, Leu-284, Asn-285, and Glu-286 (α5). Asn-266 and Gln-268 are somewhat removed from the main binding site, suggesting that a conformational change of the α3-α4 loop likely accompanies Vpr binding. The binding groove is centrally lined by hydrophobic residues, with hydrophilic residues at its periphery. A very similar binding groove has been identified on the XPCB domain of RAD23, the yeast orthologue of hHR23A, when it interacts with binding partners. For example, in the structure of the RAD23-RAD4 complex (PDB code 2QSF) (53), RAD23-XPCB interacts with a helix from RAD4 through this groove (Fig. 4B). A similar interaction is also exhibited in the RAD23·peptide·N-glycanase (PNGase) complex structure (PDB code 1X3Z) (54) (Fig. 4C). Given that RAD23-XPCB binds a helix, it is likely that hHR23A-XPCB also binds to a helical segment of Vpr. Indeed, Vpr has been shown to exhibit a predominantly helical structure under acidic conditions or in organic solvents (55).
FIGURE 4.

Structural mapping of Vpr binding sites on the XPCB and UBA2 domains. Vpr binding sites are mapped onto the NMR structures of the hHR23A XPCB domain (PDB code 1TP4) (52) (A) and the hHR23A UBA2 domain (PDB code 1QZE) (51) (D). Residues that exhibit chemical shift changes of ≥0.10 ppm and between 0.05 and 0.10 ppm are colored red and orange, respectively. They are labeled with their residue names and numbers. The RAD23 XPCB domain interactions with RAD4 and PNGase, as observed in the x-ray structures of the yeast RAD23-RAD4 complex (PDB code 2QSF) (53) (B) and the RAD23-PNGase complex (PDB code 1X3W) (C), respectively. Only the XPCB domain of RAD23 (blue) and the N-terminal helices (magenta) of RAD4 (residues 141–161, B) or of PNGase (residues 10–30, C) are shown. All XPCB domains are depicted in the same orientation. E, di-UbK48 binding site mapping onto the NMR structure of the UBA2 domain of hHR23A (PDB code 1QZE) (51) with UBA2 residues that exhibit large chemical shift changes (≥0.10 ppm), colored in magenta (from Fig. 2B in Ref. 56). CSP, chemical shift perturbations; rot, rotation.
Structural mapping of the UBA2 resonances that undergo large chemical shift changes upon Vpr binding (Fig. 3) onto the NMR structure of UBA2 (51) is provided in Fig. 4D. Fourteen resonances of UBA2 exhibit combined 1H-15N chemical shift changes of ≥0.1 ppm (Fig. 3B), affecting residues on both the front and back faces (Fig. 4D). Pro-333 has been identified previously to play a critical role in Vpr binding (50), consistent with the binding site identified here. Indeed, Pro-333 sits between the strongly affected residues Phe-332 and Leu-334. The Vpr binding site on UBA2 (Fig. 4D) identified here also overlaps with the one reported for di-UbK48 (Fig. 4E) (56) but not with that for mono-ubiquitin (Ub) (56, 57), which is predominantly located on one face. In this regard, it is worth pointing out that Vpr binding of hHR23A is likely tighter than that of Ub or di-UbK48. Resonances for free and Vpr-bound hHR23A exhibit slow exchange on the chemical shift scale (Figs. 2A and 3A), whereas binding between an isolated UBA2 domain and Ub exhibits fast exchange (Kd of ∼330 μm) (57). Binding between the UBA2 domain and di-UbK48 exhibits both slow and fast-intermediate exchange (Kd of ∼8–140 μm) (56). The tighter Vpr-hHR23A interaction, compared with complexes formed by individual domains, reflects the fact that both domains, UBA2 and XPCB, interact with Vpr.
Interaction between Vpr and Full-length hHR23A
Backbone 1H-15N resonance assignments for most (>80%) of the residues of all four structured domains and the majority (>50%) of the linker residues of the full-length hHR23A-Vpr complex were obtained by comparing its 1H-15N HSQC spectrum to that of free hHR23A, and using the assignments of the hHR23A(223–363)-Vpr complex. The differences in chemical shifts permitted detailed mapping of the Vpr binding site (Fig. 5A). XPCB and UBA2 resonances in full-length hHR23A exhibit very similar chemical shift perturbations as seen for the C-terminal construct hHR23A(223–363), and resonances of residues located in the UBA1 domain and the linkers did not exhibit any significant changes. Gratifyingly, the chemical shift perturbation data on full-length hHR23A (Fig. 5A) agree extremely well with our preliminary binding site mapping on the basis of the analysis of hHR23A 1H-15N HSQC resonance intensities upon Vpr binding (Fig. 2B). Interestingly, of the two UBA domains in hHR23A, only UBA2 is directly contacted by Vpr, not UBA1.
FIGURE 5.

Chemical shift perturbation mapping of Vpr binding to full-length hHR23A and PRE data on free full-length hHR23A. A, 1H,15N-combined chemical shift changes (calculated using the formula given in the legend for Fig. 3B) in the 1H-15N HSQC spectrum of hHR23A upon Vpr binding. Inset, schematic of intramolecular and intermolecular interactions in hHR23A upon Vpr binding. B, PRE effects on individual resonances were estimated by calculating the resonance intensity ratio from 1H-15N HSQC spectra of hHR23A with (Ipara) and without (Idia) MTSL labeling at Cys-344. Sample conditions were identical for both. Chemical shift changes (A) and PRE effects (B) are color-coded according to domain identity: UBL (orange); LNK1, LNK2, and LNK3 (gray); UBA1 (light blue); XPCB (blue); and UBA2 (green). PRE values for prolines and unassigned residues were arbitrarily set to 1 and colored according to domains using a lighter color scheme.
Significant chemical shift perturbations are also noted for resonances of a few residues that reside in the UBL domain (Fig. 5A), in particular for residues in the vicinity of Leu-10 and Gly-52. Again, this finding agrees with the observed resonance intensity changes in the 1H-15N HSQC spectrum of hHR23A upon Vpr binding (Fig. 2B). These Vpr-induced changes seen for UBL domain resonances are caused by the loss of an intramolecular interaction between UBL and UBA2 upon intermolecular complex formation with Vpr, as discussed above. The intramolecular interaction of UBL and UBA2 in full-length hHR23A is confirmed by paramagnetic relaxation enhancement (PRE) data (Fig. 5B). Upon labeling the only cysteine residue in full-length hHR23A (position 344, in the middle of the UBA2 domain) with MTSL, the most extensive broadening of resonances outside of the UBA2 domain occurs in UBL-associated resonances. In particular, the Leu-10, Gln-11, Gln-12, Ala-51, Gly-52 and Val-73 amide resonances are broadened beyond detection. Therefore, chemical shift perturbation (Fig. 5A), signal attenuation (Fig. 2B), and paramagnetic broadening (Fig. 5B) data unequivocally establish that the UBL domain interacts with the UBA2 domain in the full-length hHR23A molecule and that this interaction is lost upon Vpr binding. Similarly, the interaction between UBA2 (as well as UBA1) and UBL domains in full-length hHR23A is no longer present when hHR23A binds to S5a, a proteasome subunit fragment (51). In summary, our combined NMR data clearly show that hHR23A interacts with Vpr via the XPCB and UBA2 domains, releasing the UBA2 and UBL intramolecular interaction, as depicted schematically in Fig. 5A, inset.
Lys-48-linked Di-ubiquitin Is a Poor Competitor of Vpr for hHR23A Binding
Because our data show that the Vpr binding site on UBA2 of hHR23A overlaps with the di-UbK48 binding site (56) (Fig. 4, D versus E), we tested whether di-UbK48 is able to displace Vpr from the hHR23A-Vpr complex. Fig. 6 displays representative regions of the 1H-15N HSQC spectrum of the full-length hHR23A-Vpr complex sample upon adding di-UbK48. Fig. 6, C, G, and K, illustrates resonance changes after addition of equimolar amounts of di-UbK48 and D, H, and L after the addition of a 2-fold molar excess of di-UbK48. Identical regions of the spectra in the absence (Fig. 6, A, E, and I) or presence of Vpr (B, F, and J) are also shown for comparison. A schematic of the associated interactions between the proteins is provided in Fig. 6, M–S.
FIGURE 6.

Vpr and di-UbK48 binding to hHR23A. Selected 1H-15N HSQC resonances, representative of the UBL, UBA1, XPCB, and UBA2 domains in free hHR23A (A, E, and I), hHR23A-Vpr (B, F, and J) and hHR23A-Vpr complex samples with equimolar (C, G, and K) or 2-fold excess (D, H, and L) di-UbK48, were monitored and connected by solid (chemical shift change) or dashed (no chemical shift change) lines. Red and orange are used for Vpr- and di-UbK48-induced changes, respectively. Loss of resonances because of extensive broadening or other causes are indicated by asterisks. M–S, schematics summarizing the intramolecular and intermolecular interactions.
Upon di-UbK48 addition to the hHR23A-Vpr complex sample, UBA1 domain resonances are affected, either changing resonance frequency (e.g. Asn-189, Fig. 6F compared with G) or experiencing severe broadening (e.g. Gly-174, B compared with C and D). In contrast, resonances of Vpr-bound UBA2 residues are not perturbed by the addition of di-UbK48 (Vpr-bound Gly-331 resonance in Fig. 6B versus C and D, Ser-336 resonance in F versus G and H, and Leu-335 resonance in J versus K and L). These data clearly demonstrates that di-UbK48 can successfully compete for the UBL-binding site on the UBA1 domain but not for that on UBA2 that contacts Vpr (Fig. 6, M–P). Results obtained by the above titration experiments suggest the relative binding strength between different partners. Vpr and hHR23A (via UBA2 and XPCB) interact tightly, di-UbK48 and hHR23A (via UBA1 or UBA2) less tightly, and UBA1 (or UBA2) and UBL in the same hHR23A molecule is the weakest interaction. Interestingly, the data also revealed that a di-UbK48·hHR23A·Vpr ternary complex can be formed, with di-UbK48 binding to the UBA1 domain in the Vpr-hHR23A complex (Fig. 6, O and P).
Spectral changes in the free, full-length hHR23A protein that is present (albeit in small amounts, Fig. 2A) in the full-length hHR23A-Vpr complex sample were also monitored. As illustrated for the Vpr-free G331 resonance, severe line broadening (Fig. 6B versus C and D) or frequency shifts, in the case of the Vpr-free Leu-335 and Ser-336 resonances, are noted (J versus K and L and F versus G and H, respectively), indicating that the UBA2 domain in the free hHR23A is also capable of binding to di-UbK48.
The Gly-52 resonances of both free and Vpr-bound hHR23A also exhibited small shifts upon di-UbK48 binding (Fig. 6B versus C and D). However, these changes are most likely not caused by a direct UBL and di-UbK48 contact but result from the loss of the intramolecular contact between UBL and the UBA1 or UBA2 domains upon di-UbK48 binding to these two UBA domains, as illustrated in Fig. 6N versus O and P for the Vpr-bound hHR23A and Q versus R and S for the free hHR23A. For resonances of XPCB domain residues in hHR23A, no frequency shifts are observed upon di-UbK48 binding (Gly-263 resonance in Fig. 6B compared with C and D), in agreement with its known lack of ubiquitin binding (51).
DISCUSSION
Despite the importance of Vpr in the life cycle of HIV-1, few biochemical/biophysical studies of purified Vpr protein under physiological conditions have appeared in the literature. On the basis of our experience, this is most likely related to the challenging properties of the protein, such as low expression levels in E. coli and poor solubility under most, except extreme, conditions. Using a solubility enhancing NusA-fusion protein and coexpression with a cellular partner permitted these studies, in particular the preparation of Vpr samples at concentrations amenable for NMR and other biochemical studies. Prior to this work, the only structural NMR studies of Vpr were carried out at very low pH (pH 3.5 or 2.6) and/or in the presence of acetonitrile (55). Most proteins are adversely affected by such conditions and are unstable. For example, in the case of hHR23A, investigated here, all domains of the protein, apart from the UBL domain, unfold at pH values below 4.5 (data not shown).
The use of the above described strategy allowed us to carry out a thorough biochemical/biophysical characterization of the interaction between Vpr and hHR23A. Analytical gel filtration chromatography, SEC-MALS, and NMR results led to the identification of the Vpr binding site(s) on hHR23A and permitted the dissection of the interactions at the domain and residue levels. In contrast to yeast-two-hybrid data, which identified the UBA2 domain as the only domain of hHR23A responsible for Vpr binding (38, 39), our studies revealed that stable complex formation between Vpr and hHR23A requires two domains, UBA2 and XPCB.
On the basis of the close structural similarity between the Vpr binding site on the XPCB domain of hHR23A, identified here by NMR (Fig. 4A), and that of RAD4 (B) or PNGase (C) on the XPCB domain of RAD23, it is most likely that Vpr interacts with hHR23A in a closely related manner. Both RAD4 and PNGase bind RAD23 as a helix (53, 54). Amino acid sequence alignment by LALIGN (58) shows that residues 64–73 (LQQLLFIHFR) of Vpr, the central part of helix 3 in the low-pH NMR structure (55), are very similar to residues 15–24 (IAKMLLIKYK) of PNGase, the central part of the interacting helix. This suggests that this region of Vpr may be the binding epitope for contacting the XPCB domain of hHR23A.
Although our NMR binding site mapping identifies that similar UBA2 residues are used in the interaction with both Vpr (Fig. 4D) and di-UbK48 (E), it is difficult to predict which region of Vpr is involved in engaging the UBA2 domain of hHR23A. Di-UbK48 binds to UBA2 via its β sheet. However, no β structure has been identified in the Vpr NMR structure (55). Because both faces of the UBA2 domain constitute the large, extended binding surface for Vpr, it is very likely that a sizable portion of Vpr is involved in contacting this domain.
Severe line broadening of the Vpr resonances in the three-dimensional NMR data of the complex precluded assignment of the Vpr resonances and structural characterization of Vpr in the hHR23A(223–363) binding site(s), although a number of Vpr resonances are clearly present in the two-dimensional 1H-15N HSQC spectrum (Fig. 3A). We are currently improving the NMR data quality, using a triple 2H,13C,15N-labeled complex sample for further work.
Involvement of two different domains of hHR23A, XPCB, and UBA2, in Vpr binding suggests that diverse cellular effects can ensue. For example, Vpr binding to the XPCB domain of hHR23A may interfere with the interaction between hHR23A and XPC, affecting nucleotide excision repair. hHR23A/B are known to play a role in stabilizing XPC, preventing its rapid degradation (59, 60). In this manner, Vpr could down-regulate nucleotide excision repair by liberating XPC from the complex with hHR23A. On the other hand, given the ability of the UBA2 (and UBA1) domain to interact with poly-ubiquitinated proteins, complex formation between Vpr and the UBA2 and XPCB domains of hHR23A may modulate the role of hHR23A as a shuttle factor (61–64). Our data showing that hHR23A, Vpr, and di-UbK48 can form a ternary complex (Fig. 6, O and P) suggests that Vpr-complexed hHR23A may still shuttle ubiquitinated substrates to proteasomes. Because HIV exploits the cellular proteasome machinery to degrade host restriction factors to escape cellular defense systems, such an activity of Vpr may allow the virus to win the tug-of-war against its host.
Acknowledgments
We thank Dr. Teresa Brosenitsch for critical reading of the manuscript, Phil Greer and Doug Bevan for computer technical support, and Michael J. Delk for NMR instrumental support.
This work was supported, in whole or in part, by National Institutes of Health Grant P50GM82251 (to A. M. G.). This work was also supported by a University of Pittsburgh Center For AIDS Research Pilot grant (to J. A.).
- HIV
- human immunodeficiency virus
- HIV-1
- human immunodeficiency virus, type 1
- XPC
- xeroderma pigmentosum complementation group C
- UBL
- ubiquitin-like
- UBA
- ubiquitin-associated
- XPCB
- xeroderma pigmentosum complementation group C binding domain
- TEV
- tobacco etch virus
- MTSL
- (S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanethiosulfonate
- SEC-MALS
- size exclusion chromatography with in-line multiangle light scattering
- HSQC
- heteronuclear single quantum coherence
- PNGase
- peptide:N-glycanase
- Ub
- ubiquitin
- PRE
- paramagnetic relaxation enhancement
- di-UbK48
- Lys-48-linked di-ubiquitin.
REFERENCES
- 1. Malim M. H., Emerman M. (2008) HIV-1 accessory proteins. Ensuring viral survival in a hostile environment. Cell Host Microbe 3, 388–398 [DOI] [PubMed] [Google Scholar]
- 2. Gibbs J. S., Lackner A. A., Lang S. M., Simon M. A., Sehgal P. K., Daniel M. D., Desrosiers R. C. (1995) Progression to AIDS in the absence of a gene for Vpr or Vpx. J. Virol. 69, 2378–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lang S. M., Weeger M., Stahl-Hennig C., Coulibaly C., Hunsmann G., Müller J., Müller-Hermelink H., Fuchs D., Wachter H., Daniel M. M. (1993) Importance of Vpr for infection of rhesus monkeys with simian immunodeficiency virus. J. Virol. 67, 902–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desrosiers R. C., Lifson J. D., Gibbs J. S., Czajak S. C., Howe A. Y., Arthur L. O., Johnson R. P. (1998) Identification of highly attenuated mutants of simian immunodeficiency virus. J. Virol. 72, 1431–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simon J. H., Miller D. L., Fouchier R. A., Soares M. A., Peden K. W., Malim M. H. (1998) The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted. A role for Vif in determining virus host range and cross-species transmission. EMBO J. 17, 1259–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Varthakavi V., Smith R. M., Bour S. P., Strebel K., Spearman P. (2003) Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc. Natl. Acad. Sci. U.S.A. 100, 15154–15159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kestler H. W., 3rd, Ringler D. J., Mori K., Panicali D. L., Sehgal P. K., Daniel M. D., Desrosiers R. C. (1991) Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell 65, 651–662 [DOI] [PubMed] [Google Scholar]
- 8. Neil S., Bieniasz P. (2009) Human immunodeficiency virus, restriction factors, and interferon. J. Interferon Cytokine Res. 29, 569–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kirchhoff F. (2010) Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8, 55–67 [DOI] [PubMed] [Google Scholar]
- 10. Connor R. I., Chen B. K., Choe S., Landau N. R. (1995) Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206, 935–944 [DOI] [PubMed] [Google Scholar]
- 11. Balliet J. W., Kolson D. L., Eiger G., Kim F. M., McGann K. A., Srinivasan A., Collman R. (1994) Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef. Mutational analysis of a primary HIV-1 isolate. Virology 200, 623–631 [DOI] [PubMed] [Google Scholar]
- 12. Zhao R. Y., Li G., Bukrinsky M. I. (2011) Vpr-host interactions during HIV-1 viral life cycle. J. Neuroimmune Pharmacol. 6, 216–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li G., Bukrinsky M., Zhao R. Y. (2009) HIV-1 viral protein R (Vpr) and its interactions with host cell. Curr. HIV Res. 7, 178–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Di Marzio P., Choe S., Ebright M., Knoblauch R., Landau N. R. (1995) Mutational analysis of cell cycle arrest, nuclear localization and virion packaging of human immunodeficiency virus type 1 Vpr. J. Virol. 69, 7909–7916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. He J., Choe S., Walker R., Di Marzio P., Morgan D. O., Landau N. R. (1995) Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 69, 6705–6711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jowett J. B., Planelles V., Poon B., Shah N. P., Chen M. L., Chen I. S. (1995) The human immunodeficiency virus type 1 vpr gene arrests infected T cells in the G2 + M phase of the cell cycle. J. Virol. 69, 6304–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Planelles V., Jowett J. B., Li Q. X., Xie Y., Hahn B., Chen I. S. (1996) Vpr-induced cell cycle arrest is conserved among primate lentiviruses. J. Virol. 70, 2516–2524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Re F., Braaten D., Franke E. K., Luban J. (1995) Human immunodeficiency virus type 1 Vpr arrests the cell cycle in G2 by inhibiting the activation of p34cdc2-cyclin B. J. Virol. 69, 6859–6864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ward J., Davis Z., DeHart J., Zimmerman E., Bosque A., Brunetta E., Mavilio D., Planelles V., Barker E. (2009) HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 5, e1000613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeHart J. L., Zimmerman E. S., Ardon O., Monteiro-Filho C. M., Argañaraz E. R., Planelles V. (2007) HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol. J. 4, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan L., Ehrlich E., Yu X. F. (2007) DDB1 and Cul4A are required for human immunodeficiency virus type 1 Vpr-induced G2 arrest. J. Virol. 81, 10822–10830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wen X., Duus K. M., Friedrich T. D., de Noronha C. M. (2007) The HIV1 protein Vpr acts to promote G2 cell cycle arrest by engaging a DDB1 and Cullin4A-containing ubiquitin ligase complex using VprBP/DCAF1 as an adaptor. J. Biol. Chem. 282, 27046–27057 [DOI] [PubMed] [Google Scholar]
- 23. Belzile J. P., Duisit G., Rougeau N., Mercier J., Finzi A., Cohen E. A. (2007) HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4AVPRBP E3 ubiquitin ligase. PLoS Pathog. 3, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hrecka K., Gierszewska M., Srivastava S., Kozaczkiewicz L., Swanson S. K., Florens L., Washburn M. P., Skowronski J. (2007) Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. U.S.A. 104, 11778–11783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Le Rouzic E., Morel M., Ayinde D., Belaïdouni N., Letienne J., Transy C., Margottin-Goguet F. (2008) Assembly with the Cul4A-DDB1DCAF1 ubiquitin ligase protects HIV-1 Vpr from proteasomal degradation. J. Biol. Chem. 283, 21686–21692 [DOI] [PubMed] [Google Scholar]
- 26. Transy C., Margottin-Goguet F. (2009) HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 8, 2489–2490 [DOI] [PubMed] [Google Scholar]
- 27. Belzile J. P., Richard J., Rougeau N., Xiao Y., Cohen E. A. (2010) HIV-1 Vpr induces the K48-linked polyubiquitination and proteasomal degradation of target cellular proteins to activate ATR and promote G2 arrest. J. Virol. 84, 3320–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Le Rouzic E., Belaïdouni N., Estrabaud E., Morel M., Rain J. C., Transy C., Margottin-Goguet F. (2007) HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle 6, 182–188 [DOI] [PubMed] [Google Scholar]
- 29. Schröfelbauer B., Yu Q., Zeitlin S. G., Landau N. R. (2005) Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 79, 10978–10987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahn J., Vu T., Novince Z., Guerrero-Santoro J., Rapic-Otrin V., Gronenborn A. M. (2010) HIV-1 Vpr loads uracil DNA glycosylase-2 onto DCAF1, a substrate recognition subunit of a Cullin 4A-RING E3 ubiquitin ligase for proteasome-dependent degradation. J. Biol. Chem. 285, 37333–37341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watkins J. F., Sung P., Prakash L., Prakash S. (1993) The Saccharomyces cerevisiae DNA repair gene RAD23 encodes a nuclear protein containing a ubiquitin-like domain required for biological function. Mol. Cell. Biol. 13, 7757–7765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masutani C., Sugasawa K., Yanagisawa J., Sonoyama T., Ui M., Enomoto T., Takio K., Tanaka K., van der Spek P. J., Bootsma D. (1994) Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. EMBO J. 13, 1831–1843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. van der Spek P. J., Eker A., Rademakers S., Visser C., Sugasawa K., Masutani C., Hanaoka F., Bootsma D., Hoeijmakers J. H. (1996) XPC and human homologs of RAD23. Intracellular localization and relationship to other nucleotide excision repair complexes. Nucleic Acids Res. 24, 2551–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guzder S. N., Habraken Y., Sung P., Prakash L., Prakash S. (1995) Reconstitution of yeast nucleotide excision repair with purified Rad proteins, replication protein A, and transcription factor TFIIH. J. Biol. Chem. 270, 12973–12976 [DOI] [PubMed] [Google Scholar]
- 35. Sugasawa K., Ng J. M., Masutani C., Iwai S., van der Spek P. J., Eker A. P., Hanaoka F., Bootsma D., Hoeijmakers J. H. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 2, 223–232 [DOI] [PubMed] [Google Scholar]
- 36. Li L., Lu X., Peterson C., Legerski R. (1997) XPC interacts with both HHR23B and HHR23A in vivo. Mutat. Res. 383, 197–203 [DOI] [PubMed] [Google Scholar]
- 37. Masutani C., Araki M., Sugasawa K., van der Spek P. J., Yamada A., Uchida A., Maekawa T., Bootsma D., Hoeijmakers J. H., Hanaoka F. (1997) Identification and characterization of XPC-binding domain of hHR23B. Mol. Cell. Biol. 17, 6915–6923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Withers-Ward E. S., Jowett J. B., Stewart S. A., Xie Y. M., Garfinkel A., Shibagaki Y., Chow S. A., Shah N., Hanaoka F., Sawitz D. G., Armstrong R. W., Souza L. M., Chen I. S. (1997) Human immunodeficiency virus type 1 Vpr interacts with HHR23A, a cellular protein implicated in nucleotide excision DNA repair. J. Virol. 71, 9732–9742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gragerov A., Kino T., Ilyina-Gragerova G., Chrousos G. P., Pavlakis G. N. (1998) HHR23A, the human homologue of the yeast repair protein RAD23, interacts specifically with Vpr protein and prevents cell cycle arrest but not the transcriptional effects of Vpr. Virology 245, 323–330 [DOI] [PubMed] [Google Scholar]
- 40. Dieckmann T., Withers-Ward E. S., Jarosinski M. A., Liu C. F., Chen I. S., Feigon J. (1998) Structure of a human DNA repair protein UBA domain that interacts with HIV-1 Vpr. Nat. Struct. Biol. 5, 1042–1047 [DOI] [PubMed] [Google Scholar]
- 41. Li G., Elder R. T., Dubrovsky L., Liang D., Pushkarsky T., Chiu K., Fan T., Sire J., Bukrinsky M., Zhao R. Y. (2010) HIV-1 replication through hHR23A-mediated interaction of Vpr with 26S proteasome. PLoS ONE 5, e11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mansky L. M., Preveral S., Le Rouzic E., Bernard L. C., Selig L., Depienne C., Benarous R., Benichou S. (2001) Interaction of human immunodeficiency virus type 1 Vpr with the HHR23A DNA repair protein does not correlate with multiple biological functions of Vpr. Virology 282, 176–185 [DOI] [PubMed] [Google Scholar]
- 43. Battiste J. L., Wagner G. (2000) Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear Overhauser effect data. Biochemistry 39, 5355–5365 [DOI] [PubMed] [Google Scholar]
- 44. Piotrowski J., Beal R., Hoffman L., Wilkinson K. D., Cohen R. E., Pickart C. M. (1997) Inhibition of the 26 S proteasome by polyubiquitin chains synthesized to have defined lengths. J. Biol. Chem. 272, 23712–23721 [DOI] [PubMed] [Google Scholar]
- 45. Maciejewski M. W., Mobli M., Schuyler A. D., Stern A. S., Hoch J. C. (2012) Data sampling in multidimensional NMR. Fundamentals and strategies. Top. Curr. Chem. 316, 49–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on Unix Pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 47. Keller R. (2005) Optimizing the Process of Nuclear Magnetic Spectrum Analysis and Computer Aided Resonance Assignment. Dissertation 15947, Swiss Federal Institute of Technology, Zurich, Switzerland [Google Scholar]
- 48. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy. Development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 49. Mobli M., Maciejewski M. W., Gryk M. R., Hoch J. C. (2007) Automatic maximum entropy spectral reconstruction in NMR. J. Biomol. NMR 39, 133–139 [DOI] [PubMed] [Google Scholar]
- 50. Withers-Ward E. S., Mueller T. D., Chen I. S., Feigon J. (2000) Biochemical and structural analysis of the interaction between the UBA(2) domain of the DNA repair protein HHR23A and HIV-1 Vpr. Biochemistry 39, 14103–14112 [DOI] [PubMed] [Google Scholar]
- 51. Walters K. J., Lech P. J., Goh A. M., Wang Q., Howley P. M. (2003) DNA-repair protein hHR23a alters its protein structure upon binding proteasomal subunit S5a. Proc. Natl. Acad. Sci. U.S.A. 100, 12694–12699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kamionka M., Feigon J. (2004) Structure of the XPC binding domain of hHR23A reveals hydrophobic patches for protein interaction. Protein Sci. 13, 2370–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Min J. H., Pavletich N. P. (2007) Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 449, 570–575 [DOI] [PubMed] [Google Scholar]
- 54. Lee J. H., Choi J. M., Lee C., Yi K. J., Cho Y. (2005) Structure of a peptide:N-glycanase-Rad23 complex. Insight into the deglycosylation for denatured glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 102, 9144–9149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Morellet N., Bouaziz S., Petitjean P., Roques B. P. (2003) NMR structure of the HIV-1 regulatory protein VPR. J. Mol. Biol. 327, 215–227 [DOI] [PubMed] [Google Scholar]
- 56. Varadan R., Assfalg M., Raasi S., Pickart C., Fushman D. (2005) Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Mol. Cell 18, 687–698 [DOI] [PubMed] [Google Scholar]
- 57. Mueller T. D., Kamionka M., Feigon J. (2004) Specificity of the interaction between ubiquitin-associated domains and ubiquitin. J. Biol. Chem. 279, 11926–11936 [DOI] [PubMed] [Google Scholar]
- 58. Huang X. Q., Miller W. (1991) A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math. 12, 337–357 [Google Scholar]
- 59. Sugasawa K., Ng J. M., Masutani C., Maekawa T., Uchida A., van der Spek P. J., Eker A. P., Rademakers S., Visser C., Aboussekhra A., Wood R. D., Hanaoka F., Bootsma D., Hoeijmakers J. H. (1997) Two human homologs of Rad23 are functionally interchangeable in complex formation and stimulation of XPC repair activity. Mol. Cell. Biol. 17, 6924–6931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ng J. M., Vermeulen W., van der Horst G. T., Bergink S., Sugasawa K., Vrieling H., Hoeijmakers J. H. (2003) A novel regulation mechanism of DNA repair by damage-induced and RAD23-dependent stabilization of xeroderma pigmentosum group C protein. Genes Dev. 17, 1630–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen L., Madura K. (2002) Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell. Biol. 22, 4902–4913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Elsasser S., Chandler-Militello D., Müller B., Hanna J., Finley D. (2004) Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279, 26817–26822 [DOI] [PubMed] [Google Scholar]
- 63. Verma R., Oania R., Graumann J., Deshaies R. J. (2004) Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell 118, 99–110 [DOI] [PubMed] [Google Scholar]
- 64. Hartmann-Petersen R., Gordon C. (2004) Protein degradation: recognition of ubiquitinylated substrates. Curr. Biol. 14, R754–R756 [DOI] [PubMed] [Google Scholar]