

Background: Rich2 is a synaptic Rho-GAP (Rho-GTPase-activating protein) the target of which was unknown.
Results: We found that Rich2 controls dendritic spine morphogenesis by inhibiting Rac1 activity.
Conclusion: Rac1 is the target of Rich2 in spines.
Significance: We identified for the first time Rich2 as a Rac1-GAP protein that plays an important role in spine formation.
Keywords: Glutamate, Morphogenesis, Rac1, Rho GTPases, Synapses
Abstract
Development of dendritic spines is important for synaptic function, and alteration in spine morphogenesis is often associated with mental disorders. Rich2 was an uncharacterized Rho-GAP protein. Here we searched for a role of this protein in spine morphogenesis. We found that it is enriched in dendritic spines of cultured hippocampal pyramidal neurons during early stages of development. Rich2 specifically stimulated the Rac1 GTPase in these neurons. Inhibition of Rac1 by EHT 1864 increased the size and decreased the density of dendritic spines. Similarly, Rich2 overexpression increased the size and decreased the density of dendritic spines, whereas knock-down of the protein by specific si-RNA decreased both size and density of spines. The morphological changes were reflected by the increased amplitude and decreased frequency of miniature EPSCs induced by Rich2 overexpression, while si-RNA treatment decreased both amplitude and frequency of these events. Finally, treatment of neurons with EHT 1864 rescued the phenotype induced by Rich2 knock-down. These results suggested that Rich2 controls dendritic spine morphogenesis and function via inhibition of Rac1.
Introduction
Dendritic spines are the sites of excitatory glutamatergic synapses in the principal neurons of mammalian brain structures. Their development is essential for establishing proper adult brain connectivity. Developmental alterations in density and morphology of spines have been found in many psychiatric and neurological disorders, such as mental retardation and epilepsy (1). Understanding the role of synaptic proteins in spine development is therefore an important issue, not only in term of physiological mechanisms, but also to identify potential therapeutic targets.
Development of dendritic spines implicates rearrangement of the actin network and trafficking of postsynaptic proteins (2–5). Rho-GTPases have been identified as key regulators of cytoskeleton structural changes in many cell types (6) and play major roles in spine development (5). Rho-GTPase activity is modulated by guanine nucleotide-dissociation inhibitors (GDIs), guanine nucleotide-exchange factors (GEFs),3 and GTPase-activating proteins (GAPs). GDIs sequester Rho proteins away from their targets, while GEFs and GAPs respectively up- and down-regulate the cycle between active GTP- and inactive GDP-bound states of Rho-GTPases. These modulatory proteins thus control dynamics of actin skeleton and spine remodeling (2, 4). Recent studies have reported implication of GEFs in dendritic spine formation. For instance, kalirin-7 is a GEF for Rac1, which plays important role in spine development. Its phosphorylation promotes its activity and leads to spine enlargement in hippocampal pyramidal neurons (7). Intersectin is a GEF for Cdc42 that functions as a multidomain adaptor for proteins involved in endocytosis and regulation of the cytoskeleton. Its activity is enhanced by the numb-binding protein in vivo and controls filopodia formation (8). GAP proteins have also been involved in dendritic spine morphogenesis during development. For instance, the Rho-GAP protein oligophrenin-1 has been shown to regulate the length of dendritic spines in vivo and in hippocampal cultures (9).
Rich2 is a Rho-GAP domain containing protein that belongs to a complex implicated in the organization of the sub-apical actin network of epithelial cells (10). This protein has been cloned several years ago (11) and we have recently shown that it is a Rho-GAP protein involved in endosomal recycling and AMPA receptor GluA1 subunit exocytosis during synaptic long term potentiation (12). However, the Rho-GTPase regulated by Rich2 in neurons remained unknown. Here we found that Rich2 specifically controls spine morphogenesis of hippocampal neurons via regulation of the Rho-GTPase Rac1.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
Neuronal hippocampal cultures were prepared from 17.5 day embryonic mice and grown in Neurobasal medium supplemented with B27 and 10% fetal bovine serum (FBS). Hippocampal neurons cells were transfected at DIV-9 with Lipofectamine 2000 (Invitrogen, Cergy-Pontoise, France) according to the manufacturer's standard protocol. COS-7 cells were plated in DMEM (Invitrogen/Life Technology, Invitrogen) supplemented with 4 mm Glutamax, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS.
Antibodies and DNA Constructs
Rabbit polyclonal anti-Rich2 antibody was generated by targeting the specific Rich2 sequence, SPDMDPADRRQPEQC. Cysteine was linked to hemocyanin by sulfolink (Pierce, Thermo Fisher Scientific, Brebières, France) and the complex injected into rabbits using a previously described protocol (13). The antibody was purified by affinity chromatography using the related peptide coupled to activated-CH Sepharose (Amersham Biosciences, GE Healthcare Europe, Saclay, France) and characterized elsewhere (12). The mouse anti-Rac1 and mouse anti-Cdc42 antibodies were purchased from BD Biosciences (Le Pont-de-Claix, France, Cat. 610651 and 610929, respectively). The rabbit anti-RhoA antibody was purchased from Cell Signaling (Ozyme, Saint Quentin Yvelines, France, Cat. 67B9).
Rich2 constructs were generated from IMAGE clone of mouse Rich2 (clone 6825221) from RZPD German Resource Center for Genome Research. The wild-type Rich2 cDNA were cloned in pCMV-flag2B vector (Stratagene, Agilent Technologies). The si-Rich2 plasmid was designed and generated using the following si-RNA duplex sequence: 5′-GGUGGCAGCAGACUUCCAA-3′ from Invitrogen. Characterization of the si-Rich2 has been previously described (12). The control si-RNA was purchased from Invitrogen. For rescue experiments, we generated a si-RNA resistant Rich2 cDNA mutant (Rich2-mt) with 4 silent mutations: 5′-GATGGCAACAAACTTCTAA-3′ in pCMV-flag2B-Rich2 plasmid (12). The Sh-Rac1-GFP, Sh-Cdc42-GFP, Sh-Unr-GFP (unrelated/control) were generated in pGHSuper-GFP vector using the following sequences. Sh-Rac1: CCAATGAACCAGTCAGTAA, sh-Cdc42: GGGCAAGAGGATTATGACA, sh-Unr: ATTCTATCACTAGCGTGAC. Characterization of sh-Rac1 and sh-Cdc42 is shown in Fig. 7A. The sh-Unr and sh-Rac1 have been previously characterized (14).
FIGURE 7.
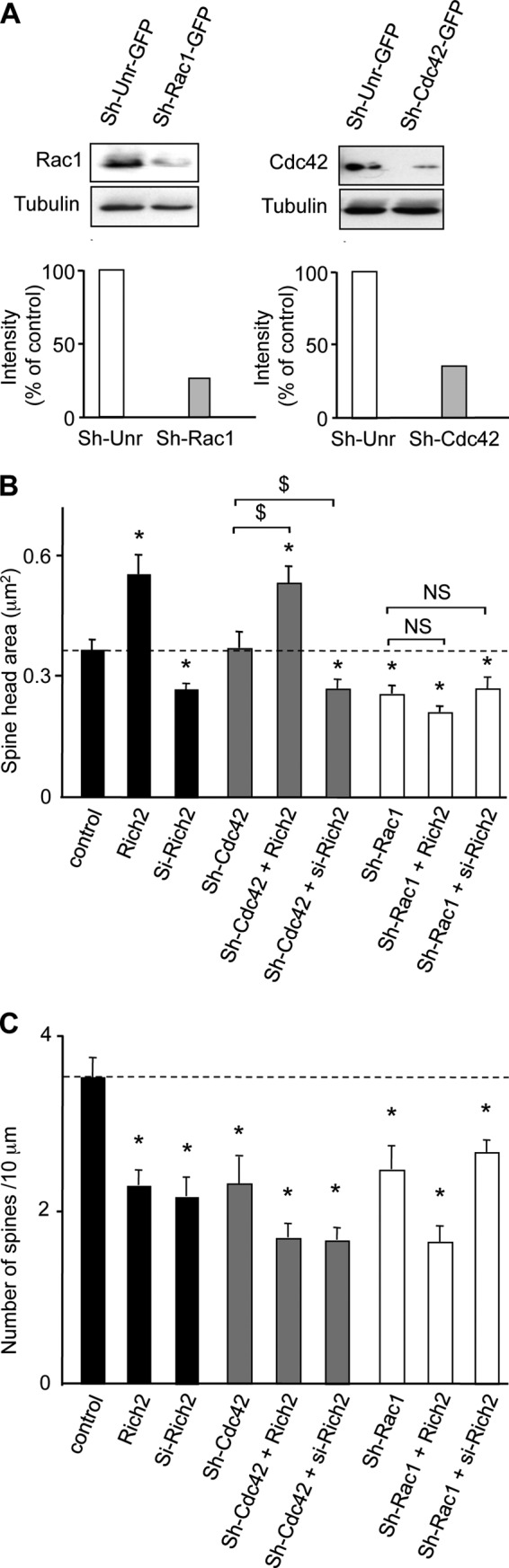
Rac1 knock-down affected Rich2-induced spine enlargement. A, Western blots obtained from HEK cells transfected with control sh-RNA (sh-Unr-GFP) or sh-Rac1-GFP or sh-Cdc42-GFP and revealed with anti-Rac1 or anti-Cdc42 or anti-tubulin antibodies. Please note that sh-Rac1-GFP and sh-Cdc42-GFP decreased Rac1 and Cdc42 protein content by roughly 70% for both. B and C, hippocampal neurons were transfected with the indicated plasmids, in combination with YFP. The histograms represent quantification of spine head size and spine density (*, p < 0.05 when compared with control; $, p < 0.05 when compared with the indicated conditions, NS, not significantly different when compared with the indicated conditions). Dotted lines represents control levels of spine head size and spine density.
GTPase Assay
For GTPase measurements, 0.1 μg of recombinant wild-type Rac, Rho, or Cdc42 was preloaded with 10 μCi of [γ-32P]GTP in 20 μl of 20 mm Tris-HCl, pH 7.5, 25 mm NaCl, 5 mm EDTA, and 0.1 mm DTT. The mixture was incubated for 10 min at 30 °C, and the reaction terminated by adding 5 μl of 0.1 m MgCl2. The resulting [γ-32P]GTP-loaded GTPase solutions were stored on ice. For the GAP assays, we used equimolar amounts of GTPases and GST-Rich2. The P50 Rho-GAP protein was purchased from Cytoskeleton (Tebu-bio, France, Cat. GAS01-B). Three μl of the [γ-32P]GTP-loaded GTPase were added to a 30-μl mixture of 20 mm Tris-HCl, pH 7.5, 1 mm non-radioactive GTP, 0.87 mg/ml bovine serum albumin, and 0.1 mm DTT with the GST-Rich2. The mixture was incubated at 30 °C, 5-μl aliquots were removed after 0, 3, 6, 9, and 12 min, and the reaction was stopped by addition of 1 ml of ice-cold buffer A (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, and 5 mm MgCl2). The samples were collected on nitrocellulose filters and washed with 10 ml of ice-cold buffer A, and the portion of [γ-32P]GTP remaining bound to the GTPases was determined by scintillation counting.
Rho-GAP Function with RAICHU Probes
The pRAICHU probes for Cdc42, RhoA, and Rac1 were generously provided by M. Matsuda (15). COS-7 cells plated in 96 wells were transfected with the different probes and stimulated with 100 nm phorbol 12-myristate 13-acetate (PMA) over 10 min to activate Rho-GTPases. Fluorescence intensity at 480 nm (CFP) and 530 nm (YFP) were measured using a Safire II fluorimeter (TECAN, Lyon, France) after CFP stimulation at 430 nm. FRET efficiency was then determined by measuring the sensitized emission of the acceptor as previously described (16). Data were expressed as the mean ± S.E. and statistical tests performed using the Student's t test.
FRET intensity was calculated as follows: FRET = YFPex430 −(CFPex430 × (YFPco/CFPfco)) − ((YFPfyo/CFPfyo) × YFPex515), where YFPex430 is YFP excited by CFP at 430 nm; CFPex430 is CFP excited at 430 nm; YFPco/CFPfco is fluorescence values for transfected CFP alone and excited at 430 nm; YFPfyo/CFPfyo is fluorescence values for transfected YFP alone and excited at 430 nm; and YFPex515 is YFP excited at 515 nm. Data were expressed as the mean ± S.E. and statistical tests performed using the Student's t test.
FRET Intensity Measurement and FRET Image Acquisition
FRET image acquisition in neurons transfected with pRAICHU probes was performed using a LSM-Meta 510 Zeiss confocal microscope (Carl Zeiss S.A.S., Le Pecq, France). CFP excitation was performed at 405 nm, and image fluorescence emission signals were collected from 415 nm to 580 nm. A fluorescence spectrum was thus generated from images acquired every 10 nm (see Fig. 3B). Ratio of images obtained at 525 nm and 480 nm provided FRET images. Calculation of FRET efficiency was obtained by dividing spectral values of individual dendritic spines obtained at 525 nm and 480 nm (Fig. 2, D and E).
FIGURE 3.

Rich2 selectively regulates Rac1. A, structure of the FRET-based Rac1 pRAICHU probe. It comprises the Rac1 interactive binding domain of p21-activated kinase (PAK) and Rac1, sandwiched between the GFP mutants YFP and CFP. Upon stimulation, the exchange of GDP to GTP on Rac1 nucleotide binding site leads to interaction between the PAK and Rac1-GTP domains, and FRET signal. Conversely, hydrolysis of GTP in the presence of a Rac1 GAP protein decreases the FRET signal. The Cdc42 pRAICHU probe displays a similar structure as the Rac1 pRAICHU probe, except for the GDP/GTP domain that is replaced by Cdc42-GDP/GTP domain, respectively. The RhoA pRAICHU probe uses human RhoA as the sensor region and Rho-binding domain of the protein kinase PKN as the ligand region. Upon GTP binding, the effector region of RhoA binds to PKN, which triggers FRET between CFP and YFP. B and C, measurement of FRET efficiency by confocal microscopy (LSM-Meta 510 Zeiss microscope) in neurons. Images were taken every 10 nm from 415 to 580 nm using the LSM-Meta 510 ZEISS confocal microscope. This resulted in a fluorescence spectrum for every pixel of the images. Scale bar in B: 1 μm. D, hippocampal neurons were co-transfected with si-Rich2 (DIV-9) or Rich2 (DIV-11) and pRAICHU-Rac1, pRAICHU-RhoA, or pRAICHU-Cdc42 constructs, and then tested at DIV-12. Images obtained from portions of dendrites of cultured hippocampal neurons transfected with either the Rac1, RhoA, or Cdc42 pRAICHU probe, in the absence (control) and presence of co-transfected si-Rich2 or Rich2. The right panels show 525/480 ratio images corresponding to FRET signals (scale bar: 2 μm) and enlarged views of the squared areas. E, histogram shows FRET efficiency obtained with Rac1, RhoA, and Cdc42 pRAICHU probes. FRET efficiency in dendritic spines was calculated from fluorescence spectrum images generated by the confocal microscope similar to those shown in D (n = 50 for Rac1, n = 65 for RhoA, n = 54 for Cdc42, n = 52 for Rac1 + Rich2, n = 55 for RhoA + Rich2, n = 59 for Cdc42 + Rich2, n = 87 for Rac1 + si-Rich2, n = 74 for RhoA + si-Rich2, n = 48 spines for Cdc42 + si-Rich2, *, p < 0.05).
FIGURE 2.

Measurements of Rich2 Rho-GAP activity in vitro. A–C, Rich2 Rho-GAP activity measured by GTPase assay. Time course of GTP hydrolysis measured on [γ-32P]GTP-loaded RhoA, Rac1, and Cdc42 GTPase, in the absence (control) and presence of the widely expressed p50 Rho-GAP (p50; Ref. 28) or Rich2. The p50 and Rich2 curves in A, p50 in B, and p50 and Rich2 in C are significantly different from their respective control curves (p < 0.001, two-way ANOVA). D, Rho-GAP function of Rich2 measured by FRET in COS-7 cells, using pRAICHU probes for Rac1, RhoA, and Cdc42. Please note that Rich2 displays a GAP activity for all members of the Rho-GTPase family. (*, p ≤ 0.01, n = 3).
Electrophysiology
Mouse hippocampal cultures were continuously perfused with the following external medium (mm): NaCl (140), MgCl2 (2), CaCl2 (2), KCl (3), HEPES (10), d-glucose (10), tetrodotoxin (0.0003), bicucullin (0.01), pH 7.4 and osmolarity of 330 mOsm. The transfected neurons were selected on the basis of their fluorescence (YFP) and recorded at room temperature using the whole-cell configuration of the patch-clamp technique. The recording pipettes had a resistance of 3–5 MOhms when filled with the following medium (mm) KCl (140), HEPES (10), d-glucose (10), pH 7.2 and osmolarity of 300 mOsm.
Miniature EPSCs (mEPCs) were recorded at −65 mV membrane potential through an Axopatch 200B amplifier (Axon Instruments, Union City, CA), filtered at 1 kHz and then digitized at 3 kHz using Axotape software (Axon Instruments). Currents were analyzed using the pClamp 9 software (Axon Instruments). All the detected events were re-examined and accepted or rejected on the basis of visual examination. Once more than 100 events had been collected from a neuron, the average frequency and amplitude of the events were measured on the total duration of the sample. Data obtained from the indicated number (n) of cells were expressed as the mean ± S.E. and analyzed using the statistical Student's t test.
Immunofluorescence Staining and Analysis
Cells were fixed for 20 min in 4% paraformaldehyde buffered in phosphate-buffered saline (PBS) followed by three washes in PBS. They were then permeabilized in PBS supplemented with 2% BSA and 0.1% Triton X-100 and incubated for 1 h at room temperature with appropriate primary antibodies in the same buffer. Mouse or rabbit secondary antibodies coupled to Alexa Fluor 488 (Molecular Probes, Invitrogen, Cergy-Pontoise, France) or cyanine 3 (Jackson ImmunoResearch Laboratories, Interchim, Montluçon, France) were incubated for 1 h at room temperature. Cells were observed under an Axio-Imager Z1 Zeiss microscope equipped with appropriate epifluorescence and filters (Carl Zeiss S.A.S., Le Pecq, France; GFP: 475 ± 40 and 530 ± 50 nm for excitation and emission respectively, cyanine 3: 545 ± 25 and 605 ± 70 nm for excitation and emission respectively) to discriminate between green and red fluorescence. Image quantification was performed using ImageJ software (National Institutes of Health). In morphological studies, spines were defined as dendritic protrusions with neck and head, whereas filopodia were defined as thin dendritic protrusions devoid of head.
Immunohistology
The brain was dissected and fixed by immersion with 4% paraformaldehyde in PBS for 24 h at 4 °C. It was then cut frontally with a vibratome into 50-μm thick sections. After careful rinsing in PBS, sections were incubated overnight at 4 °C with the rabbit anti-Rich2 antibody (1:100 dilution) in PBS, Triton X-100 0:1, and 2% BSA.
Immunoprecipitation
Immunoprecipitation experiments (IP) were performed using rabbit anti-Rich2 antibodies, and immunoprecipitated proteins were revealed by Western blot with mouse anti-Rac1 antibody. In IP, 5 μg of either rabbit antibody (control condition) or Rich2 antibody (IP) were added. Protein-A Sepharose (Amersham Biosciences) was used to immunoprecipitate IgGs.
Statistical Analyses
Values were expressed as mean ± S.E. of at least three independent experiments. In bar graphs, they were compared with control using Student's t test. For GTPase measurements, curves were compared with control using two-way ANOVA test. Values of p < 0.05 were considered as significantly different.
RESULTS
Rich2 Neuronal Localization
We found Rich2 expression in the pyramidal layers of mouse hippocampus at embryonic age 18.5 (E18.5). Double-immunolabeling with neuronal (NeuN) and glial (GFAP) markers revealed that Rich2 was highly expressed in neurons and undetectable in glial cells (Fig. 1A). In 21 days in vitro (DIV 21) cultured mouse hippocampal neurons, F-actin and Rich2 immunostaining colocalized in dendritic spines (Fig. 1B). Furthermore, Rich2 immunostaining colocalized with the synaptic marker Bassoon (Fig. 1C). Altogether, these results suggested a post-synaptic localization of Rich2 in cultured hippocampal neurons.
FIGURE 1.

Rich2 is localized in dendritic spines. A, immunostaining of E18.5 rat hippocampal CA1, CA2, and CA3 regions with anti-NeuN (green) and anti-Rich2 (red) antibodies (upper panel). In this and the following panels, right images represent merged red and green images. Middle panel: similar to the upper panel, but in an enlarged region of CA1 of a different sample. Lower panel: immunostaining of E18.5 rat hippocampal with an anti-GFAP antibody. Please note localization of Rich2 in neurons (NeuN labeling, arrows), but not glial cells (GFAP labeling; Scale bars: 100 and 40 μm, respectively). B, DIV-21 hippocampal neurons treated with phalloidin (green) and anti-Rich2 antibody (red) to visualize F-actin and endogenous Rich2, respectively. Arrows indicate co-localization of Rich2 and actin in dendritic spines (scale bars: 10 and 1 μm, respectively). C, dendritic portion of a DIV-15 cultured hippocampal neuron immunolabeled with an anti-bassoon antibody showing the synaptic localization of Rich2, (arrows, scale bar: 1 μm).
Rich2 Is a Rho-GAP for Rac1 in Neurons
Rich2 displays a Rho-GAP domain (11). We therefore addressed the issue of the Rho-GAP function of this protein. We found that Rich2 was able to stimulate GTP hydrolysis of [γ32P]GTP-loaded Rac1 and Cdc42, and to a lesser extent RhoA, in vitro (Fig. 2, A–C). We then examined if this applied to in cellulo conditions using the RAS and Interacting protein Chimeric Unit (RAICHU) probe system, (15). A RAICHU probe is a genetically encoded FRET-based probe consisting of the Rho GTPase (RhoA, Rac1, or Cdc42) conjugated to the donor fluorochrome CFP, and the GTPase-binding domain of its binding partner conjugated to the acceptor flurorochrome YFP (Fig. 3A). Interaction between the GTP-bound GTPase and the GTPase-binding domain brings CFP into close proximity of YFP, thereby generating a FRET signal upon stimulation of the donor CFP. The pRAICHU-Rac1, pRAICHU-RhoA, or pRAICHU-Cdc42 constructs were transfected separately with or without Rich2 in COS-7 cells, and their respective GTP-bound forms measured by FRET. We found that co-expression of Rich2 decreased FRET signals of all three pRAICHU probes, thus indicating that Rich2-inactivated RhoA, Rac1, and Cdc42 (Fig. 2D). These results showed that the GAP domain of Rich2 was functional and equally effective on all members of the Rho-GTPase family in COS-7 cells.
Having established the suitability of the pRAICHU probe for Rich2 GTPase activity measurement, pRAICHU-Cdc42, pRAICHU-Rac1, or pRAICHU-RhoA constructs were co-transfected with Rich2 in cultured hippocampal neurons for FRET imaging experiments. We first determined the FRET imaging efficiency by photobleaching the YFP acceptor module of the pRAICHU-Cdc42 probe. Photobleaching of YFP decreased the peak of fluorescence emission at 525 nm and increased the fluorescence emission at 480 nm, indicating that genuine FRET responses were recorded with the probe (Fig. 3, B and C). FRET imaging was then carried out in dendrites of DIV 21 cultured hippocampal neurons transfected with either Rich2 or a specific Rich2 si-RNA (si-Rich2). We found that transfected Rich2 decreased, whereas Rich2 si-RNA increased the FRET signal of the pRAICHU-Rac1 probes in spines. Conversely, the signals obtained with either pRAICHU-RhoA and pRAICHU-Cdc42 were unchanged by the modifications of Rich2 expression (Fig. 3, D and E). This indicated that Rich2 selectively up-regulated the GTPase activity and thus inhibited Rac1 in neurons. Furthermore, in cultured hippocampal neurons transfected with either wild-type Rac1 (Rac1) or the constitutively active Rac1-V12 mutant, co-transfected Rich2 partially co-localized with Rac1 and fully co-localized with Rac1-V12 (Fig. 4, A and D). On the other hand, only partial colocalization was found between Rich2 and either Cdc42, Cdc42-V12, RhoA, or RhoA-V12 (Fig. 4, B–D). This result suggested that Rich2 interacted preferentially with the active form of Rac1, as expected from a Rho-GAP specific for this GTPase. Finally, Western blots performed from postnatal day 4 (P4) mouse hippocampus, using a home-made rabbit anti-Rich2 antibody, showed that Rich2 co-immunoprecipitated with Rac1, but not Cdc42 or RhoA (Fig. 4E). This result suggested that Rac1 forms a complex with Rich2 in mouse brain. Altogether, these experiments showed that Rich2 is a Rho-GAP for Rac1 in neurons.
FIGURE 4.

Rich2 and Rac1 are part of a same complex in mouse brain. A–C, images show immunostaining of Rich2, Rac1, and Rac1-V12 (A) or Cdc42 and constitutively active Cdc42-V12 (B), or RhoA and constitutively active RhoA-V12 (C), in a dendritic portion of a DIV 12 hippocampal neuron co-transfected with the indicated proteins. The graphs below the images show fluorescence correlation between Rich2 (green) and Rac1 (red; left), and between Rich2 (green) and Rac1-V12 (red: right) measured along the white line drawn on the merged images. Scale bar: 1 μm. D, histogram represents the Pearson's Rr correlation coefficient values between Rich2 and Rac1 or Rac1-V12, Rich2, and Cdc42 or Cdc42-V12 and Rich2 and RhoA or RhoA-V12 (n = 10 in each condition, *, p < 0.05). These values translate the level of co-localization between Rich2 and Rho-GTPases and Rho-GTPase-V12 proteins. E, immunoprecipitation (IP) of Rac1, Cdc42, and RhoA with Rich2 obtained from P4 mouse hippocampus. CT is the IP obtained using a rabbit nonspecific antibody (control). IP, immunoprecipitation obtained using a rabbit anti-Rich2 antibody. Western blots revealed immunoprecipitation of Rich2 with Rac1, but neither Cdc42 nor RhoA, although inputs show the presence of Rac1, Cdc42, and RhoA in supernatants.
Rich2 Controls Dendritic Spine Morphogenesis and Function via Rac1
We then investigated whether Rich2 controlled spine morphogenesis in developing neurons and if this involved its interaction with Rac1. DIV 9 hippocampal neurons were transfected with pEGFP and either Rich2 or si-Rich2. At this stage of development, the dendrites do not yet display spines. The effects of Rich2 and si-Rich2 on dendritic spine growth were then analyzed at DIV12. Rich2 increased the size of spine head and decreased spine density, while si-Rich2 had opposite effects on spine size. Like Rich2, si-Rich2 decreased spine density (Fig. 5, A–C). The si-RNA resistant mutant of Rich2 (Rich2-mt) rescued the phenotype on spine size and density (Fig. 5, A–C). Neither Rich2 nor si-Rich2 significantly modified the density of filopodia (control = 1.25 ± 0.25, Rich2 = 1.2 ± 0.20 and si-Rich2 = 1.4 ± 0.30 filopodia per 10 μm).
FIGURE 5.

Rich2-mediated functional morphogenesis of dendritic spines. Hippocampal neurons were transfected at DIV-9 with the indicated plasmids, in combination with YFP, and tested at DIV-12. A, portions of dendrites showing spines (arrowheads) of hippocampal neurons transfected with GFP alone (control), or in combination with either Rich2, si-Rich2 or si-Rich2 + Rich2-mt (scale bar: 1 μm). The right images show enlarged views of the square areas. B and C, histograms show changes in the size of spine head and spine density in GFP-transfected neurons (control), and neurons transfected with either Rich2, si-control or si-Rich2 or si-Rich2 + Rich2-mt (*, p < 0.001). D and G, sample traces of whole-cell patch-clamp recordings obtained from cultured hippocampal neurons transfected with the indicated plasmids at DIV-9, and tested at DIV-12. E and F, frequency and amplitude histograms of mEPSCs obtained from neurons transfected with GFP alone (control) or in combination with Rich2 (n = 10 in each condition, *, p < 0.05). H and I, same legend as for E, F, but from neurons transfected with si-Rich2.
Patch-clamp recording performed in the transfected cultured hippocampal neurons showed that Rich2 increased the amplitude and decreased frequency of miniature ESPCs (mEPSCs; Fig. 5, D–F), whereas si-Rich2 decreased both amplitude and frequency of mEPSCs (Fig. 5, G–I). These functional synaptic modifications were consistent with the observed spine morphological changes. Taken together these results suggested that Rich2 controls the formation of functional dendritic spines.
As we found that Rich2 is a Rho-GAP for Rac1, we reasoned that Rac1-V12 should mimic the effect of si-Rich2. As expected, transfection of Rac1-V12 decreased spine size and density (Fig. 6, A–C), as it was the case for si-Rich2 (Fig. 5, A–C). In the same line, if the effect of Rich2 involved inhibition of Rac1 activity, overexpression of Rac1-V12 should rescue the phenotype induced by Rich2. Both Rac1-V12 and Rich2 decreased spine number. It is therefore difficult to conclude on the implication of the Rich2-Rac1 pathway on spine density regulation. However, Rac1-V12 decreased, while Rich2 increased spine size. Interestingly, in the presence of Rac1-V12, Rich2 was unable to increase spine size, thus suggesting that Rich2 and Rac1-V12 acted via a common pathway to control spine size (Fig. 6, A and B).
FIGURE 6.

The effects of Rich2 on spine morphology involved Rac1 activity. A, portions of dendrites showing spines (arrowheads) of DIV-11 hippocampal neurons transfected with YFP alone (control), or in combination with the indicated plasmids and tested at DIV-12, or treated for 24 h with 10 μm EHT-1864 (EHT). B–E, histograms represent changes in size of spine head and spine density relative to control values (spine head size: 0.30 ± 0.02 μm2 and spine density: 5.9 ± 1.0/10 μm) in neurons transfected as indicated in A (n = 14 neurons in each condition, *, p < 0.001, NS, not significantly different; scale bar: 2 μm).
The Rac1 inhibitor, ETH 1864, mimicked the effect of Rich2 transfection as it increased the size and decreased the density of dendritic spines, and this effect was partially rescued by transfection of si-Rich2 (Fig. 6, A, D, E). In addition, sh-RNAs against Rac1 but not Cdc42 (sh-Rac1 and sh-Cdc42, respectively characterized in Fig. 7A) inhibited the effect of Rich2 on spine size. (Fig. 7B). We observed a decrease in spine density in all the conditions tested (Fig. 7C). Taken together these results suggested that Rich2 controls spine morphogenesis via inhibition of Rac1 activity, but not Cdc42.
DISCUSSION
Our results show that Rich2 is a neuronal protein that is present in dendritic spines of developing hippocampal pyramidal neurons. In these neurons, Rich2 displays a GAP activity that we found to be specific for Rac1. This role had been previously suggested by knock-down of Rich2 in Caco-2 epithelial cells, which increased Rac1 activity (10). However according to previous findings (11), in non-neuronal cells as well as in vitro, Rich2 displayed a Rho-GAP activity also for Rho-A and Cdc42. We found that Rich2 controls dendritic spine morphogenesis and function via specific inhibition of Rac1. Indeed, si-Rich2, which is expected to potentiate Rac1 activity, reduced spine size and mEPSC amplitude, whereas Rich2 overexpression, which was expected to down-regulate Rac1 activity, induced spine enlargement and increased mEPSC amplitude. Consistent with these results, we observed that the constitutively active form of Rac1, Rac1-V12, reduced spine size, as it is the case in developing Purkinje neurons but not in mature hippocampal neurons where it induces opposite effect (17, 20).
Our results suggest that an optimal balance of Rac1 activity is required for adequate control of spine morphogenesis during development. As expected, Rich2 and Rac1-V12 induced opposite effects on spine size. Furthermore pharmacological inhibition of Rac1 activity mimicked Rich2 effect (spine enlargement). However Rac1 knock-down also reduced spine size. This apparent discrepancy may simply result from the fact that a sufficient amount of Rac1 is required to control spine morphogenesis.
We also found that the Rho-GAP Rich2 influenced spine number. A decrease in spine density was observed with all treatments tested, including Rich2 and Rac1-V2, as well as both knock-down and over-expression of Rich2. Therefore no firm conclusion could be drawn for the control of Rac1 activity by Rich2 and spine number. Previous studies showing that both inhibition and over-activation of Rho-GTPases similarly decrease synaptic density in developing cultured hippocampal neurons (17, 20–22) support our results. For instance both overexpression and knock-down of α1-chimaerin, a GAP for Rac1, was shown to decrease the number of spines, increase the number of filopodia, and induced formation of spines with multiple protrusions (23, 24). This suggests more complex tuning of Rac1 activity to control spine density. An alternative hypothesis could be that Rich2 displays other GAP activity on different GTPase family.
Other studies have shown that oligophrenin, a GAP for RhoA, controls spine morphogenesis (9, 25). This protein is mutated in patients affected by mental retardation (26). The GAP p190 also regulates spines density by modulating of RhoA activity via PAR-6 (partition defective 6)/αPKC complex (27). The present study further extends the spectrum of Rho-GAP proteins and their regulations that control spine morphogenesis, thus providing new insights in the mechanisms responsible of synaptogenesis.
Acknowledgments
MS analyses were carried out using facilities of the Functional Proteomic Platform (IBiSA) of Montpellier-Languedoc Roussillon. FRET confocal microscopy was carried out using facilities of the RIO imaging platform of Montpellier-Languedoc Roussillon with the help of Nicole Lautredou.
This work was supported by the European Community (Health-F2-2008-222918 - REPLACES), FUI DIATRAL, and FUI RHENEPI (to L. F.) and the Deutsche Forschungsgemeinschaft DFG (SFB 497/B8; to T. M. B.).
- GEF
- guanine nucleotide-exchange factor
- Cdc42
- cell division cycle 42 protein
- DIV
- days in vitro
- EPSC
- excitatory postsynaptic current
- GFAP
- glial fibrillary acidic protein
- GluA1
- glutamate receptor subunit A1
- NeuN
- neuronal specific nuclear protein
- RhoA
- Ras homolog family member A protein
- GAP
- GTPase-activating protein
- PMA
- phorbol 12-myristate 13-acetate.
REFERENCES
- 1. Fiala J. C., Spacek J., Harris K. M. (2002) Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54 [DOI] [PubMed] [Google Scholar]
- 2. Honkura N., Matsuzaki M., Noguchi J., Ellis-Davies G. C., Kasai H. (2008) The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 57, 719–729 [DOI] [PubMed] [Google Scholar]
- 3. Park M., Salgado J. M., Ostroff L., Helton T. D., Robinson C. G., Harris K. M., Ehlers M. D. (2006) Plasticity-induced growth of dendritic spines by exocytic trafficking from recycling endosomes. Neuron 52, 817–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sekino Y., Kojima N., Shirao T. (2007) Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem. Int. 51, 92–104 [DOI] [PubMed] [Google Scholar]
- 5. Tolias K. F., Duman J. G., Um K. (2011) Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog. Neurobiol. 94, 133–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Heasman S. J., Ridley A. J. (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690–701 [DOI] [PubMed] [Google Scholar]
- 7. Penzes P., Jones K. A. (2008) Dendritic spine dynamics–a key role for kalirin-7. Trends Neurosci. 31, 419–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishimura T., Yamaguchi T., Tokunaga A., Hara A., Hamaguchi T., Kato K., Iwamatsu A., Okano H., Kaibuchi K. (2006) Role of numb in dendritic spine development with a Cdc42 GEF intersectin and EphB2. Mol. Biol. Cell 17, 1273–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Govek E. E., Newey S. E., Akerman C. J., Cross J. R., Van der Veken L., Van Aelst L. (2004) The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat. Neurosci. 7, 364–372 [DOI] [PubMed] [Google Scholar]
- 10. Rollason R., Korolchuk V., Hamilton C., Jepson M., Banting G. (2009) A CD317/tetherin-RICH2 complex plays a critical role in the organization of the subapical actin cytoskeleton in polarized epithelial cells. J. Cell Biol. 184, 721–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richnau N., Aspenström P. (2001) Rich, a rho GTPase-activating protein domain-containing protein involved in signaling by Cdc42 and Rac1. J. Biol. Chem. 276, 35060–35070 [DOI] [PubMed] [Google Scholar]
- 12. Raynaud F., Janossy A., Dahl J., Bertaso F., Perroy J., Varrault A., Vidal M., Worley P. F., Boeckers T. M., Bockaert J., Marin P., Fagni L., Homburger V. (2013) Shank3-Rich2 interaction regulates AMPA receptor recycling and synaptic long-term potentiation. J. Neurosci. 33, 9699–9715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Benyamin Y., Roustan C., Boyer M. (1986) Anti-actin antibodies. Chemical modification allows the selective production of antibodies to the N-terminal region. J. Immunol. Methods 86, 21–29 [DOI] [PubMed] [Google Scholar]
- 14. Momboisse F., Lonchamp E., Calco V., Ceridono M., Vitale N., Bader M. F., Gasman S. (2009) betaPIX-activated Rac1 stimulates the activation of phospholipase D, which is associated with exocytosis in neuroendocrine cells. J. Cell Sci. 122, 798–806 [DOI] [PubMed] [Google Scholar]
- 15. Nakamura T., Kurokawa K., Kiyokawa E., Matsuda M. (2006) Analysis of the spatiotemporal activation of rho GTPases using Raichu probes. Methods Enzymol. 406, 315–332 [DOI] [PubMed] [Google Scholar]
- 16. Clegg R. M. (1992) Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211, 353–388 [DOI] [PubMed] [Google Scholar]
- 17. Luo L., Jan L. Y., Jan Y. N. (1997) Rho family GTP-binding proteins in growth cone signalling. Curr. Opin Neurobiol. 7, 81–86 [DOI] [PubMed] [Google Scholar]
- 18. Nakayama A. Y., Harms M. B., Luo L. (2000) Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci. 20, 5329–5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tashiro A., Minden A., Yuste R. (2000) Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex 10, 927–938 [DOI] [PubMed] [Google Scholar]
- 20. Zhang H., Webb D. J., Asmussen H., Horwitz A. F. (2003) Synapse formation is regulated by the signaling adaptor GIT1. J. Cell Biol. 161, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuhn T. B., Brown M. D., Bamburg J. R. (1998) Rac1-dependent actin filament organization in growth cones is necessary for β1-integrin-mediated advance but not for growth on poly-D-lysine. J. Neurobiol. 37, 524–540 [DOI] [PubMed] [Google Scholar]
- 22. Song H. J., Poo M. M. (1999) Signal transduction underlying growth cone guidance by diffusible factors. Curr. Opin Neurobiol. 9, 355–363 [DOI] [PubMed] [Google Scholar]
- 23. Buttery P., Beg A. A., Chih B., Broder A., Mason C. A., Scheiffele P. (2006) The diacylglycerol-binding protein α1-chimaerin regulates dendritic morphology. Proc. Natl. Acad. Sci. U.S.A. 103, 1924–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van de Ven T. J., VanDongen H. M., VanDongen A. M. (2005) The nonkinase phorbol ester receptor α1-chimerin binds the NMDA receptor NR2A subunit and regulates dendritic spine density. J. Neurosci. 25, 9488–9496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khelfaoui M., Denis C., van Galen E., de Bock F., Schmitt A., Houbron C., Morice E., Giros B., Ramakers G., Fagni L., Chelly J., Nosten-Bertrand M., Billuart P. (2007) Loss of X-linked mental retardation gene oligophrenin1 in mice impairs spatial memory and leads to ventricular enlargement and dendritic spine immaturity. J. Neurosci. 27, 9439–9450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Billuart P., Bienvenu T., Ronce N., des Portes V., Vinet M. C., Zemni R., Roest Crollius H., Carrié A., Fauchereau F., Cherry M., Briault S., Hamel B., Fryns J. P., Beldjord C., Kahn A., Moraine C., Chelly J. (1998) Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 392, 923–926 [DOI] [PubMed] [Google Scholar]
- 27. Zhang H., Macara I. G. (2008) The PAR-6 polarity protein regulates dendritic spine morphogenesis through p190 RhoGAP and the Rho GTPase. Dev Cell 14, 216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lancaster C. A., Taylor-Harris P. M., Self A. J., Brill S., van Erp H. E., Hall A. (1994) Characterization of rhoGAP. A GTPase-activating protein for rho-related small GTPases. J. Biol. Chem. 269, 1137–1142 [PubMed] [Google Scholar]