

Background: Lytic polysaccharide monooxygenases (LPMOs) are recently discovered enzymes that cleave polysaccharides.
Results: We describe a novel LPMO and use a range of analytical methods to characterize its activity.
Conclusion: Cellulose and cello-oligosaccharides are cleaved by oxidizing the sugar at the nonreducing end in the C4 position.
Significance: This study provides unequivocal evidence for C4 oxidation of the nonreducing end sugar and demonstrates a novel LPMO substrate specificity.
Keywords: Biofuel, Cellulase, Mass Spectrometry (MS), Metalloenzymes, NMR, AA10, AA9, CBM33, GH61, Lytic Polysaccharide Monooxygenase (LPMO)
Abstract
Lignocellulosic biomass is a renewable resource that significantly can substitute fossil resources for the production of fuels, chemicals, and materials. Efficient saccharification of this biomass to fermentable sugars will be a key technology in future biorefineries. Traditionally, saccharification was thought to be accomplished by mixtures of hydrolytic enzymes. However, recently it has been shown that lytic polysaccharide monooxygenases (LPMOs) contribute to this process by catalyzing oxidative cleavage of insoluble polysaccharides utilizing a mechanism involving molecular oxygen and an electron donor. These enzymes thus represent novel tools for the saccharification of plant biomass. Most characterized LPMOs, including all reported bacterial LPMOs, form aldonic acids, i.e., products oxidized in the C1 position of the terminal sugar. Oxidation at other positions has been observed, and there has been some debate concerning the nature of this position (C4 or C6). In this study, we have characterized an LPMO from Neurospora crassa (NcLPMO9C; also known as NCU02916 and NcGH61–3). Remarkably, and in contrast to all previously characterized LPMOs, which are active only on polysaccharides, NcLPMO9C is able to cleave soluble cello-oligosaccharides as short as a tetramer, a property that allowed detailed product analysis. Using mass spectrometry and NMR, we show that the cello-oligosaccharide products released by this enzyme contain a C4 gemdiol/keto group at the nonreducing end.
Introduction
In the emerging bio-economy, plant biomass will gradually substitute fossil resources for the production of fuels, chemicals, and materials. One of the main bottlenecks in such biorefining processes is the depolymerization of cellulose, a major constituent of the plant cell wall, to fermentable sugars. In nature this process is catalyzed by cellulases and the recently discovered lytic polysaccharide monooxygenases (LPMOs)2 (1). Enzymes and binding domains interacting with polysaccharides are categorized in the CAZy database, which comprises families of structurally related carbohydrate-active enzymes, such as glycoside hydrolases (GH), and carbohydrate-binding modules (CBMs) (2). LPMOs were originally classified as CBM33 (family 33 carbohydrate-binding module) or GH61 (family 61 glycoside hydrolase). However, CAZy has recently been revised, and GH61 and CBM33 are now named LPMOs and classified under the heading “auxiliary activities” (AA) as families AA9 and AA10, respectively (3).
The enzyme activities of LPMOs were first discovered in 2010 for an AA10 protein (CBP21) acting on chitin (4). Following this study, cellulose active LPMOs were found in both the AA10 (5) and AA9 (6–8) families. These copper-dependent enzymes carry out oxidative cleavage of the β-1,4-glycosidic bonds in polysaccharides, using molecular oxygen and an electron donor (1). Electrons may be supplied by small molecule reductants such as ascorbic acid and gallic acid (4, 6) or by enzymes, such as cellobiose dehydrogenase (CDH), that are co-expressed with LPMOs (8–10). The products of the reaction are oxidized oligosaccharides and native oligosaccharides containing reducing ends originally present in the polymeric substrate (1, 7) (Fig. 1). Although cleavage of polysaccharides by C1 oxidizing LPMOs, which yields aldonic acids, has been thoroughly demonstrated and analyzed (4, 5, 7, 8, 11, 12), oxidation at the nonreducing end is more difficult to analyze. Based on mass spectrometry, both oxidation at C4 and C6 have been suggested (6, 8, 13). For products generated by NcLPMO9D (also known as NCU01050 or NcGH61–4), oxidation at C4 rather than at C6 has been shown indirectly by detection of the C4 epimer of glucose, galactose, upon reduction of reaction products and by the absence of glucuronic acid upon hypoiodite oxidation of reaction products (14). However, direct evidence for the identity of the nonreducing end oxidized species is lacking.
FIGURE 1.

Overview of cellobiose with C1 and/or C4 oxidations. Oxidations are shown in red. The IUPAC names, the abbreviations, and the m/z values for lithium adducts are provided below the structures (please see Table 1 for a complete list of other adducts). In the right two structures, the IUPAC name, the abbreviation, and the m/z of the lithium adduct for the keto-form are given in parentheses.
The filamentous ascomycete Neurospora crassa is an efficient degrader of plant cell walls and produces a wide range of LPMOs and hydrolytic enzymes. The genome of N. crassa is predicted to contain 14 AA9 family LPMOs, six of which are attached to a CBM1 carbohydrate-binding module (15). These CBM1 modules contain ∼40 amino acids, typically bind cellulose, and are almost exclusively found in fungi. The activities of three of these LPMOs on cellulose have been qualitatively characterized by HPLC and MS analyses of released products. NcLPMO9E (NCU08760, NcGH61–5; attached to a CBM1) oxidizes C1, NcLPMO9D exclusively oxidizes the nonreducing end, and NcLPMO9M (NCU07898, NcGH61–13) seems to be capable of oxidizing both C1 and the nonreducing end (8, 16). In another study, using CDH as an electron donor, it was shown that three additional N. crassa AA9s, NcLPMO9C (NCU02916, NcGH61–3; attached to a CBM1), NcLPMO9F (NCU03328, NcGH61–6), and NcLPMO9J (NCU01867, NcGH61–10; attached to a CBM1), degrade cellulose, but no attempts were made to unravel details of the reaction products of these enzymes (17).
In this study, we have characterized the activity of NcLPMO9C using NMR, mass spectrometry, HPLC, and a previously described activity assay (17). Interestingly, NcLPMO9C turned out to be active on soluble substrates, which is an activity not previously described for LPMOs. Exploiting this unique property, we used NMR analysis to identify the products generated by NcLPMO9C.
EXPERIMENTAL PROCEDURES
Production and Purification of Enzymes
The AA9 encoding N. crassa gene NCU02916 was codon-optimized, cloned with its native signal sequence under control of the methanol inducible AOX1 promotor, and recombinantly produced in Pichia pastoris X-33 following a published protocol (10). The protein was purified from 0.4 liter of culture supernatant by three subsequent chromatographic steps following a published method (17). In total, 28 mg of purified NcLPMO9C was obtained, and the homogeneity was verified by SDS-PAGE. Cellobiose dehydrogenase from Myriococcum thermophilum carrying a C-terminal CBM1 (MtCDH, Uniprot accession number A9XK88) (18) was recombinantly expressed in P. pastoris using methanol for induction (19). The enzyme was purified from 1 liter of culture supernatant by two chromatographic steps according to the procedure described by Harreither et al. (20), and 180 mg of homogeneous MtCDH was obtained.
Activity Assays
Standard reaction mixtures (100–300 μl of liquid volume) contained the substrate (0.4 mg/ml cello-oligosaccharide degree of polymerization (DP) 3–6; (Megazyme), 1 mg/ml phosphoric acid swollen cellulose (PASC prepared from Avicel as described by Wood (21), 2.5 mg/ml Avicel PH-101 (Fluka) or (0.15–0.30 mg/ml) steam-exploded spruce), 4.4–8.8 μm NcLPMO9C, 5 mm ammonium acetate buffer, pH 6.0, and 2 mm reducing agent (hydroquinone, ascorbic acid, or catechin; all from Sigma-Aldrich). In some reactions, the reducing agent was replaced by 1.4 μm MtCDH. Additional substrates tested under the same conditions were: maltodextrin (DP4–14; Glucidex 9 from Roquette) mannohexaose, xylopentaose, xylohexaose, chitopentaose (all from Megazyme), α-chitin from shrimp shell (HovBio, Tromsø, Norway), and nanofibrillar α-chitin and β-chitin (10–20 nm) (22). All reactions were incubated in 2-ml screw cap microtubes (Sarstedt) at 50 °C in an Eppendorf Thermo mixer with (for insoluble substrates, 850 rpm) or without (for soluble substrates) shaking.
HPLC Analysis
From the standard reactions, samples were taken at different time points, and the reaction was stopped by adding NaOH to a final concentration of 0.05 m. After removing insoluble substrates by centrifugation, the supernatant was centrifuged and analyzed by high performance anion exchange chromatography (HPAEC) using an ICS3000 system (Dionex, Sunnivale, CA) as described previously (12). In brief, a 2-μl sample was injected on a CarboPac PA1 2 × 250 mm analytical column (Dionex) coupled to a CarboPac PA1 2 × 50 mm guard column kept at 30 °C. Cello-oligosaccharides were eluted at 0.25 ml/min using a stepwise linear gradient from 100% eluent A (0.1 m NaOH) toward 10% eluent B (1 m NaOAc in 0.1 m NaOH) 10 min after injection and 30% eluent B 25 min after injection, followed by a 5 min exponential gradient to 100% B. The column was reconditioned between each run by running initial conditions for 9 min.
Analysis by Mass Spectrometry
For time resolved product analysis, electrospray ionization mass spectrometry (ESI-MS) was used with a linear ion trap LTQ Velos Pro (Thermo Scientific, San Jose, CA USA) coupled to an UltiMate 3000 RS UHPLC from Dionex (Sunnyvale, CA USA) which delivered a constant flow and performed injection. No chromatographic separation was employed. The UHPLC delivered a flow of 0.2 ml/min of 30/70 (v/v) H2O and acetonitrile via the auto-sampler. Standard reaction mixtures were incubated in the thermostatted auto-sampler of the UHPLC at 15 °C during the entire reaction time and samples of 2 μl were injected at given time points. The electrospray was operated in positive mode at 4 kV spray current, with a sheath gas flow of 30 (arbitrary units), an auxiliary gas flow of 5 (arbitrary units) and a capillary temperature of 250 °C. The acquisition time was set to 0.2 min with a data collection time of 10 ms per acquisition. Full scans were performed in the m/z 100–1000 mass range and fragmentation was done using higher energy collisional dissociation (HCD) with N2 as the collision gas and normalized energy levels of 65, to enable observation of lower mass fragments. During fragmentation, data were collected in the m/z 100–400 mass range. The data were further processed using Xcalibur 2.2 SP1.48 (Thermo Scientific).
NMR Analysis
For NMR analysis, 1.0 mg/ml cellopentaose was dissolved in 99.996% D2O (Cambridge Isotope Laboratories, Andover, MA) containing 5 mm sodium acetate (pD 6.0) and 0.1 ‰ (v/w) 3-(trimethylsilyl)-propionic-2,2,3,3-d4 acid sodium salt (Aldrich, Milwaukee, WI) used as chemical shift reference for proton and carbon. The NMR tubes contained 500 μl of cellopentaose stock solution and 33 μl of either MtCDH (to a final concentration of 0.9 μm) or hydroquinone solution (to a final concentration of 3 or 10 mm). After addition of 17 μl NcLPMO9C (to a final concentration of 2.9 μm), the head space of the NMR tube was flushed with pure oxygen gas (YARA, Trondheim, Norway) for ∼10 s before sealing the tube. After incubation of the samples at 25 °C for 24 h, reaction products were analyzed with NMR spectroscopy.
All homo- and heteronuclear NMR experiments were recorded on a Bruker Avance 600 MHz NMR spectrometer (Bruker BioSpin AG, Fällanden, Switzerland) equipped with a 5-mm cryogenic CP-TCI z-gradient probe at 25 °C. For chemical shift assignment, the following spectra were recorded: one-dimensional proton, two-dimensional double quantum filter correlated spectroscopy (DQF-COSY), two-dimensional in-phase correlation spectroscopy (IP-COSY) (23), two-dimensional total correlation spectroscopy (TOCSY) with 70 ms of mixing time, two-dimensional 13C heteronuclear single quantum coherence (HSQC) with multiplicity editing, two-dimensional 13C HSQC-[1H,1H]TOCSY with 70 ms of mixing time on protons, and two-dimensional heteronuclear multibond correlation (HMBC) with BRID filter to suppress first order correlation. The NMR data were processed and analyzed with TopSpin 2.1 and TopSpin 3.0 software (Bruker BioSpin).
H2O2 Analysis
A fluorimetric assay based on Amplex Red and horseradish peroxidase (17) was used to measure the extent of H2O2 generation, which is a futile side reaction catalyzed by the reduced LPMO copper center. The peroxidase catalyzed conversion of Amplex Red to resorufin is proportional to H2O2 production (stoichiometry = 1). The increase of fluorescence was measured with an Enspire Multimode plate reader (PerkinElmer Life Sciences) using an excitation wavelength of 569 nm and an emission wavelength of 585 nm. The well plate assay (total volume of 200 μl, 30 °C, 6 min) was performed in 100 mm potassium phosphate buffer, pH 6.0, containing 50 μm Amplex Red, 7.1 units ml−1 horseradish peroxidase, 0.87 μm LPMO, and 30 μm ascorbate as reductant in 100 mm sodium phosphate buffer, pH 6.0. Cello-oligosaccharides (DP2–6; Sigma-Aldrich) were added to a final concentration of 5 mm.
Sequence Alignment and Modeling
A structure-guided sequence alignment to compare NcLPMO9C with two structurally characterized AA9-type LPMOs, NcLPMO9D (NCU01050; Protein Data Bank code 4EIR), and PcLPMO9D (PcGH61D; Protein Data Bank code 4B5Q), was constructed using the Expresso mode of the T-Coffee multiple sequence alignment server (24). A homology model of NcLPMO9C was made based on a structure prediction by HHpred (25) and by using Modeler (26) with the crystal structure of NcLPMO9D as a template. All structural comparisons were carried out using PyMOL (PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger, LLC). The coordinates for cellopentaose were derived from Protein Data Bank entry 2EEX (27).
RESULTS AND DISCUSSION
Enzyme Activity on Cellulose and Cello-oligosaccharides
Initial activity screening of the enzyme was done with a well established HPAEC method for the detection of C1-oxidized cello-oligosaccharide products (12). Incubation of NcLPMO9C with polymeric substrates (PASC, Avicel, steam-exploded spruce; Fig. 2A) showed release of soluble cello-oligosaccharides (DP2 and DP3), but C1 oxidized species were not detected. Instead, two dominant later eluting peaks (between 26 and 29 min) were observed that could represent a different kind of oxidized product. Similar late eluting peaks have also been observed for PASC degradation by NcLPMO9D (8). MS analysis (described in detail below) indicated that the later eluting of these two peaks represented a trimeric product, whereas the earlier peak represented a dimeric product.
FIGURE 2.

Degradation of cellulose and cello-oligosaccharides by NcLPMO9C. A shows overlaid HPAEC chromatograms of oligosaccharides released upon degradation of 1 mg/ml of PASC, Avicel, and steam-exploded spruce with 8.8 μm NcLPMO9C. B shows products released upon incubation of 0.4 mg/ml cello-oligosaccharides with 4.4 μm NcLPMO9C (Glcn traces), a standard mixture of nonoxidized cello-oligosaccharides (DP2–5, Glc2–5 trace), as well as products released upon incubation of cellopentaose with 4.4 μm NcLPMO9C and 1.4 μm MtCDH (CDH trace). All reaction mixtures contained 2 mm ascorbic acid (except the one containing CDH) and 5 mm ammonium acetate buffer, pH 6.0, and were incubated for 24 h at 50 °C. Reactions with insoluble substrates were incubated with horizontal shaking at 850 rpm. For product nomenclature, see Fig. 1. See text for further details.
Cellulose-active LPMOs characterized so far typically produce oligosaccharides with a DP up to 6 or 7 (5–7, 14). The production of relatively short oligosaccharides by NcLPMO9C could be the result of a glucanase background activity in the enzyme sample or by NcLPMO9C having activity on soluble oligosaccharides. Fig. 2B shows that NcLPMO9C indeed is able to oxidatively degrade cello-oligosaccharides. The enzyme readily degraded Glc5 and Glc6, while showing lower activity on Glc4 and no or minute activity on Glc3. There is abundant evidence for these conversions being caused by NcLPMO9C and not an impurity in the enzyme preparation: 1) the reaction generates oxidized species (see below for detailed characterization), 2) product formation was not observed in the absence of a reducing agent (results not shown), and 3) product formation was not observed in the presence of reducing agent only (results not shown). The oxidized products released were mainly DP2 from Glc4 and Glc5 and a mix of DP2 and DP3 from Glc6 (Fig. 2B).
The degradation patterns shown in Fig. 2 (A and B) were independent of the reductant used (we tested hydroquinone, ascorbic acid, and catechin in concentrations varying from 1.5 to 10 mm). Fig. 2B (CDH trace) further shows that use of MtCDH as electron donor for degradation of Glc5 led to a clear change in the product profile. As expected, native oligosaccharides were no longer observed because they were oxidized to aldonic acids. The oxidized species observed between 26 and 29 min for the reactions with a reducing agent present were not seen, but a new peak appeared at ∼40 min that may represent double oxidized species, an interpretation that is supported by mass spectrometry and NMR analysis of the samples (see below). This shift clearly shows that the products eluting between 25 and 30 min cannot be aldonic acids, because the CDH oxidizes reducing ends.
MS analysis confirmed the presence of only two major products upon incubating NcLPMO9C with Glc5, namely an oxidized dimer and a nonoxidized trimer (see below for details). Nevertheless, the chromatograms (Fig. 2) show additional peaks eluting in between the native and the main oxidized products. We propose that this is due to tautomerization, because keto groups on ring carbons are prone to this process at the elevated pH values used during the HPAEC runs. Migration of the 4-keto group to C3 and maybe C2 will change the elution behavior of the oligosaccharides.
NcLPMO9C is the first LPMO unequivocally shown to be active on soluble cello-oligosaccharides. The fact that the pentamer is degraded faster than the tetramer and the clear preference for releasing an oxidized dimer from Glc5 indicates the presence of at least five subsites on the enzyme running from −3 to +2 (subsites numbered according to the nomenclature used for glycoside hydrolases) (28). Degradation of Glc6 yielded both DP2 and DP3 oxidized species, indicating binding to −4 to +2 and −3 to +3. Notably, the location of these subsites and the orientation of the productively bound substrate relative to the catalytic center are currently unknown.
To test whether the cello-oligosaccharide oxidizing activity of NcLPMO9C is specific or an unspecific side reaction, the enzyme was incubated with other oligosaccharides (Man6, Xyl5, Xyl6, chitopentaose, and maltodextrin) and both crystalline α-chitin and nanofibrillar α- and β-chitin. No activity was observed on any of these substrates. Thus, NcLPMO9C seems to be specific for β-1–4-linked glucose units.
Product Identification by MS
MS analysis of product mixtures showed that the main products from degradation of Glc5 were two species with m/z values of 527 and 381, corresponding to the sodium adducts of Glc3 and a cellobiose with a gemdiol (i.e., a hydrated keto group) at the nonreducing end, respectively. Minor amounts of cellobiose and oxidized cellotriose were also detected. To investigate this further, the time course of the enzyme reaction was studied in H218O. This resulted in the same signal of m/z 527, whereas a new peak at m/z 383 appeared (Fig. 3A). This shows that the oxygen of the glycosidic linkage remains in the Glc3 product as the OH group at C1, whereas the oxidized product acquires one oxygen atom from water and one from oxygen as schematically shown in Fig. 3B. As indicated in Fig. 3A, a m/z 385 peak appears after some time, which is due to the lactone-gemdiol equilibrium leading to exchange with water and, thus, to the eventual incorporation of two 18O atoms.
FIGURE 3.

A, ESI-MS analysis of products generated over time during degradation of Glc5 (m/z 851) in H218O. Reaction products were analyzed after 0, 5, 15, 25, 90, and 1440 min of incubation, and the main picture shows the full spectrum for the 90-min sample. The insets show the signal over time for key products (from 0 to 1440 min, from left to right; the blue signal represents the 90-min sample). The m/z values 851, 527, and 365 correspond to the sodium adducts of Glc5, Glc3, and Glc2, respectively (m/z 829 is [M+H]+ for Glc5), whereas m/z 383 and the minor m/z 543 correspond to [M+Na]+ of gemdiol products (Glc4GemGlc and Glc4GemGlc2) carrying one 18O (see Fig. 1; the mass difference between sodium and lithium is 16 Da). The 383 signal is accompanied by a signal at m/z 385 because of exchange with the solvent, which gradually leads to incorporation of two 18O atoms. The mass of 363 corresponds to the keto-form of the oxidized dimer ([M+Na]+), and its intensity corresponds to ∼10% of the intensity of the gemdiol. B, schematic presentation of enzymatic cleavage of the glycosidic bond and introduction of molecular oxygen in the nonreducing end, followed by water incorporation transforming the keto group into a gemdiol. The incorporation of water leads to uptake of 18O in the products.
Because the glycosidic bond in cello-oligosaccharides (and cellulose) links the C1 of one glucose to the C4 of the adjacent glucose, it is logical to suggest that the oxidation carried out by NcLPMO9C takes place at the C4 position of the nonreducing end moiety. The resulting keto sugar will be in equilibrium with the C4 gemdiol in water solution (a feature that is common to keto saccharides (29)). Generally, it is not straightforward to prove the position of LPMO generated oxidations using mass spectrometry because the masses of various possible products are identical and because the mass difference between sodium and potassium adducts equals the mass of an oxygen atom. As shown in Fig. 1, the aldonic acid and gemdiol forms have identical masses, as do the corresponding lactone and keto forms. Thus, MS analysis alone cannot determine the type of LPMO activity. Exploiting the low complexity product mixtures obtainable thanks to the activity of NcLPMO9C on soluble substrates, we have addressed this issue by carrying out MS/MS analyses of products after lithium doping, which facilitates reducing end cross-ring fragmentation (30), as well as by NMR analyses.
Fig. 4 shows the result of MS/MS analysis of the m/z 365 [M+Li]+ products (oxidized dimers) generated by NcLPMO9C and C1 oxidizing PcLPMO9D (7). The spectra show differences including the occurrence of fragment ions that are diagnostic for C1 or C4 oxidations. Cellobionic acid (fragments indicated in blue in Fig. 4) readily loses mass corresponding to one carboxyl group (m/z 319). The C4 oxidized dimer (fragments indicated in black in Fig. 4) readily loses masses corresponding to both one and two water molecules (m/z 347 and 329) but does not generate ions corresponding to the loss of a carboxyl group. In addition, the most prevalent Y1 and Z1 ions (31) from glycosidic bond cleavage will be different for the two oxidized compounds. The Y1 and Z1 ions for glycosidic bond cleavage of cellobionic acid have m/z values of 203 and 185, respectively, species that include the carboxylic acid. The Y1 and Z1 ions for the C4 oxidized dimer have m/z values of 187 and 169, respectively, meaning no oxidation in the reducing end. The corresponding B1 and C1 ions (for the C4 oxidized dimer) are m/z 185 and 203, respectively, and tend to lose a water molecule, generating m/z 167 and 185 species, respectively. Loss of a water molecule from the B1 and C1 ions seems a likely event considering the presence of the gemdiol group at C4.
FIGURE 4.

MS/MS analysis of reaction products. The picture shows overlaid MS/MS spectra obtained for the cellobionic acid generated by PcLPMO9D (left structure; blue trace and labeling) and the oxidized dimer from an NcLPMO9C catalyzed reaction (right structure; black trace and labeling). The analysis was done with lithium doping and all labeled signals reflect lithium adducts.
Notably, comparison of the blue and black spectra in Fig. 4 shows that MS/MS analysis can reveal whether an LPMO oxidizes the reducing or the nonreducing end of the substrate. Distinct m/z ions for cellobionic acid are 319, 203, and 157. For Glc4gemGlc, distinct m/z ions are 347, 329, and 287. A relatively high m/z 187 signal is also typical for Glc4gemGlc. In the case CDH is used as the electron donor for a C4-oxidizing LPMO, double oxidized products will emerge (Fig. 2B, CDH trace) with yet another characteristic MS/MS pattern, as illustrated in Fig. 5. Lithium and sodium adducts of MS/MS fragments obtained for the different LPMO products are summarized in Table 1.
FIGURE 5.

MS/MS analysis of a double oxidized dimer (Glc4gemGlc1A) generated by treating cellopentaose with NcLPMO9C and MtCDH. The analysis was done with lithium doping, and all labeled signals reflect lithium adducts.
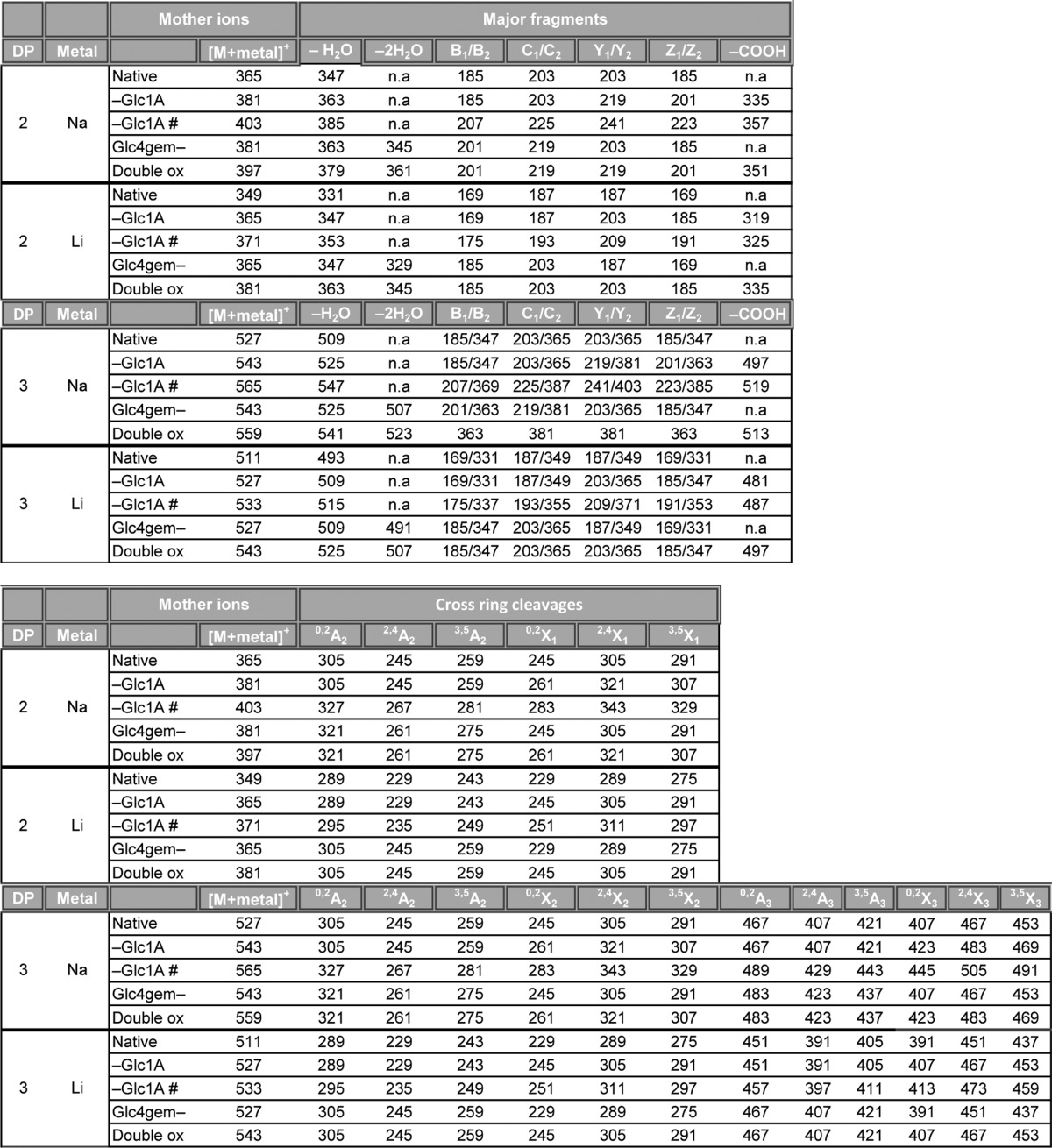
TABLE 1.
Fragmentation table for MS/MS analysis of native and oxidized cellobiose and cellotriose compounds
The table shows the m/z values of sodium and lithium adducts of the different fragments. n.a., not analyzed. # represents [M+2Metal-H]+. Double ox represents a double oxidized oligosaccharide with a gemdiol in the nonreducing end and an aldonic acid in the downstream end. Fragmentation nomenclature according to Domon and Costello (31).
Product Identification by NMR
To obtain proof for the identity of the oxidized reaction products, NMR spectroscopy was used to analyze products generated from cellopentaose by NcLPMO9C in the presence of either CDH or hydroquinone (Fig. 6). The individual monosaccharide residues were assigned by starting at the anomeric signal and/or at the primary alcohol group at C6 and then following the proton-proton connectivity using TOCSY, DQF-COSY/IP-COSY, and 13C HSQC-[1H,1H]TOCSY, whereas connectivity between the individual monosaccharide residues was obtained from the HMBC spectrum (see Table 2 for assignment of chemical shifts). These experiments showed the presence of dimeric and trimeric products in both reactions, as expected from the MS analyses described above. An overlay of the 13C HSQC spectra of product mixtures obtained in the presence of hydroquinone or CDH (Fig. 6A) readily shows different occurrence of oxidations. C4 oxidation occurs in both reactions, whereas signals reflecting C1 oxidation are only observed in the reaction with CDH (red signals in Fig. 6A). There were no indications of oxidation of C6 because this part of the 13C HSQC spectrum was essentially identical to that observed for nontreated cellopentaose and because no novel signals possibly reflecting additional oxidations were observed.
FIGURE 6.

NMR analysis of reaction products. A, overlay of 13C HSQC spectra for reaction products generated by treating 0.9 mg/ml cellopentaose with 2.9 μm NcLPMO9C in the presence of 10 mm hydroquinone (black signals) or 0.9 μm MtCDH (red signals). The samples were both in 99.996% D2O with 5 mm sodium acetate, pH 6.0, and spectra were recorded at 25 °C. Peaks in the proton/carbon signals of the C4 oxidized monosaccharide residue are marked by H/C#, where # refers to the ring carbon number (overlapping red and black signals). Peaks in the proton/carbon signals of the C1 oxidized monosaccharide residue are marked by H/C#* (only red signals). For the sake of simplicity, peaks related to nonoxidized monosaccharide residues are not marked (a full assignment of chemical shifts is provided in Table 2). B, two details of an overlay of a 13C HSQC spectrum and a 13C HMBC spectrum recorded for products obtained in a reaction with both NcLPMO9C and CDH. The left panel shows a correlation (indicated by a vertical line) from the H/C2* peak in HSQC (red) to a peak with a carbon chemical shift of 181.1 ppm in HMBC (blue), corresponding well to the (expected) presence of a carboxylate group at position C1. The right panel shows correlation from the H/C3 and H/C5 peaks in HSQC (red) to carbon peaks with a carbon chemical shift at C4 of 95.9 and 175.2 ppm in HMBC (blue), corresponding to the presence of a gemdiol and a keto group at C4, respectively.
TABLE 2.
Assignment of chemical shifts
The individual monosaccharide residues were assigned by starting at the anomeric signal and/or at the primary alcohol group at C6 and then following the proton-proton connectivity using TOCSY, DQF-COSY/IP-COSY, and 13C HSQC-TOCSY spectra. 13C HSQC was used for assigning the carbon chemical shifts. The 13C HMBC spectrum provided long range bond correlations, allowing identification of the connectivity between the sugar units and allowing identification of the carboxyl group at C1, as well as the gem-diol group and keto group at C4. Chemical shifts were assigned for the major products resulting from treatment of 0.9 mg/ml cellopentaose with 2.9 μm NcLPMO9C in the presence of 0.9 μm MtCDH or 10 mm hydroquinone in 99.996% D2O with 5 mm sodium acetate pD 6.0, for spectra recorded at 25 °C. Numbers 1–6 represent ring carbon numbers to which the chemical shift values (1H, 13C or 1H, 1H, 13C for C6) are assigned for a nonreducing end glucose (NR), a second after NR glucose (SNR), the α anomer of glucose (α), the β anomer of glucose (β), the aldonate glucose (C1), and the keto/gemdiol glucose (C4). The chemical shifts reported are with accuracy of 0.01 ppm for 1H and 0.1 ppm for 13C, based on spectral resolution.
| Glucose unit/carbon number | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Hydroquinone, Glc3 | ||||||
| NR | 4.51; 105.4 | 3.32; 76.0 | 3.49; 78.6 | 3.51; 78.4 | 3.42; 72.3 | 3.93; 3.73; 63.5 |
| SNR | 4,54; 105.1 | 3.37; 75.8 | 3.67; 77.0 | 3.62; 81.2 | 3.61; 77.7 | 3.97; 3.83; 62.8 |
| α | 5.22; 94.7 | 3.57; 74.1 | 3.83; 74.1 | 3.65; 81.5 | 3.95; 73.0 | 3.96; 3.83; 62.8 |
| β | 4.66; 98.6 | 3.29; 76.8 | 3.65; 77.6 | 3.61; 81.3 | 3.63; 77.1 | 3.98; 3.87; 62.8 |
| Hydroquinone, Glc4gemGlc | ||||||
| C4 | 4.55; 105.5 | 3.41; 75.3 | 3.55; 78.5 | 175.2/95.9 | 3.58; 80.4 | 4.02; 3.81; 61.7 |
| α | 5.22; 94.7 | 3.57; 74.1 | 3.83; 74.1 | 3.65; 81.5 | 3.95; 73.0 | 3.96; 3.83; 62.8 |
| β | 4.66; 98.6 | 3.29; 76.8 | 3.65; 77.6 | 3.61; 81.3 | 3.63; 77.1 | 3.98; 3.87; 62.8 |
| MtCDH, Glc2Glc1A | ||||||
| NR | 4.51; 105.3 | 3.32; 76.1 | 3.42; 78.4 | 3.50; 78.3 | 3.49; 72.3 | 3.91; 3.74; 63.4 |
| SNR | 4.65; 105.6 | 3.41; 76.1 | 3.66; 76.9 | 3.67; 81.4 | 3.60; 77.8 | 3.97; 3.82; 62.8 |
| C1 | 181.1 | 4.18; 75.2 | 4.10; 74.4 | 4.01; 84.51 | 3.99; 74.7 | 3.85; 3.76; 64.8 |
| MtCDH, Glc4gemGlc1A | ||||||
| C4 | 4.53; 105.3 | 3.44; 75.5 | 3.55; 78.6 | 175.2/95.9 | 3.65; 80.3 | 3.82; 3.98; 61.8 |
| C1 | 181.1 | 4.18; 75.2 | 4.10; 74.4 | 4.01; 84.51 | 3.99; 74.7 | 3.85; 3.76; 64.8 |
13C HMBC spectra provided further insight into the nature of the products. This is illustrated by Fig. 6B, showing an overlay of the 13C HSQC and the 13C HMBC spectra of products obtained in the reaction with NcLPMO9C and CDH. The overlay shows correlations from H/C-5 and H/C-3 to peaks with a carbon chemical shift at C4 of 95.9 and 175.2 ppm, corresponding well to the chemical shift of a gemdiol (29) and keto group, respectively. The gemdiol and keto groups account for ∼80 and 20% of the signal intensity, respectively. Although documentation on the keto:gemdiol ratio in literature is limited, the gemdiol would be expected to dominate at pH 6.0 because the keto group is easily hydrated (29) in aqueous medium. The overlay also shows a correlation from the H/C-2 to a peak with a carbon chemical shift of 181.1 ppm that corresponds well to the shift expected when a carboxylate group is present at position C1 (12). Even though there are two different Glc1A groups in the product mixture (one in a trimeric and one in a dimeric product), their chemical shifts are very similar, meaning that the peaks appear nearly at the same position in the spectra, looking like one broad peak (see Table 2 for more details). Thus, for the first time, NMR has been used to prove that the products generated by a nonreducing end active LPMO are oxidized in the C4 position and that the products primarily exist as a gemdiol.
Suppression of H2O2 Production
In the absence of a cellulosic substrate, activated LPMOs produce H2O2 (17). We employed this ability to demonstrate binding of cello-oligomeric substrates to NcLPMO9C. Fig. 7 shows that production of H2O2 was diminished in the presence of cello-oligosaccharides with a DP >4. Although only minor inhibition was observed in the presence of Glc4, almost complete inhibition of H2O2 formation was observed in the presence of Glc6. Thus, the data in Fig. 7 show that cello-oligomers with a minimal length of five sugars form complexes with the enzyme that are sufficiently strong to suppress the futile H2O2 generating side reaction. This corresponds very well with the HPLC data, which showed high activity for Glc5 and Glc6 and much lower activity for Glc4 (Fig. 2B). As mentioned above, NcLPMO9C is not active on Xyl5 or Man6, and Fig. 7 shows that these substrates do not suppress the H2O2 production.
FIGURE 7.
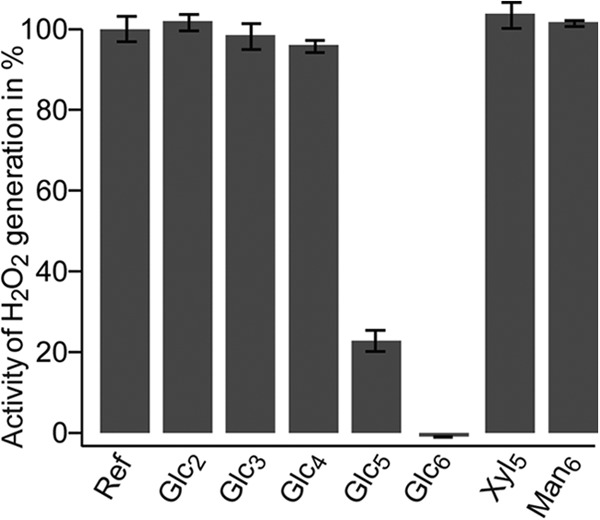
Generation of H2O2 by NcLPMO9C. The enzyme was incubated with reductant and the production of H2O2 (here referred to as activity) was measured as described under “Experimental Procedures.” The figure shows that the enzyme generates H2O2 in the absence of oligosaccharides (Ref) and in the presence of Xyl5, Man6, or short cello-oligosaccharides, whereas cello-oligosaccharide with DP > 4 diminish H2O2 production, indicative of productive binding to the LPMO. The reported results are mean values of five experiments. Control experiments without the reductant or the LPMO showed no H2O2 production, regardless of the presence of cello-oligosaccharides.
Sequence and Structural Model of NcLPMO9C
For comparison, the sequence of NcLPMO9C was aligned to a C1 oxidizing and a nonreducing end oxidizing LPMO. The AA9 domain of NcLPMO9C shares 36.6% sequence identity with C1-oxidizing PcLPMO9D (7) and 47.5% with nonreducing end oxidizing NcLPMO9D (8, 16) (Fig. 8A). The metal coordinating residues (His-1 and His-83 in NcLPMO9C) as well as four residues surrounding the copper site (Asn-26, His-155, Gln-164, and Tyr-166 in NcLPMO9C) are conserved in all three AA9s. Looking at the LPMO structures available, AA9s tend to contain several surface-exposed aromatic residues that seem to be aligned to interact with a cellulose chain that would then traverse the catalytic center (32). Indeed, these aromatic residues have roughly the same spatial orientation as residues that, by experiment, have been shown to interact with chitin in an AA10 LPMO (33). This adds confidence to the notion that these aromatic residues interact with the substrate, possibly analogous to what is been suggested for PcLPMO9D (32).
FIGURE 8.

Sequence and structural comparisons of PcLPMO9D, NcLPMO9D and NcLPMO9C. A, structure-guided sequence alignment of PcLPMO9D, NcLPMO9D, and NcLPMO9C with fully conserved residues shaded in gray. Residues involved in coordination of the copper are marked with asterisks above the sequence (His-1, His-83 and Tyr-166 in NcLPMO9C), whereas aromatic surface residues and protruding polar surface residues potentially involved in substrate binding are colored in red (all highlighted in B). Cysteines forming disulfide bonds are indicated in boxes with numbers above showing which cysteines are connected. B, side chains on the substrate binding surface of PcLPMO9D (Protein Data Bank code 4B5Q) and NcLPMO9D (Protein Data Bank code 4EIR) compared with the modeled structure of NcLPMO9C. Copper coordinating residues are colored dark gray, aromatic and protruding polar residues putatively involved in substrate binding are colored yellow and violet, respectively, and the copper is shown as an orange sphere. For illustration purposes only, a cellopentaose (coordinates derived from Protein Data Bank code 2EEX; shown in green with oxygens in red) is placed above the surface of the modeled structure of NcLPMO9C. The view in the right panels is rotated 90° relative to the view in the left panels, looking down at the flat surface containing the copper binding site.
Based on the crystal structure of NcLPMO9D a model was built for NcLPMO9C. Comparison of the surface-exposed residues potentially involved in substrate binding (Fig. 8B) shows that there are more aromatic residues on the surfaces of NcLPMO9D and PcLPMO9D compared with NcLPMO9C. Because of this difference, the length of the substrate-binding surface seems shorter in NcLPMO9C. Even if one considers the contribution of protruding polar residues on the surface, the binding surface of NcLPMO9C seems less extended (Fig. 8B) and possibly more adapted to binding shorter substrates compared with PcLPMO9D and NcLPMO9D. Interestingly, the structural model of NcLPMO9C indicates that this enzyme has a cluster of three asparagine residues (Asn-25, Asn-26, and Asn-27) in a location that could potentially be a +2 subsite. One could speculate that these asparagines interact with the reducing end sugar. Indeed, as illustrated in Fig. 8B, a cellopentaose would span the putative binding surface of NcLPMO9C, when its reducing end is positioned near these asparagines. Such an orientation would be in accordance with the observation that cellopentaose primarily is cleaved into a cellotriose and a Glc4gemGlc.
Conclusions
In this article, we have shown that NcLPMO9C cleaves both crystalline cellulose as well as cello-oligosaccharides yielding products oxidized in the nonreducing end. By applying NMR, we unambiguously showed that the nonreducing end sugar was oxidized at the C4 position and that this sugar primarily exists as a gemdiol in solution. We have also shown that MS/MS fragmentation analysis of oxidized products can be applied to differentiate between C1 and C4 oxidizing LMPOs. A range of different substrates were tested, but NcLPMO9C was only active on β-1,4-linked glucose units, and the enzyme seemed to require a minimum stretch of at least four glucose units. It has not escaped our attention that such an activity on short cello-oligosaccharides could imply that the enzyme is active on hemicellulose structures containing β-1,4-linked glucose units.
Acknowledgments
We thank Prof. Johannes Vliegenthart and Dr. Derek Horton for suggesting IUPAC nomenclature for the enzyme products. We thank Morten Skaugen for help in managing our HPLC and MS systems.
This work was supported by Norwegian Research Council Projects 193817, 203402, 214613, 216162, and 217708 and by the European Commission through the FP7-KBBE-2013-7-613549 project INDOX.
- LPMO
- lytic polysaccharide monooxygenase
- CBM
- carbohydrate-binding module
- GH
- glycoside hydrolase
- AA
- auxiliary activities
- DP
- degree of polymerization
- CDH
- cellobiose dehydrogenase
- PASC
- phosphoric acid swollen cellulose
- HPAEC
- high performance anion exchange chromatography
- DQF-COSY
- double quantum filter correlation spectroscopy
- IP-COSY
- in-phase COSY
- TOCSY
- total correlation spectroscopy
- HSQC
- heteronuclear single quantum coherence
- HMBC
- heteronuclear multibond correlation
- Glc4gemGlc
- 4-hydroxy-β-d-xylo-hexopyranosyl-(1→4)-β-d-glucopyranosyl, 4-hydroxy-β-d-xylo-Hexp-(1→4)-β-d-Glcp
- Glc4KGlc2
- β-d-xylo-hexos-4-ulopyranosyl-(1→4)-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl, β-d-xylo-Hex4ulop-(1→4)-β-d-Glcp-(1→4)-β-d-Glcp
- Glc4gemGlc2
- 4-hydroxy-β-d-xylo-hexopyranosyl-(1→4)-β-d-glucopyranosyl-(1→4)-β-d-glucopyranosyl, 4-hydroxy-β-d-xylo-Hexp-(1→4)-β-d-Glcp-(1→4)-β-d-Glcp
- Glc4KGlcGlc1A
- β-d-xylo-hexos-4-ulopyranosyl-(1→4)-β-d-glucopyranosyl-(1→4)-d-gluconic acid, β-d-xylo-Hex4ulop-(1→4)-β-d-Glcp-(1→4)-d-Glc1A
- Glc4gemGlcGlc1A
- 4-hydroxy-β-d-xylo-hexopyranosyl-(1→4)-β-d-glucopyranosyl-(1→4)-d-gluconic acid, 4-hydroxy-β-d-xylo-Hexp-(1→4)-β-d-Glcp-(1→4)-d-Glc1A.
REFERENCES
- 1. Horn S. J., Vaaje-Kolstad G., Westereng B., Eijsink V. G. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cantarel B. L., Coutinho P. M., Rancurel C., Bernard T., Lombard V., Henrissat B. (2009) The carbohydrate-active enzymes database (CAZy). An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Levasseur A., Drula E., Lombard V., Coutinho P. M., Henrissat B. (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 5. Forsberg Z., Vaaje-Kolstad G., Westereng B., Bunæs A. C., Stenstrøm Y., MacKenzie A., Sørlie M., Horn S. J., Eijsink V. G. (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci. 20, 1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Westereng B., Ishida T., Vaaje-Kolstad G., Wu M., Eijsink V. G., Igarashi K., Samejima M., Ståhlberg J., Horn S. J., Sandgren M. (2011) The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS One 6, e27807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Phillips C. M., Beeson W. T., Cate J. H., Marletta M. A. (2011) Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 6, 1399–1406 [DOI] [PubMed] [Google Scholar]
- 9. Langston J. A., Shaghasi T., Abbate E., Xu F., Vlasenko E., Sweeney M. D. (2011) Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 77, 7007–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sygmund C., Kracher D., Scheiblbrandner S., Zahma K., Felice A. K., Harreither W., Kittl R., Ludwig R. (2012) Characterization of the two Neurospora crassa cellobiose dehydrogenases and their connection to oxidative cellulose degradation. Appl. Environ. Microbiol. 78, 6161–6171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hemsworth G. R., Taylor E. J., Kim R. Q., Gregory R. C., Lewis S. J., Turkenburg J. P., Parkin A., Davies G. J., Walton P. H. (2013) The copper active site of CBM33 polysaccharide oxygenases. J. Am. Chem. Soc. 135, 6069–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Westereng B., Agger J. W., Horn S. J., Vaaje-Kolstad G., Aachmann F. L., Stenstrøm Y. H., Eijsink V. G. (2013) Efficient separation of oxidized cello-oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J. Chromatogr. A 1271, 144–152 [DOI] [PubMed] [Google Scholar]
- 13. Bey M., Zhou S., Poidevin L., Henrissat B., Coutinho P. M., Berrin J.-G., Sigoillot J.-C. (2013) Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (Family GH61) from Podospora anserina. Appl. Environ. Microbiol. 79, 488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beeson W. T., Phillips C. M., Cate J. H., Marletta M. A. (2012) Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 134, 890–892 [DOI] [PubMed] [Google Scholar]
- 15. Tian C., Beeson W. T., Iavarone A. T., Sun J., Marletta M. A., Cate J. H., Glass N. L. (2009) Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc. Natl. Acad. Sci. U.S.A. 106, 22157–22162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X., Beeson W. T., 4th, Phillips C. M., Marletta M. A., Cate J. H. (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kittl R., Kracher D., Burgstaller D., Haltrich D., Ludwig R. (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zámocký M., Schumann C., Sygmund C., O'Callaghan J., Dobson A. D., Ludwig R., Haltrich D., Peterbauer C. K. (2008) Cloning, sequence analysis and heterologous expression in Pichia pastoris of a gene encoding a thermostable cellobiose dehydrogenase from Myriococcum thermophilum. Protein Expr. Purif. 59, 258–265 [DOI] [PubMed] [Google Scholar]
- 19. Flitsch A., Prasetyo E. N., Sygmund C., Ludwig R., Nyanhongo G. S., Guebitz G. M. (2013) Cellulose oxidation and bleaching processes based on recombinant Myriococcum thermophilum cellobiose dehydrogenase. Enzyme Microb. Technol. 52, 60–67 [DOI] [PubMed] [Google Scholar]
- 20. Harreither W., Sygmund C., Augustin M., Narciso M., Rabinovich M. L., Gorton L., Haltrich D., Ludwig R. (2011) Catalytic properties and classification of cellobiose dehydrogenases from Ascomycetes. Appl. Environ. Microbiol. 77, 1804–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wood T. M. (1988) Preparation of crystalline, amorphous, and dyed cellulase substrates. Methods Enzymol. 160, 19–25 [Google Scholar]
- 22. Ifuku S., Saimoto H. (2012) Chitin nanofibers. Preparations, modifications, and applications. Nanoscale 4, 3308–3318 [DOI] [PubMed] [Google Scholar]
- 23. Xia Y., Legge G., Jun K. Y., Qi Y., Lee H., Gao X. (2005) IP-COSY, a totally in-phase and sensitive COSY experiment. Magn. Reson. Chem. 43, 372–379 [DOI] [PubMed] [Google Scholar]
- 24. Armougom F., Moretti S., Poirot O., Audic S., Dumas P., Schaeli B., Keduas V., Notredame C. (2006) Expresso. Automatic incorporation of structural information in multiple sequence alignments using 3D-coffee. Nucleic Acids Res. 34, W604–W608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Söding J., Biegert A., Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 27. Kitago Y., Karita S., Watanabe N., Kamiya M., Aizawa T., Sakka K., Tanaka I. (2007) Crystal structure of Cel44A, a glycoside hydrolase family 44 endoglucanase from Clostridium thermocellum. J. Biol. Chem. 282, 35703–35711 [DOI] [PubMed] [Google Scholar]
- 28. Davies G. J., Wilson K. S., Henrissat B. (1997) Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 321, 557–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kopper S., Freimund S. (2003) The composition of keto aldoses in aqueous solution as determined by NMR spectroscopy. Helv. Chim. Acta 86, 827–843 [Google Scholar]
- 30. Cancilla M. T., Wong A. W., Voss L. R., Lebrilla C. B. (1999) Fragmentation reactions in the mass spectrometry analysis of neutral oligosaccharides. Anal. Chem. 71, 3206–3218 [DOI] [PubMed] [Google Scholar]
- 31. Domon B., Costello C. E. (1988) A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconj. J. 5, 397–409 [Google Scholar]
- 32. Wu M., Beckham G. T., Larsson A. M., Ishida T., Kim S., Payne C. M., Himmel M. E., Crowley M. F., Horn S. J., Westereng B., Igarashi K., Samejima M., Ståhlberg J., Eijsink V. G., Sandgren M. (2013) Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the basidiomycota fungus Phanerochaete chrysosporium. J. Biol. Chem. 288, 12828–12839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aachmann F. L., Sørlie M., Skjåk-Braek G., Eijsink V. G., Vaaje-Kolstad G. (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc. Natl. Acad. Sci. U.S.A. 109, 18779–18784 [DOI] [PMC free article] [PubMed] [Google Scholar]