

Background: Prion strains are believed to be enciphered by distinct conformations of misfolded prion protein (PrP).
Results: Strains of PrP amyloid with different conformational stabilities were found to have identical β-sheet core regions but different steric zipper interfaces.
Conclusion: Strain-specific differences in PrP amyloid stability are dictated by a packing polymorphism.
Significance: These findings have implications for understanding the structural basis of prion strains.
Keywords: Amyloid, Neurodegenerative Diseases, Prions, Protein Aggregation, Protein Misfolding
Abstract
Mammalian prion strains are believed to arise from the propagation of distinct conformations of the misfolded prion protein PrPSc. One key operational parameter used to define differences between strains has been conformational stability of PrPSc as defined by resistance to thermal and/or chemical denaturation. However, the structural basis of these stability differences is unknown. To bridge this gap, we have generated two strains of recombinant human prion protein amyloid fibrils that show dramatic differences in conformational stability and have characterized them by a number of biophysical methods. Backbone amide hydrogen/deuterium exchange experiments revealed that, in sharp contrast to previously studied strains of infectious amyloid formed from the yeast prion protein Sup35, differences in β-sheet core size do not underlie differences in conformational stability between strains of mammalian prion protein amyloid. Instead, these stability differences appear to be dictated by distinct packing arrangements (i.e. steric zipper interfaces) within the amyloid core, as indicated by distinct x-ray fiber diffraction patterns and large strain-dependent differences in hydrogen/deuterium exchange kinetics for histidine side chains within the core region. Although this study was limited to synthetic prion protein amyloid fibrils, a similar structural basis for strain-dependent conformational stability may apply to brain-derived PrPSc, especially because large strain-specific differences in PrPSc stability are often observed despite a similar size of the PrPSc core region.
Introduction
Transmissible spongiform encephalopathies, or prion diseases, are a group of fatal neurodegenerative disorders that afflict many mammalian species (1). Even though these diseases are clinically distinct, all of them appear to be intimately associated with conformational conversion of the normal cellular prion protein (PrPC)6 into a disease-causing aggregated isoform known as PrPSc (1–5). It is generally believed that PrPSc itself represents the infectious prion agent, which self-propagates by first binding to PrPC and then inducing conformational conversion of the protein into the PrPSc state. This “protein-only” model is supported by wealth of experimental data (1–6), including the recent success in generating infectious prions in vitro from bacterially expressed recombinant PrP (7–11).
One of the most confounding aspects of prion diseases is the existence of multiple “strains” that give rise to different disease phenotypes (distinguished by clinical signs, incubation time, and distinct neuropathology) that are faithfully maintained upon repeated passaging in laboratory animals (1, 3, 4, 12). Infectious PrPSc isolates from humans or animals associated with distinct disease phenotypes show several strain-specific characteristics, differing with respects to global and/or local secondary structure (13, 14), resistance to proteinase K digestion (15, 16), glycosylation profile, exposure of surface-exposed epitopes (17), and, most notably, conformational stability (18–23). Rationalized within the framework of the protein-only model, these different biochemical and biophysical characteristics have led to the hypothesis that multiple prion strains result from distinct conformational states of the PrPSc aggregate (1, 3, 4, 12).
Although progress in a detailed understanding of the molecular basis of mammalian prion strains has been slow (largely due to technical difficulties associated with structural studies using brain-derived PrPSc), some light has been shed on this phenomenon by recent studies using distinct strains of amyloid fibrils formed from the recombinant mouse PrP that induce prion diseases in transgenic mice overexpressing PrPC (21, 22). Interestingly, the conformational stability of prion isolates from these mice was found to correlate with the stability of amyloid fibrils used to generate them. Furthermore, a correlation was also found between the conformational stability of these synthetic prions and the incubation period of the disease, with less stable PrPSc aggregates corresponding to shorter incubation times (22). A similar phenomenon has been observed for the yeast prion [PSI+] associated with aggregation of the Sup35 protein, where a weaker prion state is induced by a Sup35 amyloid strain with higher conformational stability (24). In this case, structural studies revealed that this more stable amyloid strain is characterized by a marked expansion of the amyloid β-sheet core region as compared with that found in the less conformationally stable strain (25). However, no information is yet available regarding the structural basis for different stabilities of mammalian prions.
To bridge this gap, we generated two strains of recombinant human PrP (rPrP) amyloid with dramatically different conformational stabilities and characterized them by a host of biophysical techniques. In sharp contrast to previously studied Sup35 amyloid strains, we found that differences in rPrP amyloid conformational stability do not correspond to β-sheet core size but are dictated by differences in the assembly of the steric zipper interfaces within the core region.
EXPERIMENTAL PROCEDURES
Protein Purification
Full-length rPrP used in this study was prepared and purified according to previously described protocols (26). After purification, the protein was stored at −80 °C in 10 mm acetate buffer, pH 4.0. Protein concentration was determined spectrophotometrically using a molar extinction coefficient (ϵ280) of 57,900 m−1 cm−1.
Preparation of rPrP Amyloid Fibrils
rPrP amyloid fibrils were generated by incubating the protein (0.5 mg/ml) in 50 mm phosphate buffer, pH 6.5, containing either 2 or 4 m guanidinium chloride (GdmCl). The reaction was performed at 37 °C with continuous rotation (8 rpm). The progress of the reaction was monitored using a fluorometric thioflavin T assay (26, 27).
Fibril Denaturation Assay
For chemical denaturation assays, freshly prepared samples of amyloid fibrils prepared in buffer containing either 2 or 4 m GdmCl were washed with 50 mm phosphate buffer and 2 m GdmCl, pH 6.5. Samples were then resuspended to a concentration of 25 μm in 50 mm phosphate buffer, pH 6.5, containing either 0.5 m GdmCl (for GdmCl denaturation experiments) or guanidinium thiocyanate (GdmThCN; for GdmThCN denaturation experiments). These solutions were diluted 1:4 in buffer containing varying concentrations of GdmCl or GdmThCN and then incubated for 60 min at room temperature. Samples were mixed 1:20 with thioflavin T-containing buffer (50 mm phosphate and 20 μm thioflavin T, pH 6.5) that contained GdmCl or GdmThCN at the same concentration as in the incubation mixture. Immediately after mixing, thioflavin T fluorescence was measured at 480 nm using an excitation wavelength of 450. Fractional loss of signal at increasing denaturant concentrations corresponds to the fraction of rPrP dissociated from amyloid fibrils.
Atomic Force Microscopy (AFM)
For AFM experiments, 10 μl of the sample were deposited on freshly cleaved mica and left to adsorb for 1 min, and the sample was rinsed with 1 ml of water and dried gently using airflow. Imaging was performed in a tapping mode on a Digital Instruments MultiMode microscope equipped with a NanoScope IV controller (Veeco, Santa Barbara, CA) using RTESP7 silicon tips.
FTIR Spectroscopy
Amyloid fibril samples for infrared spectroscopy were washed twice with D2O buffer containing 10 mm phosphate (pD 6.5). Samples were placed between calcium fluoride windows separated with a 50-μm Teflon spacer. For each FTIR spectrum, 256 scans were collected and Fourier-transformed to yield a resolution of 2 cm−1. Spectra were corrected by subtracting a spectrum of buffer alone.
Backbone Amide Hydrogen/Deuterium (H/D) Exchange Mass Spectrometry (HXMS) Experiments
Amyloid fibrils were collected by centrifugation and washed three times with 10 mm phosphate buffer, pH 7.0, to remove any residual GdmCl. To initiate deuterium labeling, pellets were resuspended in 100 μl of 10 mm phosphate buffer, pH 7.0, prepared in D2O. After 24 h of incubation at room temperature, samples were collected by centrifugation at 16,000 × g for 5 min and rapidly dissociated into monomers by adding a solution of 6 m GdmThCN in exchange quenching buffer (0.1 m phosphate, pH 2.5) containing a reducing agent (0.1 m tris(2-carboxyethyl)phosphine hydrochloride). After 30 s of incubation at room temperature, the samples were diluted 50 times with ice-cold 0.05% trifluoroacetic acid in H2O and digested for 5 min with pepsin. The peptic fragments were collected in a peptide microtrap, washed to remove salts, and eluted on a C18 HPLC column using a gradient of 2–35% acetonitrile at a flow rate of 50 μl/min. Peptides separated on the column were analyzed using a Finnigan LTQ mass spectrometer (Thermo Electron Corp., San Jose, CA). The trap and the column were immersed in ice to minimize back-exchange. The extent of deuterium incorporation in each peptic fragment was determined as described previously (28, 29).
His-HXMS Experiments
These experiments were performed as described above for HXMS measurements with the exception that amyloid fibrils were incubated in D2O buffer (10 mm phosphate, pH 9.0) for 5 days at 37 °C. The pseudo-first-order rate constant (k) of the His hydrogen exchange reaction was determined by the following equation: k = −ln(1 − ((R(t) − R(0))/(1 + R(t) − R(0))) × 1/P)/t, where P is the fractional D2O content in the solvent, and R is the ratio of the M+1/M isotopic peak of a given peptide before (time = 0) and after (time = t) the hydrogen exchange reaction. The half-life (t½, days) of the His hydrogen exchange reaction was calculated using the following equation: t½ (day) = ln 2/k/24, where k (hour−1) is the rate constant under alkaline conditions, pH 9 (30, 31).
X-ray Fiber Diffraction
Fibrils were pelleted by low-speed centrifugation, washed with water to eliminate salts, and repelleted. The pellet was subsequently resuspended in a small volume of water and pipetted into a space between the ends of two glass rods. As the pellet dried, fibrils aligned themselves between the rods. Diffraction images were collected at the Cleveland Center of Membrane and Structural Biology using a Rigaku MicroMax-007 HF copper anode x-ray source and a Rigaku Saturn 944+ CCD detector.
RESULTS
rPrP amyloid fibrils are typically prepared under conditions that induce destabilization and/or partial unfolding of the native PrP structure, i.e. in the presence of moderate concentrations of chemical denaturants or at mildly acidic pH (26, 32). In previous work, we characterized such amyloid fibrils formed in the presence of 2 m GdmCl (28, 33). Here, we further probed conditions under which the conformational conversion of rPrP takes place, finding that fibrils can also be formed when rPrP is incubated in the presence of 4 m GdmCl, i.e. under conditions in which monomeric rPrP substrate is fully unfolded. When followed by the standard thioflavin T fluorescence assay (27), both fibrillization reactions display lag and growth phases, consistent with a nucleated polymerization mechanism for amyloid formation. Although the lag phases of these two reactions are similar, the reaction in 4 m GdmCl is characterized by a much slower growth phase, occurring on the timescale of days rather than hours as observed for conversion in the presence of 2 m GdmCl (Fig. 1).
FIGURE 1.
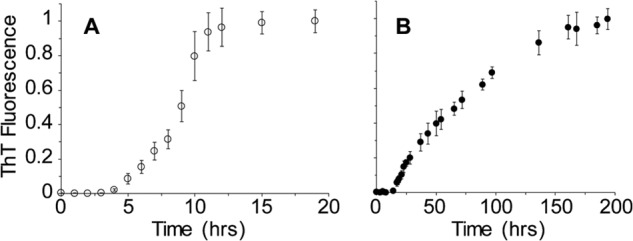
Kinetics of rPrP amyloid fibril formation in the presence of 2 and 4 m GdmCl. A, time course of the reaction in the presence of 2 m GdmCl. B, time course of the reaction in 4 m GdmCl. Data are expressed as a fraction of maximal thioflavin T (ThT) fluorescence.
The finding that fibrils can be formed in a 4 m GdmCl-containing solution is intriguing, as rPrP amyloid generated in the presence of 2 m GdmCl dissociates into constitutive monomers when incubated in 4 m GdmCl. Thus, it appears that a distinct and remarkably stable amyloid fold has been adopted by the protein under these strongly denaturing conditions. To further explore this, we probed the stability of rPrP fibrils formed in 2 and 4 m GdmCl (rPrP-A2M and rPrP-A4M, respectively) against chaotropic salts. Although rPrP-A2M fibrils were characterized by midpoint denaturation at ∼3.5 m GdmCl, rPrP-A4M fibrils could not be fully denatured using even 7.5 m GdmCl, consistent with their remarkably high stability (Fig. 2A). To better visualize the difference in conformational stability, a denaturation assay using a more strongly chaotropic salt, GdmThCN, was performed. As shown in Fig. 2B, also in this case, rPrP-A2M fibrils displayed a substantially lower midpoint of denaturation compared with rPrP-A4M fibrils (2.0 and 2.9 m GdmThCN, respectively).
FIGURE 2.
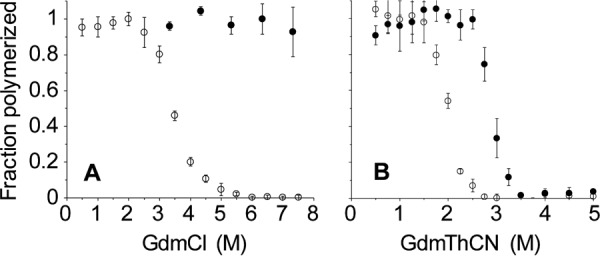
rPrP amyloid fibrils formed in 2 and 4 m GdmCl have different conformational stabilities. A, denaturation profiles in GdmCl. B, denaturation profiles in GdmThCN. In each case, the open and closed circles represent fibrils formed in 2 and 4 m GdmCl, respectively.
Conformational stability defined by resistance to denaturation with chaotropic agents has been frequently used as an operational parameter to discriminate between different strains of brain-derived PrPSc (18–23). The availability of two types of rPrP fibrils with dramatically different conformational stabilities provides an opportunity to explore the structural basis of this phenomenon. To this end, we probed both fibril types using a number of complementary biophysical techniques, including AFM, backbone amide HXMS, FTIR spectroscopy, His-HXMS, and x-ray fiber diffraction.
AFM
When analyzed by AFM, rPrP-A2M fibrils imaged at the midpoint of the fibrillization reaction displayed a right-handed ribbon-like helical morphology with an apparent periodicity of 80–100 nm and a fibril height of 3–4 nm (6–7 nm in the thicker “turn” regions) (Fig. 3, A and C). Upon longer incubation, these fibrils tended to clump together, and for most of them, helical morphology was no longer evident (Fig. 3E). The morphological hallmark of rPrP-A4M fibrils was lateral self-association of individual filaments into well defined pairs, in which these filaments occasionally twisted one around another (Fig. 3, B and D). Furthermore, in contrast to fibrils formed in 2 m GdmCl, rPrP-A4M fibrils appeared to have little tendency to clump into large aggregates, even after prolonged incubation times (Fig. 3F).
FIGURE 3.

AFM images of rPrP fibrils formed under different conditions. A and C, fibrils formed in 2 m GdmCl for 8 h. B and D, fibrils formed in 4 m GdmCl for 3 days. E, fibrils formed in 2 m GdmCl for 2 days. F, fibrils formed in 4 m GdmCl for 6 days. Scale bars = 250 nm.
Backbone Amide H/D Exchange
H/D exchange of backbone amide groups combined with mass spectrometry (HXMS) has recently emerged as a powerful method for structural characterization of different types of PrP aggregates formed in vitro and in vivo (28, 29, 34). This approach takes advantage of the rapid exchange of backbone amide hydrogens within unstructured regions of proteins compared with the relatively slow exchange of those involved in systematically hydrogen-bonded structures such as β-sheets or α-helices. These exchange rates are especially slow for amide protons in β-sheet “cores” of ordered protein aggregates, allowing mapping of these core regions (35). Here, we used this technique to probe potential structural differences between the two strains of rPrP amyloid fibrils.
Consistent with previous data for amyloid generated from rPrP(90–231) in 2 m GdmCl (28), fibrils formed under the same conditions using full-length rPrP showed long-term protection against deuterium labeling only for peptic fragments derived from the C-terminal region starting at residue ∼169 (Fig. 4). Partial protection observed for fragment 218–224 allowed us to map the end of this exchange-protected region to residues ∼220–221. As discussed previously for rPrP(90–231) fibrils (28), the exchange-protected ∼169–220 region defines the β-sheet core of rPrP amyloid, consistent with structural studies using site-directed spin labeling (33). To our surprise, essentially identical profiles of protection against H/D exchange were found for rPrP-A2M and rPrP-A4M fibrils, indicating very similar core regions. Thus, the higher stability of amyloid formed in 4 m GdmCl does not result from a longer β-sheet core region in the latter fibrils compared with those formed in 2 m GdmCl.
FIGURE 4.
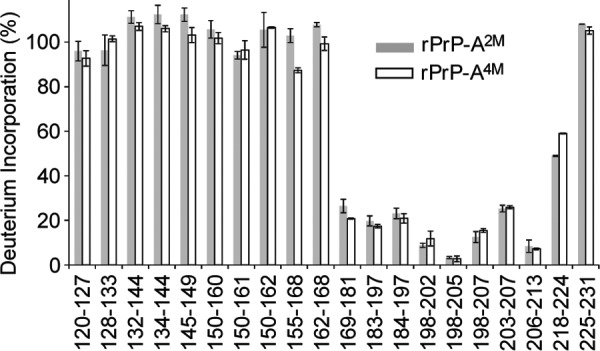
Backbone amide H/D exchange for rPrP fibrils formed in the presence of 2 m GdmCl (gray bars) and 4 m GdmCl (white bars). Fibrils were incubated in D2O buffer for 24 h at room temperature, and deuterium incorporation for each peptic fragment derived from these fibrils was assessed based on masses calculated from the overall centroids of the isotopic envelopes in mass spectra.
FTIR Spectroscopy
The two fibril types were further analyzed by FTIR spectroscopy, an established technique for studying the global secondary structure of proteins (36). This method is particularly well suited for probing subtle differences in β-sheet structures, with frequencies of amide I bands associated with these structures depending on factors such as specific packing of β-strands, length of β-strands, and solvent exposure (36, 37). Fig. 5 shows FTIR spectra in the amide I region for rPrP-A2M and rPrP-A4M fibrils. Both spectra displayed a prominent band at ∼1620 cm−1, which corresponds to β-sheet structure, and shoulders that likely represent random coil and turn structures. Although a quantitative assessment of protein secondary structure based on infrared spectra is full of pitfalls (36), qualitatively similar curve shapes suggest similar overall secondary structure in both fibril types, consistent with the HXMS data discussed above. It should be noted, however, that the predominant β-sheet band in rPrP-A2M was at 1623 cm−1, whereas in rPrP-A4M fibrils, it shifted to 1620 cm−1. This small shift in band frequency suggests subtle strain-specific structural differences, possibly at the level of β-strand packing and/or their solvent exposure.
FIGURE 5.
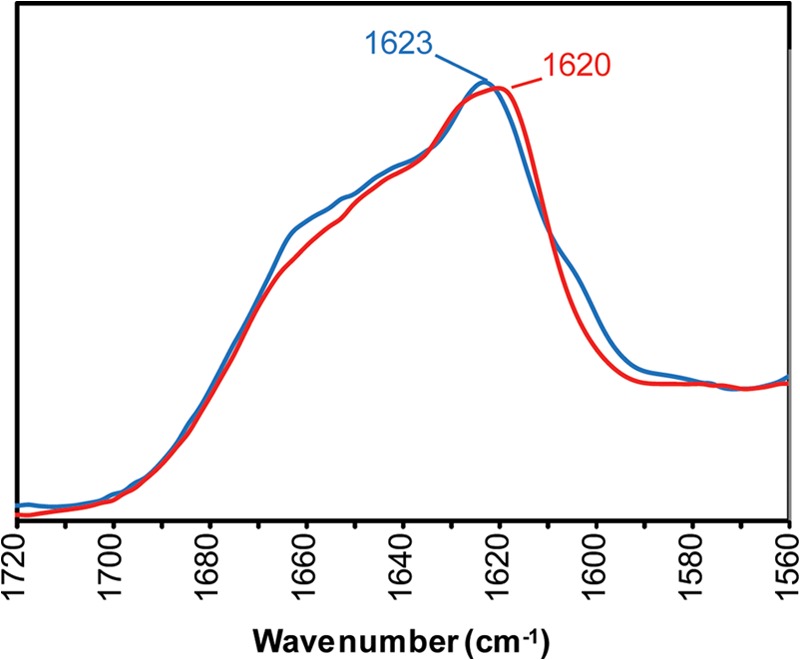
FTIR spectra of rPrP amyloid fibrils formed in the presence of 2 m GdmCl (blue line) or 4 m GdmCl (red line).
X-ray Fiber Diffraction Studies
Analysis of amyloid by x-ray fiber diffraction yields a specific pattern of reflections (referred to as “cross-β” (38)) that represent specific repeating structural features in the fibrils. The diffraction patterns for rPrP-A2M and rPrP-A4M fibrils are shown in Fig. 6, comparing the reflections found for each fibril type along the meridian and equator. A sharp meridional reflection at 4.75 Å was observed in both rPrP-A2M and rPrP-A4M strains. This common reflection is interpreted as the spacing between strands that run perpendicular to the length of the fibrils, with the β-sheet hydrogen bonding network extending parallel to this axis (38). However, distinguishing features between the two fibril types appear in equatorial reflections that are attributed to spacing between β-sheets. For rPrP-A2M fibrils, diffuse reflections centered at ∼10.5 Å with a shoulder up to 14 Å and another low-resolution reflection at 27 Å were observed. By contrast, rPrP-A4M fibrils showed a sharper reflection at ∼9.6 Å, with weaker reflections at ∼5.5 and 21.5 Å. Although meridional reflections are fixed by distances of hydrogen bonding between the peptide backbone, equatorial reflections have a variable nature in amyloid fibrils. Because these reflections correspond to the spacing between stacked β-sheets, they are dictated by side chains and the nature of interactions between them. Thus, the differences observed in equatorial reflections for rPrP-A2M and rPrP-A4M fibrils are likely caused by differences in the packing of β-sheets within the structure of the fibrils. These packing differences could account for the small differences observed in FTIR spectra as discussed above.
FIGURE 6.
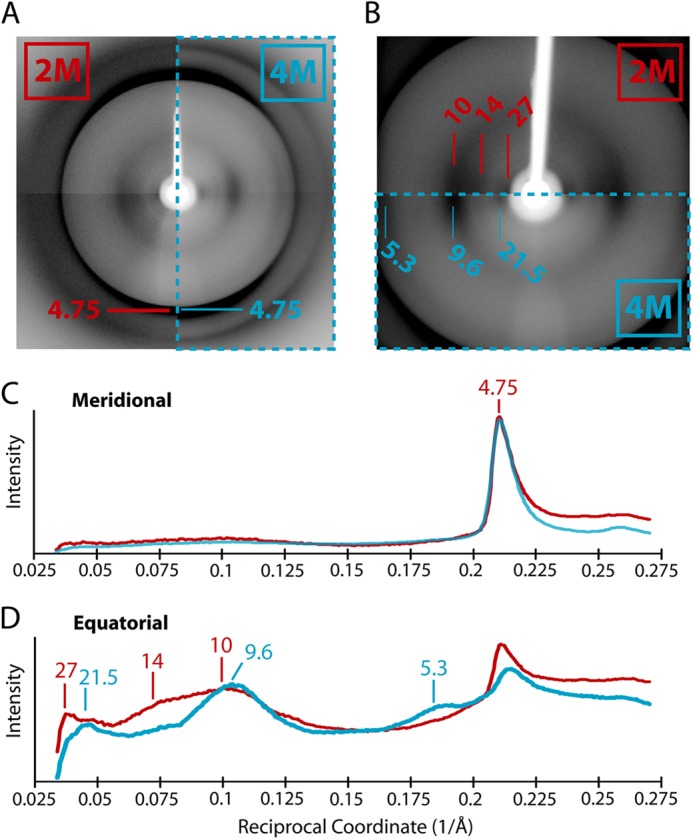
X-ray fiber diffraction of fibrils formed in 2 and 4 m GdmCl. A, the x-ray diffraction patterns of rPrP-A2M (left) and rPrP-A4M (right, enclosed in dashed box) fibrils are compared. Both fibril types have a prominent 4.75-Å meridional reflection corresponding to the spacing between β-strands along this axis. B, diffraction images of lower resolution reflections point to the differences along the equator between rPrP-A2M (upper) and rPrP-A4M (lower, enclosed in a dashed box) fibril diffraction patterns. C, a comparison of the intensity profiles of reflections along the meridian for rPrP-A2M (red line) and rPrP-A4M (blue line) fibrils shows that both have a similar 4.75-Å reflection. D, a comparison of the intensity profiles along the equator shows that rPrP-A2M and rPrP-A4 M fibrils have different peak reflections, indicating packing differences within the amyloid core. In C and D, peaks are labeled in units of Å.
His-HXMS
Structural differences between the two fibril types were further probed using the recently developed method of His-HXMS (30, 31, 39). In this approach, the rate of H/D exchange for C2 protons in histidine imidazole rings is measured. Even for unprotected histidines, this exchange is relatively slow (on the order of ∼2 days), allowing convenient monitoring by mass spectrometry. If the His side chain is buried in a water-protected environment, this exchange time is increased to many weeks or even months (30, 31). Amide HXMS and His-HXMS methods are highly complementary: whereas the former probes protein structure at the level of the polypeptide backbone, the latter provides information regarding the microenvironment (water accessibility) of specific His side chains. Therefore, His-HXMS is potentially very useful as a tool to provide site-specific information about the nature of steric zipper interfaces within amyloids.
There are only two His residues within the experimentally determined β-core region of rPrP amyloid (His-177 and His-187), with two additional histidines present in the segment adjacent to the core (His-140 and His-155). As expected for the region outside the core, exchange rates for His-140 and His-155 in both fibril types are characterized by half-times of 2–3 days (Fig. 7), i.e. within the range observed for unprotected histidines. By contrast, both His-177 and His-187 in rPrP-A4M fibrils show very strong protection from deuterium labeling, with half-times for exchange of 264 and 436 days, respectively. Such high levels of protection from exchange indicate that these side chains are in a highly dehydrated environment, suggesting that they are involved in “dry” steric zipper interfaces of the fibril (40, 41). Although some protection against hydrogen exchange is also observed for these side chains in rPrP-A2M fibrils, the level of protection (exchange half-times of 14 and 10 days for His-177 and His-187, respectively) is much lower compared with that observed for rPrP-A4M fibrils. Thus, it appears that rPrP-A2M and rPrP-A4M fibrils differ considerably with respect to the precise nature of steric zipper interfaces in the regions involving amino acid residues 177 and 187.
FIGURE 7.

Histidine H/D exchange for fibrils formed in 2 m GdmCl (gray bars) and 4 m GdmCl (white bars). The parameter t½ represents the half-time of the exchange reaction for individual His residues.
DISCUSSION
One of the most intriguing properties of amyloidogenic proteins is their ability to form different strains of amyloid fibrils. Such a structural polymorphism has been observed in amyloids formed by proteins as varied as lysozyme, insulin, amyloid-β, α-synuclein, tau, β2-microglobulin, recombinant mammalian PrP, and the yeast prion protein Sup35 (24, 35, 42–55). However, the molecular level details giving rise to amyloid strains may vary with the constitutive protein. In the well studied yeast prion protein Sup35, for example, distinct prion strains generated by varying the temperature at which polymerization is carried out differ with respect to the length of the amyloid core (25). However, in the case of amyloid-β and α-synuclein, solid-state NMR has revealed that the residues participating in the amyloid core region of the heterogeneous fibril types are quite similar, indicating that differences in packing of β-strands may also underlie the strain phenomenon (44, 46). Recent crystallographic studies of steric zipper segments derived from amyloidogenic proteins have discussed two basic mechanisms that may lead to amyloid polymorphism (56). In segmental polymorphisms, different parts of a protein sequence can form distinct amyloid structures. On the other hand, packing polymorphisms may result when different amyloid structures arise from alternative arrangements of the same sequence.
Structural polymorphism of ordered protein aggregates is of particular importance in the context of prion diseases, as distinct phenotypes of this disorder appear to be encoded by conformationally different strains of PrPSc. Furthermore, strain-dependent PrPSc structure is one of the major determinants of prion transmissibility barriers between different mammalian species (12). In the absence of high-resolution structural data, one of the key surrogate parameters used to characterize different strains of PrPSc has been the conformational stability as defined by resistance to denaturation by temperature and/or chemical agents such as GdmCl (18, 19, 21–23). In a recent study, Prusiner and co-workers (22) inoculated transgenic mice overexpressing PrPC with structurally distinct preparations of recombinant mouse PrP amyloid fibrils, finding that the conformational stability of each prion isolate from these mice correlated closely to the conformational stability of the amyloid preparation used to generate it. Even more intriguingly, a correlation was found between the conformational stability of these synthetic prion strains and the incubation period for the second passage, with less stable PrPSc isolates corresponding to shorter incubation times (22). These observations are reminiscent of findings for the yeast prion protein Sup35, in which amyloid fibrils with higher stability (Sc37; formed at 37 °C) induce a weaker prion phenotype in yeast than those with lower stability (Sc4; formed at 4 °C) (24). The stronger phenotype associated with less stable Sc4 fibrils is believed to result from their greater propensity to undergo Hsp104 chaperone-mediated fragmentation, which creates more surfaces for the recruitment and templated conversion of the Sup35 monomer (57). However, the picture for mammalian prions appears to be more complicated, as, in contrast to murine prion strains, a reverse correlation between PrPSc stability and incubation period was reported for hamster-adapted prions (19). Furthermore, no homolog of the disaggregating chaperone Hsp104 has yet been identified in mammals. Unlike for structurally well characterized yeast prions, no information is available regarding the structural basis of differences in the stability of mammalian prion strains.
In this study, we have created two strains of full-length rPrP amyloid fibrils that show drastic differences in conformational stability. Although the strain formed in the presence of 2 m GdmCl (rPrP-A2M) is characterized by midpoint denaturation at 3.5 m GdmCl, fibrils formed in 4 m GdmCl (rPrP-A4M) remain stable even in the presence of 7.5 m GdmCl. Such a large difference in stability against dissociation by chaotropic salts implies some degree of structural differences between the two fibril types. However, these differences were not readily detectable by conventional biophysical techniques used to probe the structure of amyloid fibrils. In particular, only small differences could be detected in FTIR spectra, indicating that the overall secondary structure of the two amyloid strains is similar, in each case rich in β-sheet structure. Consistent with the FTIR data, backbone amide H/D exchange experiments showed essentially identical protection maps against deuterium incorporation, demonstrating very similar β-sheet core regions in both fibril types. This latter finding is particularly intriguing in light of previous studies with the yeast prion protein Sup35. Here, large differences in the size of the amyloid core are known to underlie strain-dependent stability differences, with the core of the more stable Sc37 strain being some 30 residues longer relative to that of the less stable Sc4 strain (25). Thus, it appears that the structural basis of differential stability for mammalian PrP amyloid fibrils is fundamentally different from that underlying stability differences of Sup35 amyloid strains.
The H/D exchange experiments revealed that, in both rPrP-A2M and rPrP-A4M fibrils, the amyloid core is demarcated by stable secondary structural elements spanning residues ∼169–220. This is in agreement with prior H/D exchange (28) and site-directed spin labeling (33) studies performed on fibrils formed from rPrP(90–231). Data from the latter study supported modeling of the rPrP(90–231) amyloid core as a parallel and in-register β-structure (33) based upon the steric zipper motif (40, 41). To investigate if there are differences in the packing of the steric zippers cores of rPrP-A2M and rPrP-A4M fibrils, we employed x-ray fiber diffraction. X-ray diffraction studies of the two fibril types showed cross-β patterns typical of amyloid. However, distinct equatorial reflections were observed for rPrP-A2M and rPrP-A4M fibrils, suggesting structural differences in the assembly of their steric zipper interfaces. This finding was further supported by His-HXMS data showing a large strain-dependent difference in the environment (solvent accessibility) of the side chains of two His residues (positions 177 and 187) present within the core of rPrP amyloid, suggesting that these residues are present in either the wet or dry interface in rPrP-A2M and rPrP-A4M fibrils, respectively. These strain-specific differences in the exposure of specific amino acid residues may contribute to the distinct pairing and/or clumping propensities of rPrP-A2M and rPrP-A4M fibrils as observed by AFM. Given the same size and amino acid sequence of amyloid cores in rPrP-A2M and rPrP-A4M fibrils, we infer that different packing arrangements within the core region (i.e. steric zipper interfaces) are the main factor responsible for the large stability differences between the two fibril types. High-resolution structural data would be needed to determine the specific nature of these interfaces in both fibril types. However, high-resolution structural studies with full-length PrP amyloid fibrils are hampered by major technical difficulties, and no such data have been reported to date. Nevertheless, it has been established in crystallographic studies with short peptide fragments of amyloidogenic proteins that steric zipper motifs may vary by features such as surface complementarity, degree of side chain interdigitation, and side chain hydrogen bonding (40, 41, 56). Thus, fibril stability is likely to be strongly affected by a combination of van der Waals and electrostatic forces at the steric zipper interfaces. Within the context of a parallel in-register β-structure as proposed for rPrP amyloid, a particularly important factor might be the orientation of charged side chains, as stacking of these side chains within the dry interfaces would be expected to have a strong destabilizing effect.
The preference for one aggregation pathway over another may be tied to the folding state of the precursor monomer and/or the final amyloid structure. It has been proposed that the direct monomeric precursor of PrP that is recruited into the aggregated state is a partially structured folding intermediate (58), although the fully unfolded state is also a potential candidate for this monomeric precursor. Different populations of natively folded PrP, partially folded intermediates(s), and the fully unfolded state are likely to be present in the presence of 2 and 4 m GdmCl (59, 60). In the context of fibril formation, it is plausible that each of these species is able to give rise to unique fibrillar structures by promoting the formation of different steric zipper interfaces that have the ability to nucleate further growth of similarly misfolded PrP aggregates.
In conclusion, we have created two strains of rPrP amyloid fibrils that show dramatic differences in stability against chemical denaturation, and we have characterized them by a number of biophysical methods. In contrast to previously characterized strains of amyloid formed from the yeast prion protein Sup35 (25), the difference in stability between the two strains of rPrP amyloid does not result from differences in the size of the core region. Instead, it appears to be linked to specific differences in the packing arrangement within the amyloid core. Although this study was limited to synthetic PrP amyloid fibrils, a similar structural basis for strain-dependent conformational stability may apply to brain-derived PrPSc, especially because large differences in PrPSc stability are often observed for prion strains despite a similar size of the proteinase K-resistant core (22, 23).
This work was supported, in whole or in part, by National Institutes of Health Grants NS044158 and NS074317 (to W. K. S.).
- PrPC
- cellular prion protein
- PrPSc
- scrapie PrP
- rPrP
- recombinant human prion protein
- GdmCl
- guanidinium chloride
- GdmThCN
- guanidinium thiocyanate
- AFM
- atomic force microscopy
- HXMS
- hydrogen/deuterium exchange mass spectrometry
- H/D
- hydrogen/deuterium.
REFERENCES
- 1. Prusiner S. B. (1998) Prions. Proc. Natl. Acad. Sci. U.S.A. 95, 13363–13383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aguzzi A., Polymenidou M. (2004) Mammalian prion biology: one century of evolving concepts. Cell 116, 313–327 [DOI] [PubMed] [Google Scholar]
- 3. Collinge J. (2001) Prion diseases of humans and animals: their causes and molecular basis. Annu. Rev. Neurosci. 24, 519–550 [DOI] [PubMed] [Google Scholar]
- 4. Cobb N. J., Surewicz W. K. (2009) Prion diseases and their biochemical mechanisms. Biochemistry 48, 2574–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kraus A., Groveman B. R., Caughey B. (2013) Prions and the potential transmissibility of protein misfolding diseases. Annu. Rev. Microbiol. 67, 543–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castilla J., Saá P., Hetz C., Soto C. (2005) In vitro generation of infectious scrapie prions. Cell 121, 195–206 [DOI] [PubMed] [Google Scholar]
- 7. Wang F., Wang X., Yuan C. G., Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Legname G., Baskakov I. V., Nguyen H. O., Riesner D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2004) Synthetic mammalian prions. Science 305, 673–676 [DOI] [PubMed] [Google Scholar]
- 9. Makarava N., Kovacs G. G., Bocharova O., Savtchenko R., Alexeeva I., Budka H., Rohwer R. G., Baskakov I. V. (2010) Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 119, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kim J. I., Cali I., Surewicz K., Kong Q., Raymond G. J., Atarashi R., Race B., Qing L., Gambetti P., Caughey B., Surewicz W. K. (2010) Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J. Biol. Chem. 285, 14083–14087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deleault N. R., Piro J. R., Walsh D. J., Wang F., Ma J., Geoghegan J. C., Supattapone S. (2012) Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 109, 8546–8551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Collinge J., Clarke A. R. (2007) A general model of prion strains and their pathogenicity. Science 318, 930–936 [DOI] [PubMed] [Google Scholar]
- 13. Caughey B., Raymond G. J., Bessen R. A. (1998) Strain-dependent differences in β-sheet conformations of abnormal prion protein. J. Biol. Chem. 273, 32230–32235 [DOI] [PubMed] [Google Scholar]
- 14. Baron G. S., Hughson A. G., Raymond G. J., Offerdahl D. K., Barton K. A., Raymond L. D., Dorward D. W., Caughey B. (2011) Effect of glycans and the glycophosphatidylinositol anchor on strain-dependent conformations of scrapie prion protein: improved purifications and infrared spectra. Biochemistry 50, 4479–4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bessen R. A., Marsh R. F. (1994) Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J. Virol. 68, 7859–7868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuczius T., Groschup M. H. (1999) Differences in proteinase K resistance and neuronal deposition of abnormal prion proteins characterize bovine spongiform encephalopathy (BSE) and scrapie strains. Mol. Med. 5, 406–418 [PMC free article] [PubMed] [Google Scholar]
- 17. Safar J., Wille H., Itri V., Groth D., Serban H., Torchia M., Cohen F. E., Prusiner S. B. (1998) Eight prion strains have PrPSc molecules with different conformations. Nat. Med. 4, 1157–1165 [DOI] [PubMed] [Google Scholar]
- 18. Bett C., Kurt T. D., Lucero M., Trejo M., Rozemuller A. J., Kong Q., Nilsson K. P., Masliah E., Oldstone M. B., Sigurdson C. J. (2013) Defining the conformational features of anchorless, poorly neuroinvasive prions. PLoS Pathog. 9, e1003280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayers J. I., Schutt C. R., Shikiya R. A., Aguzzi A., Kincaid A. E., Bartz J. C. (2011) The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 7, e1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peretz D., Williamson R. A., Legname G., Matsunaga Y., Vergara J., Burton D. R., DeArmond S. J., Prusiner S. B., Scott M. R. (2002) A change in the conformation of prions accompanies the emergence of a new prion strain. Neuron 34, 921–932 [DOI] [PubMed] [Google Scholar]
- 21. Legname G., Nguyen H. O., Peretz D., Cohen F. E., DeArmond S. J., Prusiner S. B. (2006) Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc. Natl. Acad. Sci. U.S.A. 103, 19105–19110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Colby D. W., Giles K., Legname G., Wille H., Baskakov I. V., DeArmond S. J., Prusiner S. B. (2009) Design and construction of diverse mammalian prion strains. Proc. Natl. Acad. Sci. U.S.A. 106, 20417–20422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bett C., Joshi-Barr S., Lucero M., Trejo M., Liberski P., Kelly J. W., Masliah E., Sigurdson C. J. (2012) Biochemical properties of highly neuroinvasive prion strains. PLoS Pathog. 8, e1002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka M., Chien P., Naber N., Cooke R., Weissman J. S. (2004) Conformational variations in an infectious protein determine prion strain differences. Nature 428, 323–328 [DOI] [PubMed] [Google Scholar]
- 25. Toyama B. H., Kelly M. J., Gross J. D., Weissman J. S. (2007) The structural basis of yeast prion strain variants. Nature 449, 233–237 [DOI] [PubMed] [Google Scholar]
- 26. Apetri A. C., Vanik D. L., Surewicz W. K. (2005) Polymorphism at residue 129 modulates the conformational conversion of the D178N variant of human prion protein 90–231. Biochemistry 44, 15880–15888 [DOI] [PubMed] [Google Scholar]
- 27. Naiki H., Higuchi K., Hosokawa M., Takeda T. (1989) Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye, thioflavin T1. Anal. Biochem. 177, 244–249 [DOI] [PubMed] [Google Scholar]
- 28. Lu X., Wintrode P. L., Surewicz W. K. (2007) β-Sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc. Natl. Acad. Sci. U.S.A. 104, 1510–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smirnovas V., Kim J. I., Lu X., Atarashi R., Caughey B., Surewicz W. K. (2009) Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J. Biol. Chem. 284, 24233–24241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miyagi M., Nakazawa T. (2008) Determination of pKa values of individual histidine residues in proteins using mass spectrometry. Anal. Chem. 80, 6481–6487 [DOI] [PubMed] [Google Scholar]
- 31. Miyagi M., Wan Q., Ahmad M. F., Gokulrangan G., Tomechko S. E., Bennett B., Dealwis C. (2011) Histidine hydrogen-deuterium exchange mass spectrometry for probing the microenvironment of histidine residues in dihydrofolate reductase. PLoS ONE 6, e17055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bocharova O. V., Breydo L., Parfenov A. S., Salnikov V. V., Baskakov I. V. (2005) In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrPSc. J. Mol. Biol. 346, 645–659 [DOI] [PubMed] [Google Scholar]
- 33. Cobb N. J., Sönnichsen F. D., McHaourab H., Surewicz W. K. (2007) Molecular architecture of human prion protein amyloid: a parallel, in-register β-structure. Proc. Natl. Acad. Sci. U.S.A. 104, 18946–18951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Smirnovas V., Baron G. S., Offerdahl D. K., Raymond G. J., Caughey B., Surewicz W. K. (2011) Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 18, 504–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Toyama B. H., Weissman J. S. (2011) Amyloid structure: conformational diversity and consequences. Annu. Rev. Biochem. 80, 557–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Surewicz W. K., Mantsch H. H., Chapman D. (1993) Determination of protein secondary structure by Fourier transform infrared spectroscopy: a critical assessment. Biochemistry 32, 389–394 [DOI] [PubMed] [Google Scholar]
- 37. Zandomeneghi G., Krebs M. R., McCammon M. G., Fändrich M. (2004) FTIR reveals structural differences between native β-sheet proteins and amyloid fibrils. Protein Sci. 13, 3314–3321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Serpell L. C., Fraser P. E., Sunde M. (1999) X-ray fiber diffraction of amyloid fibrils. Methods Enzymol. 309, 526–536 [DOI] [PubMed] [Google Scholar]
- 39. Tran D. T., Banerjee S., Alayash A. I., Crumbliss A. L., Fitzgerald M. C. (2012) Slow histidine H/D exchange protocol for thermodynamic analysis of protein folding and stability using mass spectrometry. Anal. Chem. 84, 1653–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nelson R., Sawaya M. R., Balbirnie M., Madsen A. Ø., Riekel C., Grothe R., Eisenberg D. (2005) Structure of the cross-β spine of amyloid-like fibrils. Nature 435, 773–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 42. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 43. Shewmaker F., McGlinchey R. P., Wickner R. B. (2011) Structural insights into functional and pathological amyloid. J. Biol. Chem. 286, 16533–16540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Petkova A. T., Leapman R. D., Guo Z., Yau W. M., Mattson M. P., Tycko R. (2005) Self-propagating, molecular-level polymorphism in Alzheimer's β-amyloid fibrils. Science 307, 262–265 [DOI] [PubMed] [Google Scholar]
- 45. Paravastu A. K., Leapman R. D., Yau W. M., Tycko R. (2008) Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 18349–18354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Heise H., Hoyer W., Becker S., Andronesi O. C., Riedel D., Baldus M. (2005) Molecular-level secondary structure, polymorphism, and dynamics of full-length α-synuclein fibrils studied by solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 102, 15871–15876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dinkel P. D., Siddiqua A., Huynh H., Shah M., Margittai M. (2011) Variations in filament conformation dictate seeding barrier between three- and four-repeat tau. Biochemistry 50, 4330–4336 [DOI] [PubMed] [Google Scholar]
- 48. Yamaguchi K., Katou H., Hoshino M., Hasegawa K., Naiki H., Goto Y. (2004) Core and heterogeneity of β2-microglobulin amyloid fibrils as revealed by H/D exchange. J. Mol. Biol. 338, 559–571 [DOI] [PubMed] [Google Scholar]
- 49. Chatani E., Yagi H., Naiki H., Goto Y. (2012) Polymorphism of β2-microglobulin amyloid fibrils manifested by ultrasonication-enhanced fibril formation in trifluoroethanol. J. Biol. Chem. 287, 22827–22837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Debelouchina G. T., Platt G. W., Bayro M. J., Radford S. E., Griffin R. G. (2010) Magic angle spinning NMR analysis of β2-microglobulin amyloid fibrils in two distinct morphologies. J. Am. Chem. Soc. 132, 10414–10423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Anderson M., Bocharova O. V., Makarava N., Breydo L., Salnikov V. V., Baskakov I. V. (2006) Polymorphism and ultrastructural organization of prion protein amyloid fibrils: an insight from high resolution atomic force microscopy. J. Mol. Biol. 358, 580–596 [DOI] [PubMed] [Google Scholar]
- 52. Jones E. M., Surewicz W. K. (2005) Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell 121, 63–72 [DOI] [PubMed] [Google Scholar]
- 53. Jones E. M., Wu B., Surewicz K., Nadaud P. S., Helmus J. J., Chen S., Jaroniec C. P., Surewicz W. K. (2011) Structural polymorphism in amyloids. New insights from studies with Y145Stop prion protein fibrils. J. Biol. Chem. 286, 42777–42784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dzwolak W., Smirnovas V., Jansen R., Winter R. (2004) Insulin forms amyloid in a strain-dependent manner: an FT-IR spectroscopic study. Protein Sci. 13, 1927–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chien P., Weissman J. S., DePace A. H. (2004) Emerging principles of conformation-based prion inheritance. Annu. Rev. Biochem. 73, 617–656 [DOI] [PubMed] [Google Scholar]
- 56. Wiltzius J. J., Landau M., Nelson R., Sawaya M. R., Apostol M. I., Goldschmidt L., Soriaga A. B., Cascio D., Rajashankar K., Eisenberg D. (2009) Molecular mechanisms for protein-encoded inheritance. Nat. Struct. Mol. Biol. 16, 973–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tanaka M., Collins S. R., Toyama B. H., Weissman J. S. (2006) The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589 [DOI] [PubMed] [Google Scholar]
- 58. Surewicz W. K., Apostol M. I. (2011) Prion protein and its conformational conversion: a structural perspective. Top. Curr. Chem. 305, 135–167 [DOI] [PubMed] [Google Scholar]
- 59. Hosszu L. L., Wells M. A., Jackson G. S., Jones S., Batchelor M., Clarke A. R., Craven C. J., Waltho J. P., Collinge J. (2005) Definable equilibrium states in the folding of human prion protein. Biochemistry 44, 16649–16657 [DOI] [PubMed] [Google Scholar]
- 60. Apetri A. C., Maki K., Roder H., Surewicz W. K. (2006) Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J. Am. Chem. Soc. 128, 11673–11678 [DOI] [PMC free article] [PubMed] [Google Scholar]