

Background: SREBP transcription factor controls sterol homeostasis.
Results: Fission yeast casein kinase 1 (CK1) binds and accelerates degradation of active SREBP, inhibiting expression of target genes and decreasing ergosterol.
Conclusion: CK1 controls nuclear SREBP stability to regulate sterol homeostasis in fission yeast.
Significance: CK1 is the first kinase shown to regulate fungal SREBP and functions in the control of SREBP and sterol homeostasis.
Keywords: Protein Stability, Serine/Threonine Protein Kinase, Sterol, Transcription Factors, Yeast, CK1, Casein Kinase 1, SREBP
Abstract
Sterol homeostasis is tightly controlled by the sterol regulatory element-binding protein (SREBP) transcription factor that is highly conserved from fungi to mammals. In fission yeast, SREBP functions in an oxygen-sensing pathway to promote adaptation to decreased oxygen supply that limits oxygen-dependent sterol synthesis. Low oxygen stimulates proteolytic cleavage of the SREBP homolog Sre1, generating the active transcription factor Sre1N that drives expression of sterol biosynthetic enzymes. In addition, low oxygen increases the stability and DNA binding activity of Sre1N. To identify additional signals controlling Sre1 activity, we conducted a genetic overexpression screen. Here, we describe our isolation and characterization of the casein kinase 1 family member Hhp2 as a novel regulator of Sre1N. Deletion of Hhp2 increases Sre1N protein stability and ergosterol levels in the presence of oxygen. Hhp2-dependent Sre1N degradation by the proteasome requires Hhp2 kinase activity, and Hhp2 binds and phosphorylates Sre1N at specific residues. Our results describe a role for casein kinase 1 as a direct regulator of sterol homeostasis. Given the role of mammalian Hhp2 homologs, casein kinase 1δ and 1ϵ, in regulation of the circadian clock, these findings may provide a mechanism for coordinating circadian rhythm and lipid metabolism.
Introduction
Maintenance of cholesterol homeostasis in mammals is imperative for proper cell membrane fluidity and structure, regulation of membrane protein function, and the biosynthesis of bile acids and steroids (1). Altered levels of cholesterol result in hypercholesterolemia and contribute to the manifestation of several human diseases (2, 3). Thus, maintaining tight control of cholesterol levels is important for health and cell survival. A master regulator of cellular cholesterol levels is the family of proteins known as sterol regulatory element-binding proteins (SREBPs)2; the specific isoforms SREBP-2 and -1a are important for cholesterol metabolism (4, 5). SREBPs are highly conserved from fungi to mammals, and under normal sterol conditions they exist as inactive endoplasmic reticulum transmembrane proteins. Depletion of cellular sterol results in translocation of SREBP to the Golgi where it undergoes proteolytic cleavage, liberating the N terminus which then localizes to the nucleus to function as a transcription factor (4, 6). Once in the nucleus, active SREBP promotes expression of genes required for sterol homeostasis by binding to specific sterol regulatory elements (SREs) in the promoters of target genes.
SREBPs modulate cellular sterol levels in response to extracellular cues that may alter or compromise sterol metabolism, including changes in oxygen and iron availability, exposure to insulin, and light-dark cycles (7–12). Oxygen is an essential substrate for sterol synthesis and many other metabolic pathways (13). In Schizosaccharomyces pombe, reduction in environmental oxygen supply activates the SREBP homolog Sre1 to restore sterol synthesis and cell growth under low oxygen (8, 14). We previously demonstrated that low oxygen stimulates cleavage of Sre1, increasing production of the active N-terminal Sre1N transcription factor and expression of its target genes (15, 16). In addition, low oxygen stimulates Sre1N DNA binding activity and prevents Sre1N degradation by the proteasome (17–19). Finally, the sre1+ promoter contains SREs, allowing Sre1 to induce its own gene expression via a positive feedback loop (14). Through these regulatory mechanisms, yeast SREBP controls sterol homeostasis and cell growth in response to low environmental oxygen.
Proper transcriptional regulation of sterol homeostasis necessitates the integration of information from multiple pathways, such as energy supply, nutrient availability, and cell growth signals. To date, only the candidate prolyl hydroxylase Ofd1 and its regulator Nro1 have been identified as modulators of Sre1N activity (17, 18). Ofd1 alters Sre1N function through two oxygen-dependent mechanisms, promotion of Sre1N protein degradation and inhibition of Sre1N DNA binding (17–20). Other factors controlling Sre1N function are not known. To elucidate the mechanisms by which Sre1N is regulated, we performed a high copy plasmid screen in the presence of oxygen using an S. pombe cDNA library and isolated clones that decreased Sre1N activity. From our screen, we identified the casein kinase 1 Hhp2 as a negative regulator of Sre1N function.
Hhp2 is a member of the casein kinase 1 (CK1) family, a highly conserved group of serine/threonine kinases present in eukaryotes spanning yeast to humans (21). Hhp2 is the S. pombe homolog of mammalian CK1 isoforms δ and ϵ. Hhp2 has been implicated in the response to DNA damage and is required for chromosomal segregation during meiosis (22, 23). In mammals, CK1 isoforms δ and ϵ have well described roles in the control of circadian rhythm and Wnt signaling (21, 24). Recently, CK1s have been shown to influence cell metabolism through control of the transcriptional co-activator peroxisome proliferator-activated receptor γ co-activator-1α and hypoxia-inducible factor (25, 26). Here, we demonstrate a novel requirement for CK1 in maintaining sterol homeostasis and control of SREBP degradation.
EXPERIMENTAL PROCEDURES
Reagents
Edinburgh 20 minimal medium (EMM) was obtained from MP Biologicals; yeast extract was from BD Biosciences; amino acids and protease inhibitors (leupeptin, PMSF, and pepstatin A) were from Sigma; horseradish peroxidase-conjugated affinity-purified donkey anti-rabbit and anti-mouse IgG were from Jackson ImmunoResearch, oligonucleotides were from Integrated DNA Technologies; dithiobis(succinimidyl propionate) (DSP) cross-linker was from Thermo Scientific; mouse monoclonal Myc antibody (9E10) and β-actin antibody (C4) were from Santa Cruz Biotechnology; and rabbit polyclonal Myc antibody (06-549) was from Upstate. Sre1 antibody has been previously described (8); cholesterol was from Steraloids; [γ-32P]ATP was from PerkinElmer Life Sciences, and bortezomib was from LC Laboratories.
Yeast Strains
Wild-type haploid S. pombe strain KGY425 (h-, his3-D1, leu1–32, ura4-D18, and ade6-M210) was obtained from American Type Culture Collection (27). S. pombe strains sre1Δ, sre1N, sre1N-MP, and sre1N ubr1Δ have been described previously (19). S. pombe strains sre1N hhp2Δ, sre1 hhp2Δ, sre1N ubr1Δ hhp2-myc, and sre1NΔ hhp2-myc ubr1Δ were generated by homologous recombination from haploid KGY425 yeast using established techniques (28). Yeast strains expressing plasmids were generated by transformation.
Strains were grown to exponential phase at 30 °C in rich medium (YES) or EMM plus appropriate supplements for selection (225 μg/ml each of histidine, uracil, leucine, adenine, and lysine) using established techniques (8). An In Vivo2 400 workstation (Biotrace Inc.) was used to maintain anaerobic conditions as described previously (8, 14). EMM containing 0.1% 5-fluoroorotic acid (5-FOA) (Toronto Research Chemicals) was made as described (29).
Plasmids
hhp2, ofd1, and tom70 constructs driven by the adh promoter were isolated from our screen and were from an S. pombe cDNA library (30). Plasmids expressing wild-type or kinase-dead (K41N) Hhp2 from the thiamine-repressible nmt promoter were generated by subcloning wild-type or mutant hhp2+ cDNA fragments into pSLF172 and pSLF272 (31). Bacterial constructs expressing His6-tagged wild-type or kinase-dead Hhp2 and Sre1N (amino acids 1–440) were produced by inserting appropriate S. pombe cDNAs into pET15b (Novagen). The His6-tagged Cnp3-N (amino acids 1–277) bacterial construct was generated by subcloning cnp3-N+ S. pombe cDNA into pProExHTb (Invitrogen).
Plasmid cDNA Library Screen
sre1N 4×SRE-ura4+ cells were transformed with the SPLE-1 S. pombe cDNA library (30, 32). Expression of the cDNA library was driven by the adh promoter in the pLEV3 vector that contains the S. cerevisiae LEU2 gene for plasmid selection. Transformed cells were plated onto EMM plates lacking leucine and containing 0.1% 5-FOA. A total of 2.4 × 105 transformants were screened for growth on selection plates for 6 days and isolated by single colony purification. Plasmid DNA was extracted from positive clones, amplified in Escherichia coli, and sequenced as described previously (18).
Northern and Western Blotting
Total RNA was isolated, and Northern blotting was performed as described previously (8). Design and synthesis of radiolabeled RNA probes for sre1N+, hem13+, and tub1+ were performed as described previously (14, 19). Whole-cell yeast protein extraction and Western blotting were performed as described previously using primary antibodies and appropriate horseradish peroxidase-conjugated affinity-purified immunoglobulin (8). Where mentioned, lysates were treated with 1 unit/μl alkaline phosphatase (Roche Applied Science) for 1 h at 37 °C prior to Western blot analysis.
Measurement of Protein Half-life
Experiments were performed as described previously (18). Briefly, yeast cells were grown in the absence of oxygen for 4 h to exponential phase prior to being treated with cycloheximide (200 μg/ml) and shifted to normoxic conditions. Samples were harvested at 0, 5, 10, 15, and 20 min post-cycloheximide addition. Whole-cell lysis was performed followed by Western blotting. Blots were imaged with the VersaDoc Imaging System (Bio-Rad), and band intensities were quantified using Quantity One software (Bio-Rad). Sre1N protein half-life was calculated using the equation t½ = (ln 2)/k, where k is the slope of the line calculated using an exponential trendline (33).
Co-immunoprecipitation
Immunoprecipitation of endogenous yeast protein was performed as described previously (8). Briefly, cells were grown for 4 h in the presence of oxygen and then treated with 2 mm DSP cross-linker in PBS for 30 min. Cross-linking was stopped by treatment with 20 mm Tris-HCl, pH 7.5, for 15 min. Exponentially growing cells (5 × 107 cells) were lysed with glass beads (0.5 mm) in 100 μl of Nonidet P-40 lysis buffer (50 mm HEPES-HCl, pH 7.4, 100 mm NaCl, 1.5 mm MgCl2, 1% Nonidet P-40) plus 2× protease inhibitors (PMSF, leupeptin, and pepstatin A) for 14 min. Insoluble material was removed by centrifugation at 14,000 rpm for 2 min. The supernatant was saved and incubated with 5 μg of monoclonal Myc antibody (9E10) in 1 ml of Nonidet P-40 lysis buffer for 15 min prior to the addition of 40 μl of protein A-Sepharose beads (Repligen). Samples were rotated for 2.5 h at 4 °C followed by three washes with Nonidet P-40 lysis buffer and resuspended in 1× SDS-PAGE loading buffer prior to Western blot analysis. Immunoprecipitation of exogenously expressed proteins was performed as above except cells were grown for 4 h in the absence of oxygen prior to treatment with DSP under normoxic conditions.
In Vitro Kinase Assay
Plasmids expressing recombinant His6-tagged Hhp2, Hhp2-K41N, and Sre1N were transformed into E. coli BL21-CodonPlus (DE3) cells (Agilent). The His6-tagged Cnp3-N plasmid was transformed into E. coli BL21(DE3) ompT. Protein expression in E. coli was induced by treatment of cells with 0.6 mm isopropyl β-d-thiogalactopyranoside at an A600 of 0.5 and shifted to 30 °C for 1–3 h. Cells were lysed by sonication in lysis buffer (50 mm NaH2PO4, 300 mm NaCl, and 10 mm imidazole at pH 8.0) with 1× Protease Complete tablets (Roche Applied Science). Tagged proteins were purified by nickel affinity chromatography using nickel-nitrilotriacetic acid-agarose beads per the manufacturer's instructions (Qiagen). Purified recombinant proteins (50 ng of kinase and 0.5 μg of substrate) were incubated in kinase buffer (50 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 1% Triton X-100, 100 μm ATP, 0.1 μg/μl BSA) at 30 °C for 1 h with 1 μCi of [γ-32]ATP. Reactions were stopped by addition of cold SDS-loading dye followed by boiling for 5 min. Proteins were resolved by SDS-PAGE, and incorporation of radioactive phosphate was detected by autoradiography.
Mass Spectrometry
In vitro kinase assay was carried out as described above but using unlabeled ATP. Proteins were visualized by staining gels with GelCode Blue (Thermo Scientific), and bands corresponding to substrates and kinases were excised and analyzed by MS/MS at the Johns Hopkins University Mass Spectrometry and Proteomics Facility and the Taplin Mass Spectrometry Facility. A radiolabeled kinase assay was performed in parallel to confirm the reaction efficiency.
Sterol Analysis
Sterols were extracted from cells as described previously with the following modifications (34). Cell pellets (1 × 108 cells) were resuspended in 1 ml of methanol and transferred to disposable glass tubes to which 8 ml of methanol and 4.5 ml of 60% (w/v) KOH were added along with 5 μg of cholesterol as a recovery standard. Samples were vortexed and sterol esters were saponified at 75 °C for 2 h with gentle agitation. Samples were cooled to room temperature, and nonsaponifiable sterols were extracted with 4 ml of petroleum ether (Sigma) and dried under nitrogen gas. Samples were resuspended in 300 μl of heptane, and 2 μl was injected into an automatic liquid sampler on an Agilent 6850 gas chromatograph equipped with an HP-1 column and flame ionization detector. Sterol species were identified, and peak areas were normalized to the cholesterol standard.
RESULTS
Identification of Sre1N Negative Regulators
Decreased oxygen or ergosterol synthesis stimulates proteolytic cleavage of Sre1 to produce the active Sre1N transcription factor (Fig. 1A) (8, 35). We previously demonstrated that Sre1N activity is negatively regulated in the presence of oxygen by the candidate prolyl hydroxylase Ofd1 (17–19). To identify additional genes involved in Sre1N regulation, we conducted a genetic overexpression screen in S. pombe using an integrated reporter gene regulated by Sre1N activity (Fig. 1B). The reporter gene consisted of four tandem Sre1N DNA-binding sites driving expression of ura4+ that codes for orotidine 5-monophosphate decarboxylase and is required for growth in the absence of uracil (18, 36). Orotidine 5-monophosphate decarboxylase also enables the cells to metabolize the compound 5-FOA to the toxin 5-fluorouracil; thus, cells that highly express ura4+ fail to grow in the presence of 5-FOA (29). Previously, we isolated positive regulators of Sre1N using a related reporter gene carrying two SRE sites and selecting for growth on medium lacking uracil (18).
FIGURE 1.
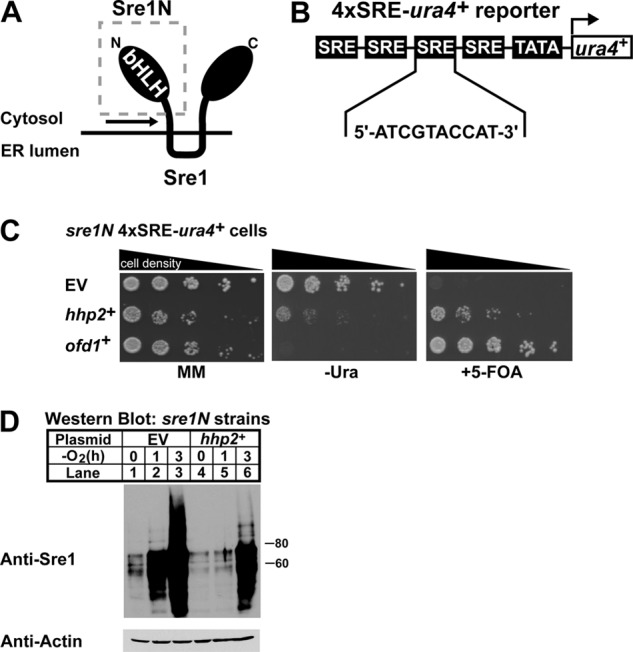
Genetic screen identifies negative regulators of Sre1N. A, schematic of Sre1 with soluble Sre1N (gray box) and region of cleavage (black arrow) highlighted. ER, endoplasmic reticulum. B, diagram of the integrated 4×SRE-ura4+ reporter gene containing four consecutive SRE sequences upstream of a minimal promoter and the ura4+ gene. C, growth assay of sre1N 4×SRE-ura4+ reporter strains containing empty vector (EV), hhp2+, or ofd1+ constructs plated onto minimal medium (MM), minimal medium lacking uracil (−Ura), and minimal medium containing 5-FOA. Cells were plated in 5-fold serial dilutions. D, Western blot analysis of Sre1N expression in the sre1N 4×SRE-ura4+ reporter strain expressing adh-driven empty vector or hhp2+ constructs. Cells were grown in the absence of oxygen for the indicated times. Actin served as a loading control.
To identify negative regulators of Sre1N, we generated a strain that expressed the 4×SRE-ura4+ reporter construct and Sre1N (amino acids 1–440) from the endogenous sre1+ locus (18). Expression of Sre1N was achieved by insertion of a stop codon upstream of the first transmembrane domain segment of Sre1 (Fig. 1A). This approach enabled the expression of Sre1N independently of proteolytic cleavage. The sre1N 4×SRE-ura4+ reporter strain was able to grow in complete medium and in medium lacking uracil; however, the strain failed to grow in the presence of 5-FOA (Fig. 1C) (18). In contrast, we previously reported that sre1N 3×SRE-ura4+ reporter cells grew on 5-FOA medium, suggesting that modest inhibition of Sre1N activity might rescue growth of sre1N 4×SRE-ura4+ cells on 5-FOA medium (18).
To screen for negative regulators of Sre1N, we transformed sre1N 4×SRE-ura4+ cells with the SPLE-1 S. pombe plasmid cDNA library (32). Each cDNA plasmid was driven by the alcohol dehydrogenase promoter (adh) and contained the S. cerevisiae LEU2 gene, which allowed for selection of transformants on medium lacking leucine (32). A total of 2.4 × 105 transformants were screened for growth on leucine-free medium containing 5-FOA. Cells expressing cDNAs that decreased Sre1N transcriptional activity would grow in the presence of 5-FOA, whereas those containing cDNAs that either enhanced or had no effect on Sre1N activity would fail to grow. Clones growing in the presence of 5-FOA were isolated, and plasmids were extracted and sequenced to identify the expressed cDNAs. Several clones were isolated that contained plasmids coding for the same gene. From our screen we isolated several genes that promoted growth in the presence of 5-FOA. Notably ofd1+ and hhp2+ were isolated multiple times, and tom70+ was isolated once (Table 1). ofd1+ is a transcriptional target and negative regulator of Sre1N, thus validating our screen, and tom70+ is a predicted subunit of the mitochondrial TOM complex (14, 17). hhp2+, a homolog of the mammalian casein kinase 1 (CK1) family, was isolated four times (22, 37). Hhp2, like Sre1N, localizes to the nucleus and has been shown to function in response to DNA damage and to be required for chromosomal segregation during meiosis (22, 23). A role for Hhp2 in sterol homeostasis has not been described.
TABLE 1.
cDNAs isolated from genetic screen
| Name | Description | No. of clones isolated |
|---|---|---|
| hhp2+ | Casein kinase 1 family protein | 4 |
| ofd1+ | 2-Oxoglutarate and Fe(II) dioxygenase domain containing protein 1 | 8 |
| tom70+ | Mitochondrial TOM complex subunit Tom70 | 1 |
Overexpression of hhp2+ Reduces Sre1N Protein
We verified that the 5-FOA-resistant phenotype of the hhp2+-expressing clones was plasmid-dependent by retransforming the hhp2+ library plasmid into the sre1N 4×SRE-ura4+ strain and plating cells onto minimal medium, minimal medium lacking uracil, and minimal medium with 5-FOA (Fig. 1C). Strains containing appropriate empty or ofd1+ plasmids served as controls. Cells with empty vector grew on both minimal medium and minimal medium lacking uracil plates, but cells failed to grow on plates that contained 5-FOA. Cells expressing hhp2+ showed reduced growth in the absence of uracil compared with cells containing empty vector but more growth relative to cells expressing ofd1+. Additionally, like Ofd1, expression of hhp2+ promoted growth on medium containing 5-FOA relative to empty vector cells. These data suggest that hhp2+ expression negatively affects Sre1N activity, resulting in decreased Ura4 production compared with empty vector cells, allowing for growth in the presence of 5-FOA. Compared with ofd1+-expressing cells, hhp2+-expressing cells grew better on uracil-free medium but more slowly on 5-FOA medium.
To test if hhp2+ altered Sre1N levels, we measured Sre1N by Western blot in sre1N 4×SRE-ura4+ cells expressing either empty vector or hhp2+ grown under normoxia or low oxygen (Fig. 1D). Empty vector cells expressed Sre1N in the presence of oxygen, and upon oxygen depletion, the Sre1N increased. However, hhp2+-expressing cells contained less Sre1N than empty vector cells both in the presence of oxygen and upon being shifted to low oxygen. These data, along with the growth assay, indicate that hhp2+ is a negative regulator of Sre1N.
We performed a parallel study to verify the phenotype for tom70+. Compared with cells with empty vector, tom70+-expressing cells were resistant to 5-FOA (Fig. 2A) and contained less Sre1N in the absence of oxygen relative to cells with empty vector (Fig. 2B). These data suggest that, like hhp2+, tom70+ negatively regulates Sre1N. However, tom70+-expressing cells exhibited slow growth relative to cells carrying empty vector, necessitating additional experiments to determine whether these effects are direct. These studies are ongoing.
FIGURE 2.
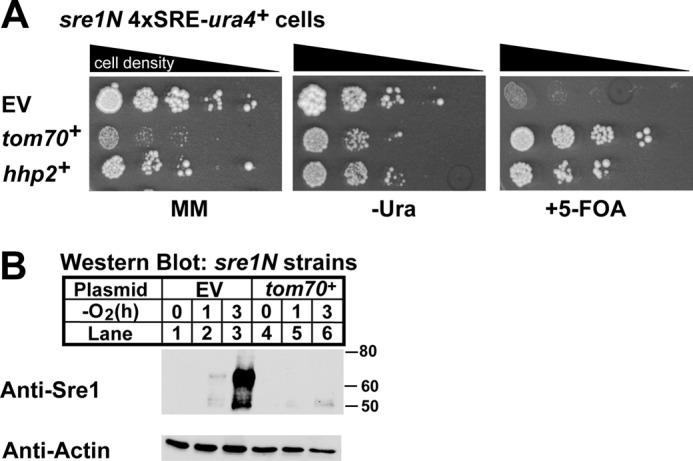
Verification of tom70+ as a negative regulator of Sre1N. A, growth assay of sre1N 4×SRE-ura4+ reporter strains containing empty vector (EV), tom70+, or hhp2+ constructs plated as in Fig. 1C. B, Western blot analysis of Sre1N expression in the sre1N 4×SRE-ura4+ reporter strain expressing adh-driven empty vector or tom70+ constructs. Cells were treated as in Fig. 1D. MM, minimal medium.
Hhp2 Regulates Sre1N and Sterol Homeostasis
Overexpression of hhp2+ decreased Sre1N. To confirm the role of Hhp2 in Sre1N regulation, we generated an sre1N strain with a deletion of hhp2+ and assessed Sre1N levels in sre1N and sre1N hhp2Δ cells by Western blot under normoxic and low oxygen conditions (Fig. 3A, top panel). In the presence of oxygen, Sre1N levels were low in sre1N cells and increased upon shifting cells to low oxygen conditions. However, sre1N hhp2Δ cells showed elevated levels of Sre1N in the presence of oxygen and a slight increase during low oxygen.
FIGURE 3.
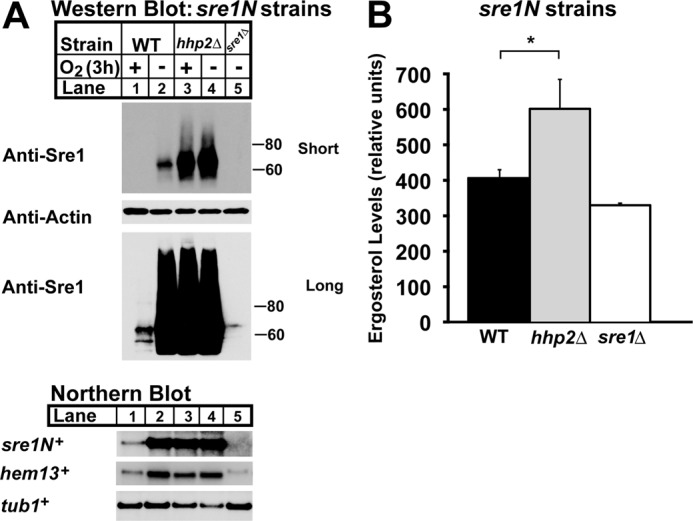
Hhp2 regulates Sre1N and sterol homeostasis. A, Western (top panels) and Northern blot (bottom panel) analyses of sre1N, sre1N hhp2Δ, and sre1Δ cells grown in the presence or absence of oxygen for 3 h. Whole-cell lysates were subjected to Western blotting to assess Sre1N and β-actin expression. Total RNA was analyzed by Northern blot and probed for sre1N+, hem13+, and tub1+ expression. Blots shown are representative of three independent experiments. B, sterol levels of sre1N, sre1N hhp2Δ, and sre1Δ cells grown under normoxic conditions were quantified by gas chromatography. Recovery was normalized to an internal cholesterol standard. All data are expressed as mean ± S.E. of three independent experiments. *, p < 0.03.
Sre1N directly activates its own transcription through a positive feedback loop (14). To determine whether the Sre1N present in sre1N hhp2Δ cells was functional, we examined Sre1N activity. We assessed mRNA levels of the Sre1N target genes hem13+ and sre1N+ by Northern blot and observed that, relative to sre1N cells, sre1N hhp2Δ cells exhibited higher levels of target mRNAs under normoxic conditions (Fig. 3A, bottom panel). Because Sre1N induces expression of genes required for ergosterol biosynthesis, we also measured ergosterol levels of sre1N and sre1N hhp2Δ cells in the presence of oxygen (Fig. 3B) (14). Compared with sre1N cells, ergosterol was 47% higher in sre1N hhp2Δ cells. Together, these results confirm our overexpression studies showing that Hhp2 is a negative regulator of Sre1N, and we demonstrate that deletion of hhp2+ increases functional Sre1N and ergosterol.
Hhp2 Controls Sre1N Stability
Oxygen regulates both the stability of Sre1N and its DNA binding activity (17, 19). Given that sre1N expression involves a positive feedback loop, changes at multiple regulatory steps could affect Sre1N expression. To test whether Hhp2 regulates Sre1N stability, we utilized a previously characterized sre1N-MP (mutant promoter) strain in which the Sre1 DNA-binding sites (SREs) in the sre1N promoter have been mutated, inhibiting the Sre1N-positive feedback loop (17). Consequently, changes in Sre1N are due to post-transcriptional regulation in sre1N-MP cells. Under low oxygen, sre1N-MP cells showed increased Sre1N, but no change in sre1N mRNA, reflecting the oxygen regulation of Sre1N stability (Fig. 4A) (17). sre1N-MP hhp2Δ cells showed increased Sre1N in both the presence and absence of oxygen relative to sre1N-MP cells (Fig. 4A, lanes 3 and 4). sre1N+ mRNA was equal under all conditions, consistent with Hhp2 post-transcriptional regulation of Sre1N. As expected, increased Sre1N resulted in elevated mRNA for the Sre1N target gene hem13+, indicating that the Sre1N accumulating in sre1N-MP hhp2Δ cells was functional.
FIGURE 4.

Hhp2 accelerates Sre1N degradation. A, Western (top panel) and Northern (bottom panel) blot analyses of sre1N-MP and sre1N-MP hhp2Δ strains grown in the presence or absence of oxygen for 3 h. Whole-cell lysates and total RNA were analyzed as in Fig. 3. Blots shown are representative of three independent experiments. B, cycloheximide chase experiment of sre1N and sre1N hhp2Δ cells grown in the absence of oxygen for 4 h, treated with cycloheximide (CHX) (200 μg/ml) at t = 0, and shifted to normoxic conditions for the indicated time. Graph shows percentage of Sre1N remaining at each time point. Blot and graph are representative of three independent experiments.
To test further whether Hhp2 controls Sre1N stability, we measured Sre1N half-life using a cycloheximide chase assay (Fig. 4B) (17). sre1N and sre1N hhp2Δ cells were grown in the absence of oxygen for 4 h to accumulate Sre1N. Cells were then treated with the translation inhibitor cycloheximide and shifted to normoxic conditions, and whole-cell lysates were harvested every 5 min. Western blot analysis indicated that the half-life of Sre1N in wild-type cells was ∼8 min, similar to what has been reported previously (17, 18). In contrast, hhp2Δ cells had an average Sre1N half-life of ∼27 min (Fig. 4B). These results coupled with our studies in the sre1N-MP strain show that Hhp2 regulates Sre1N by accelerating its degradation.
Hhp2 Kinase Activity Is Required to Accelerate Sre1N Degradation
Next, we sought to elucidate the mechanism by which Hhp2 regulates Sre1N stability. Hhp2 is the S. pombe homolog of mammalian casein kinase 1δ (CK1δ) and CK1ϵ that have been shown to phosphorylate and subsequently promote the degradation of target proteins; therefore, we hypothesized that Hhp2 may regulate Sre1N stability through Sre1N phosphorylation (22, 25, 37–39).
To investigate whether Hhp2 kinase activity was required for Sre1N regulation, we performed Western blot analysis of whole-cell lysates from sre1N hhp2Δ strains expressing empty vector, hhp2+-myc, or kinase-dead hhp2+-K41N-myc (generated by a lysine to asparagine mutation at residue 41) (Fig. 5A). sre1N hhp2Δ cells containing an empty vector exhibited highly elevated levels of Sre1N relative to control sre1N cells, and expression of hhp2+ in the mutant resulted in a significant decrease of Sre1N. However, when kinase-dead Hhp2 K41N was expressed to an equivalent level in the mutant, we observed no change in Sre1N compared with empty vector hhp2Δ cells. The inability of Hhp2 K41N to alter levels of Sre1N indicated that Hhp2 kinase activity is required to accelerate Sre1N degradation. Additionally, the data further confirm Hhp2 as a negative regulator of Sre1N protein, because expression of hhp2+ largely complemented the hhp2Δ strain. Incomplete complementation was most likely due to cell-to-cell variation in plasmid copy number due to the fact that S. pombe expression systems lack centromere-based plasmids (40).
FIGURE 5.
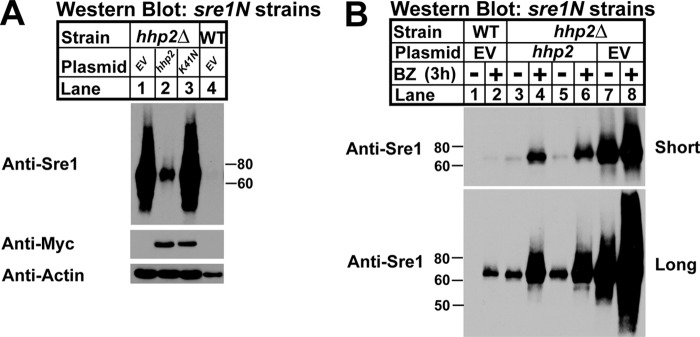
Hhp2 kinase activity is required for Sre1N degradation. A, Western blot of whole-cell lysates from sre1N hhp2Δ cells carrying empty vector, hhp2+, or hhp2-K41N constructs and sre1N cells with empty vector. Blots were probed for Sre1N, Myc, and β-actin expression and are representative of three independent experiments. B, Western blot of whole-cell lysates from sre1N or sre1N hhp2Δ cells carrying empty vector or hhp2+ plasmid treated with DMSO or 1 mm bortezomib (BZ) for 3 h in the presence of oxygen. Lanes 3–6 show two independent plasmid transformants. Blots were probed for Sre1N expression and are representative of three independent experiments.
Sre1N is degraded via the proteasome in the presence of oxygen, and thus we sought to determine whether Hhp2 accelerated Sre1N degradation through this pathway (17, 18). We assayed Sre1N levels in hhp2Δ strains carrying empty vector or expressing hhp2+-myc after treatment for 3 h with the proteasome inhibitor bortezomib (BZ) or vehicle (DMSO); sre1N cells containing empty vector were included as a positive control (Fig. 5B). Two independent hhp2-myc hhp2Δ strains were analyzed. In sre1N vehicle-treated cells, Sre1N was undetectable by Western blot, and treatment with BZ resulted in accumulation of Sre1N indicating proteasome inhibition (Fig. 5B, lanes 1 and 2). hhp2Δ cells showed highly elevated levels of Sre1N in the absence of BZ (Fig. 5B, lane 7). Expression of hhp2+ in hhp2Δ cells decreased Sre1N as expected due to Sre1N accelerated degradation. Importantly, the Hhp2-dependent degradation was blocked by addition of BZ, consistent with Hhp2 promoting Sre1N degradation by the proteasome (Fig. 5B, lanes 3–6). BZ treatment of hhp2Δ cells carrying hhp2+ vector increased Sre1N, but levels did not equal that of the BZ-treated hhp2Δ strain carrying empty vector (Fig. 5B, lanes 4, 6, and 8). The failure of BZ treatment to completely block the effects of Hhp2 on Sre1N suggested incomplete complementation from a lack of centromere-based plasmids in S. pombe or that Hhp2 may have proteasome-independent effects on Sre1N (40). Interestingly, the hhp2Δ strain carrying empty vector contained high levels of Sre1N, and upon treatment with BZ, the Sre1N levels increased further (Fig. 5B, lanes 7 and 8), suggesting the existence of an Hhp2-independent mechanism for Sre1N degradation.
Hhp2 Binds and Phosphorylates Sre1N
Sre1N exists as a hyperphosphorylated protein that contains at least 22 phosphorylated serine and threonine residues3 (8). This fact, along with the requirement for Hhp2 kinase to accelerate Sre1N degradation, suggested that Sre1N may be an Hhp2 substrate. To address this, we first tested whether Hhp2 interacts with Sre1N by performing a co-immunoprecipitation assay using sre1N cells that expressed either untagged or Myc-tagged Hhp2 at the hhp2+ locus (Fig. 6A). Sre1N is rapidly degraded under normoxic conditions through the action of the E3 ubiquitin ligase Ubr1 (17, 19). To improve our ability to detect an interaction between Hhp2 and Sre1N, we used a ubr1Δ strain that accumulates Sre1N in the presence of oxygen. To test whether Hhp2 interacted with the precursor Sre1 or the activated transcription factor Sre1N, we performed these studies in cells that expressed endogenous full-length, cleavable Sre1. Immunoprecipitation of Hhp2 from cross-linker-treated cells specifically pulled down Sre1N, but not Sre1 precursor, indicating that Hhp2 interacts with Sre1N in vivo in the presence of oxygen (Fig. 6A).
FIGURE 6.

Hhp2 binds and phosphorylates Sre1N. A, co-immunoprecipitation (IP) of Hhp2-Myc and Sre1 from ubr1Δ, ubr1Δ hhp2-myc, and ubr1Δ hhp2-myc sre1Δ cells treated with 2 mm DSP cross-linker in PBS for 30 min was carried out using anti-Myc antibody and Nonidet P-40-solubilized whole-cell lysates. Input and bound (20× overloaded) fractions were analyzed by Western blot using anti-Myc and anti-Sre1. Blots are representative of three independent experiments. P and N denote the precursor and nuclear forms of Sre1, respectively. B, co-immunoprecipitation of Hhp2 and Sre1N in sre1N hhp2Δ cells carrying the indicated plasmids was carried out as in A except cells were grown for 4 h in the absence of oxygen, switched to normoxic conditions, and treated with 2 mm DSP cross-linker in PBS for 30 min prior to lysis. Anti-Myc bound is 10× overloaded. Blots are representative of three independent experiments. C, Western blot of whole-cell lysates from sre1N and sre1N hhp2Δ cells grown in the presence of oxygen and treated with 1 mm bortezomib (BZ) for 3 h. Lysates in lanes 3 and 4 were treated with alkaline phosphatase (AP). sre1N samples (lanes 1 and 3) were 12× overloaded relative to mutant samples to enable visualization of Sre1N protein. D, in vitro kinase assay was performed with the indicated purified recombinant Hhp2 and either Sre1N or Cnp3-N as substrate. Kinase reaction products were resolved by SDS-PAGE, and autoradiograms are shown. E, sequence of Sre1N showing mass spectrometry results of Sre1N-Hhp2 in vitro kinase reaction. Peptides identified are underlined. Phosphorylated residues are boxed.
We next investigated whether Hhp2 kinase activity was required for binding to Sre1N. Co-immunoprecipitation assays were performed using hhp2Δ cells carrying either empty vector, hhp2+-myc, or kinase-dead hhp2+-K41N-myc plasmids (Fig. 6B). Sre1N co-immunoprecipitated with both Hhp2 and Hhp2-K41N demonstrating that kinase activity is not required for the Sre1N-Hhp2 interaction. However, relative to the input lane, kinase-dead Hhp2 bound less Sre1N compared with the wild-type enzyme (Fig. 6B, lane 3), suggesting that kinase activity or the Lys-41 side chain is required for optimal Sre1N binding.
To determine whether Sre1N is an Hhp2 substrate, we tested the effect of hhp2+ deletion on Sre1N phosphorylation by Western blot (Fig. 6C). Western blot analysis of sre1N cell lysates showed Sre1N as a smear (Fig. 6C, lane 1). Treatment of cell lysates with alkaline phosphatase collapsed the smear to a single band indicating that Sre1N is hyperphosphorylated as reported previously (Fig. 6C, lane 3) (8). Sre1N mobility was unaltered in hhp2Δ cells, and the hyperphosphorylated species collapsed to a single band upon alkaline phosphatase treatment (Fig. 6C, lanes 2 and 4). These data suggest that Hhp2 is not the sole Sre1N kinase and that Hhp2 likely phosphorylates Sre1N at either a single or a small number of residues whose effect on the mass of Sre1N cannot be seen due to the heavily phosphorylated state of Sre1N.
We next decided to employ a direct approach to test whether Hhp2 phosphorylated Sre1N by performing an in vitro kinase assay using purified, bacterially expressed Hhp2 and Sre1N (Fig. 6D). Recombinant Hhp2 was active insomuch as it phosphorylated the known substrate Cnp3-N that served as a positive control (Fig. 6D, lane 3, bottom panel) (23). No kinase activity was detected in the reaction containing kinase-dead Hhp2-K41N and Sre1N (Fig. 6D, lane 4, bottom panel). Hhp2 undergoes autophosphorylation, and we observed this in the Hhp2-only reaction (Fig. 6D, lane 1) (41). This signal was absent from the Cnp3-N panel due to the different masses of Hhp2 (∼55 kDa) and Cnp3-N (∼30 kDa). Incubation of Sre1N with Hhp2 resulted in robust phosphorylation of Sre1N that required Hhp2 kinase activity (Fig. 6D, lanes 3 and 4). Hhp2-phosphorylated Sre1N species did not display the same extent of mobility shift as the smear observed in Fig. 6C, suggesting that Hhp2 phosphorylates a smaller number of Sre1N residues.
To confirm the in vitro phosphorylation of Sre1N, we performed tandem mass spectrometry analysis of excised gel slices from Sre1N–Hhp2 and Sre1N–Hhp2-K41N in vitro kinase assays. Purified Hhp2 and Sre1N have similar molecular masses and thus co-migrated on SDS-PAGE. Mass spectrometry analysis confirmed that the bands visualized by autoradiography in the Sre1N–Hhp2 sample corresponded to phosphorylated Sre1N peptides. Peptide coverage of Sre1N was 65%, and we identified four phosphorylated Sre1N residues as follows: Thr-197, Ser-245, Ser-247, and Thr-249 that are all upstream of the basic helix-loop-helix DNA binding domain of Sre1N (Fig. 6E). Several serine and threonine residues are present in the undetected Sre1N peptides; therefore, it is possible that not all phosphorylated residues were identified. We were unable to identify autophosphorylated Hhp2 residues in the Sre1N–Hhp2 sample, possibly due to the low abundance of phosphorylated Hhp2. No phosphorylated peptides for either Sre1N or Hhp2-K41N were identified in the Sre1N–Hhp2-K41N sample. Together, these data demonstrate that Hhp2 binds and phosphorylates Sre1N.
Hhp2 Is a Regulator of Sterol Homeostasis
Thus far, our studies have been conducted in cells expressing Sre1N but not the endogenous full-length Sre1 protein. To evaluate the function of Hhp2 in sterol regulation under physiological conditions, we generated an hhp2 deletion strain from wild-type S. pombe that expresses a full-length Sre1. Measurement of ergosterol levels in the hhp2Δ cells grown in the presence of oxygen revealed a 29% increase in ergosterol relative to wild-type cells (Fig. 7A). We next assessed Sre1N levels in these cells and observed an accumulation of Sre1N in the hhp2Δ strain relative to wild-type cells, consistent with a role for Hhp2 in Sre1N degradation (Fig. 7B). We also observed an increase in Sre1 precursor in hhp2Δ cells, likely due to increased Sre1N and positive feedback on the sre1+ promoter. Together, these data demonstrate that Hhp2 is a key negative regulator of Sre1N that is required to maintain sterol homeostasis under nonstress conditions. In the presence of oxygen, Hhp2 functions in the degradation of Sre1N that results from a constitutive, low level of Sre1 cleavage.
FIGURE 7.
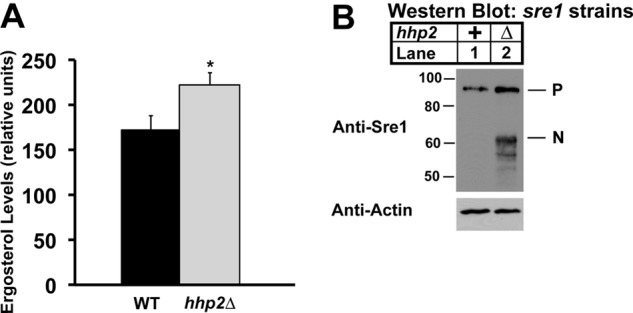
Hhp2 regulates sterol homeostasis. A, ergosterol level in wild-type and hhp2Δ cells was measured by gas chromatography. Recovery was normalized to an internal cholesterol standard. All data are expressed as mean ± S.E. of three independent experiments. *, p < 0.04. B, Western blot of whole-cell lysates from wild-type and hhp2Δ cells. Blots probed for Sre1 and β-actin expression are representative of three independent experiments. Uncleaved Sre1 precursor (P) and cleaved Sre1N nuclear form (N) are designated.
DISCUSSION
SREBPs regulate sterol homeostasis in yeast and mammals (42). The initial characterization of the fission yeast SREBP pathway revealed that the active Sre1N transcription factor was hyperphosphorylated, suggesting potential regulation of sterol homeostasis by kinases/phosphatases (8). Using a high copy plasmid screen, we identified the mammalian casein kinase 1δ/ϵ homolog, Hhp2, as a novel regulator of Sre1N stability and sterol homeostasis in fission yeast. Hhp2 binds Sre1N and accelerates its degradation via the proteasome. Hhp2 phosphorylates Sre1N in vitro, and kinase activity is required for Sre1N degradation, suggesting that Hhp2 phosphorylation of Sre1N targets it for degradation. Consistent with the role of Sre1N in activating sterol synthesis, deletion of Hhp2 increased sterols in wild-type cells and cells expressing only the truncated Sre1N. Importantly, Hhp2 functions independently from changes in oxygen, providing a novel mechanism for control of SREBP activity.
The SREBP pathway is highly conserved in pathogenic fungi that cause infections in immunocompromised individuals (43). Indeed, Sre1 is required for virulence of the fungal pathogens Cryptococcus neoformans and Aspergillus fumigatus in mouse models of disease (44–46). As a regulator of Sre1, Hhp2 may also be required for virulence of these clinically important pathogens. Current antifungal drugs target the ergosterol pathway (47). Given that Hhp2 regulates ergosterol homeostasis, modulation of Hhp2 activity may improve the efficacy of existing antifungals.
Using mass spectrometry, we identified four Sre1N residues phosphorylated by Hhp2 in vitro (Fig. 6E), and these data validated that Hhp2 binds Sre1N directly. To test whether these residues were endogenous substrates required for Sre1N stability, we mutated all four amino acids to alanine. Stability of this mutant Sre1N was unchanged, indicating that the residues are in vitro substrates but likely not Hhp2 sites in vivo (data not shown). CK1s prefer pre-phosphorylated substrates that have been primed by action of another kinase and recognize the consensus sequence (pS/pT)XX(S/T) (21). The Sre1N substrate used in our studies was purified from E. coli and likely lacked any priming phosphorylation that occurs in fission yeast cells. Future studies will determine the endogenous Hhp2 phosphorylation sites to dissect the role of Hhp2-dependent degradation in the overall control of the Sre1 pathway. Importantly, the requirement for priming phosphorylation for Hhp2 substrate recognition provides an opportunity for additional kinase regulation of Sre1N turnover.
Sre1N is a short lived protein with a half-life of ∼8 min in wild-type cells (17). This rapid turnover of Sre1N is mediated by the E2 ubiquitin-conjugating enzyme Rhp6 and the E3 ubiquitin ligase Ubr1 that target Sre1N for proteasomal degradation (19). Hhp2 accelerates Sre1N proteasomal degradation, and hhp2+ deletion increased Sre1N half-life to ∼27 min (Fig. 4B). Sre1N degradation in hhp2Δ cells is still faster than in ubr1Δ cells (∼100 min), and addition of proteasome inhibitor to hhp2Δ cells further stabilized Sre1N (Fig. 5B). Together, these data indicate that Sre1N is degraded by both Hhp2-dependent and Hhp2-independent pathways. The candidate prolyl hydroxylase Ofd1 accelerates Sre1N in an oxygen-regulated manner (17, 20). Future studies will investigate the relationship between Hhp2- and Ofd1-dependent mechanisms for Sre1N degradation through Rhp6/Ubr1. The presence of Hhp2-dependent and -independent pathways would enable cells to fine-tune ergosterol metabolism in response to various stimuli such as changes in oxygen concentration and nutrient availability. In addition to ergosterol biosynthesis, Sre1N regulates genes required for heme and sphingolipid biosynthesis, electron transport, and amino acid metabolism, suggesting that different Sre1N regulatory pathways may exist to modulate specific arms of metabolism (14). Here, we describe a mechanism of Hhp2-dependent degradation of Sre1N, yet it is possible that Hhp2 may also alter Sre1N activity at the level of sre1N translation. Because Hhp2 binds to Sre1N and based on reports demonstrating the direct role of CK1s on protein stability, it is unlikely Hhp2 impacts Sre1N solely at the level of translation (25, 38, 39).
The ability of Hhp2 to accelerate Sre1N degradation provides a mechanism for integration of non-oxygen signals into the control of sterol metabolism. But the signals that control Hhp2 activity are unknown. Autophosphorylation increases Hhp2 activity in vitro, whereas CK1δ/ϵ autophosphorylation is inhibitory (41, 48–50). One possibility is that Hhp2 could be controlled by a protein phosphatase through removal of regulatory phosphorylation. In addition, it was recently reported that mammalian CK1ϵ enzymatic activity is activated upon binding to the DEAD box RNA helicase DDX3 (51). Although the precise mechanism behind this regulation is not known, discovery of DDX3 as a CK1 enhancer suggests that Hhp2 activity may also be regulated in this fashion. Additional genetic screens may identify factors that regulated Hhp2 activity, providing clues to the upstream signals that control Sre1N turnover.
In addition to Hhp2, S. pombe codes for a second CK1δ/ϵ homolog called Hhp1 that shares 75% amino acid sequence identity with Hhp2 (22). Both kinases have been shown to serve redundant cellular functions (22, 23, 52). However, previously, we identified Hhp1 in a high copy plasmid screen to identify positive regulators of Sre1N activity (18). It will be interesting to test whether Sre1N is also a substrate for Hhp1 and whether the two kinases have opposing effects on Sre1N activity.
The CK1 Hhp2 is the first kinase reported to function in the fungal SREBP pathway, but multiple kinases have been implicated in the regulation of mammalian SREBPs, including GSK3, CDK8, and AMP-activate protein kinase (53–56). Interestingly, like Hhp2, GSK3 and CDK8 phosphorylate SREBP, promoting its degradation via the proteasome (53, 55). The high degree of SREBP pathway conservation between S. pombe and mammals and the existence of mammalian Hhp2 homologs CK1δ and CK1ϵ suggest that CK1s may also regulate cholesterol homeostasis by controlling SREBP stability in humans. Indeed, a recent high throughput cDNA overexpression screen for SREBP regulators identified CK1ϵ as a negative regulator of SREBP activity in HEK-293 cells (57). Should CK1 regulation of SREBP be conserved, this finding would have important implications for regulation of lipid metabolism. Multiple lines of evidence link circadian rhythm to control of cell metabolism (25, 58–61). Mammalian CK1δ/ϵ are key regulators of the mammalian molecular circadian clock; mutation of CK1ϵ is responsible for the short period mutation in the tau mutant Syrian hamster, and a missense mutation in CK1δ results in human familial advanced sleep phase syndrome (62–64). Identification of CK1s as regulators of mammalian SREBPs would provide a mechanism for the coordinate regulation of lipid and circadian-dependent metabolism. Studies are underway to test whether SREBP regulation by CK1 is conserved.
Acknowledgments
We kindly thank Charlie Hoffman (Boston College) for providing the S. pombe cDNA plasmid library and the Taplin Mass Spectrometry Facility and the Johns Hopkins University Mass Spectrometry and Proteomics Facility for mass spectrometry analyses. We also thank Diedre Ribbens for reviewing the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL077588 (to P. J. E.) and HL106971 (to R. T. B.).
B. T. Hughes and P. J. Espenshade, unpublished observations.
- SREBP
- sterol regulatory element-binding protein
- CK1
- casein kinase 1
- SRE
- sterol regulatory element
- 5-FOA
- 5-fluoroorotic acid
- BZ
- bortezomib
- EMM
- Edinburgh 20 minimal medium
- DSP
- dithiobis(succinimidyl propionate).
REFERENCES
- 1. Ikonen E. (2008) Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 125–138 [DOI] [PubMed] [Google Scholar]
- 2. Tabas I. (2002) Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J. Clin. Invest. 110, 905–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maxfield F. R., Tabas I. (2005) Role of cholesterol and lipid organization in disease. Nature 438, 612–621 [DOI] [PubMed] [Google Scholar]
- 4. Goldstein J. L., DeBose-Boyd R. A., Brown M. S. (2006) Protein sensors for membrane sterols. Cell 124, 35–46 [DOI] [PubMed] [Google Scholar]
- 5. Horton J. D., Goldstein J. L., Brown M. S. (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Osborne T. F., Espenshade P. J. (2009) Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev. 23, 2578–2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blatzer M., Barker B. M., Willger S. D., Beckmann N., Blosser S. J., Cornish E. J., Mazurie A., Grahl N., Haas H., Cramer R. A. (2011) SREBP coordinates iron and ergosterol homeostasis to mediate triazole drug and hypoxia responses in the human fungal pathogen Aspergillus fumigatus. PLoS Genet. 7, e1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hughes A. L., Todd B. L., Espenshade P. J. (2005) SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell 120, 831–842 [DOI] [PubMed] [Google Scholar]
- 9. Matsumoto E., Ishihara A., Tamai S., Nemoto A., Iwase K., Hiwasa T., Shibata S., Takiguchi M. (2010) Time of day and nutrients in feeding govern daily expression rhythms of the gene for sterol regulatory element-binding protein (SREBP)-1 in the mouse liver. J. Biol. Chem. 285, 33028–33036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peterson T. R., Sengupta S. S., Harris T. E., Carmack A. E., Kang S. A., Balderas E., Guertin D. A., Madden K. L., Carpenter A. E., Finck B. N., Sabatini D. M. (2011) mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hatori M., Hirota T., Iitsuka M., Kurabayashi N., Haraguchi S., Kokame K., Sato R., Nakai A., Miyata T., Tsutsui K., Fukada Y. (2011) Light-dependent and circadian clock-regulated activation of sterol regulatory element-binding protein, X-box-binding protein 1, and heat shock factor pathways. Proc. Natl. Acad. Sci. U.S.A. 108, 4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kotzka J., Lehr S., Roth G., Avci H., Knebel B., Muller-Wieland D. (2004) Insulin-activated Erk-mitogen-activated protein kinases phosphorylate sterol regulatory element-binding protein-2 at serine residues 432 and 455 in vivo. J. Biol. Chem. 279, 22404–22411 [DOI] [PubMed] [Google Scholar]
- 13. Rosenfeld E., Beauvoit B. (2003) Role of the non-respiratory pathways in the utilization of molecular oxygen by Saccharomyces cerevisiae. Yeast 20, 1115–1144 [DOI] [PubMed] [Google Scholar]
- 14. Todd B. L., Stewart E. V., Burg J. S., Hughes A. L., Espenshade P. J. (2006) Sterol regulatory element binding protein is a principal regulator of anaerobic gene expression in fission yeast. Mol. Cell. Biol. 26, 2817–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stewart E. V, Nwosu C. C., Tong Z., Roguev A., Cummins T. D., Kim D. U., Hayles J., Park H. O., Hoe K. L., Powell D. W., Krogan N. J., Espenshade P. J. (2011) Yeast SREBP cleavage activation requires the Golgi Dsc E3 ligase complex. Mol. Cell 42, 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Stewart E. V., Lloyd S. J., Burg J. S., Nwosu C. C., Lintner R. E., Daza R., Russ C., Ponchner K., Nusbaum C., Espenshade P. J. (2012) Yeast sterol regulatory element-binding protein (SREBP) cleavage requires Cdc48 and Dsc5, a ubiquitin regulatory X domain-containing subunit of the Golgi Dsc E3 ligase. J. Biol. Chem. 287, 672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hughes B. T., Espenshade P. J. (2008) Oxygen-regulated degradation of fission yeast SREBP by Ofd1, a prolyl hydroxylase family member. EMBO J. 27, 1491–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee C. Y., Stewart E. V, Hughes B. T., Espenshade P. J. (2009) Oxygen-dependent binding of Nro1 to the prolyl hydroxylase Ofd1 regulates SREBP degradation in yeast. EMBO J. 28, 135–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee C. Y., Yeh T. L., Hughes B. T., Espenshade P. J. (2011) Regulation of the Sre1 hypoxic transcription factor by oxygen-dependent control of DNA binding. Mol. Cell 44, 225–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Porter J. R., Lee C. Y., Espenshade P. J., Iglesias P. A. (2012) Regulation of SREBP during hypoxia requires Ofd1-mediated control of both DNA binding and degradation. Mol. Biol. Cell 23, 3764–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheong J. K., Virshup D. M. (2011) Casein kinase 1: Complexity in the family. Int. J. Biochem. Cell Biol. 43, 465–469 [DOI] [PubMed] [Google Scholar]
- 22. Dhillon N., Hoekstra M. F. (1994) Characterization of two protein kinases from Schizosaccharomyces pombe involved in the regulation of DNA repair. EMBO J. 13, 2777–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ishiguro T., Tanaka K., Sakuno T., Watanabe Y. (2010) Shugoshin-PP2A counteracts casein-kinase-1-dependent cleavage of Rec8 by separase. Nat. Cell Biol. 12, 500–506 [DOI] [PubMed] [Google Scholar]
- 24. Price M. A. (2006) CKI, there's more than one: casein kinase I family members in Wnt and Hedgehog signaling. Genes Dev. 20, 399–410 [DOI] [PubMed] [Google Scholar]
- 25. Li S., Chen X.-W., Yu L., Saltiel A. R., Lin J. D. (2011) Circadian metabolic regulation through crosstalk between casein kinase 1δ and transcriptional coactivator PGC-1α. Mol. Endocrinol 25, 2084–2093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kalousi A., Mylonis I., Politou A. S., Chachami G., Paraskeva E., Simos G. (2010) Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 123, 2976–2986 [DOI] [PubMed] [Google Scholar]
- 27. Burke J. D., Gould K. L. (1994) Molecular cloning and sequence characterization of the Schizosaccharomyces pombe his3 gene for use as a selectable marker. Mol. Gen. Genet. 242, 169–176 [DOI] [PubMed] [Google Scholar]
- 28. Bähler J., Wu J. Q., Longtine M. S., Shah N. G., McKenzie A., 3rd., Steever A. B., Wach A., Philippsen P., Pringle J. R. (1998) Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943–951 [DOI] [PubMed] [Google Scholar]
- 29. Boeke J. D., Trueheart J., Natsoulis G., Fink, G. R. (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 154, 164–175 [DOI] [PubMed] [Google Scholar]
- 30. Okazaki K., Okazaki N., Kume K., Jinno S., Tanaka K., Okayama H. (1990) High-frequency transformation method and library transducing vectors for cloning mammalian cDNAs by trans-complementation of Schizosaccharomyces pombe. Nucleic Acids Res. 18, 6485–6489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Forsburg S. L., Sherman D. A. (1997) General purpose tagging vectors for fission yeast. Gene 191, 191–195 [DOI] [PubMed] [Google Scholar]
- 32. Janoo R. T., Neely L. A., Braun B. R., Whitehall S. K., Hoffman C. S. (2001) Transcriptional regulators of the Schizosaccharomyces pombe fbp1 gene include activation complex. Genetics 157, 1205–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Belle A., Tanay A., Bitincka L., Shamir R., O'Shea E. K. (2006) Quantification of protein half-lives in the budding yeast proteome. Proc. Natl. Acad. Sci. U.S.A. 103, 13004–13009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hughes A. L., Lee C. Y., Bien C. M., Espenshade P. J. (2007) 4-Methyl sterols regulate fission yeast SREBP-Scap under low oxygen and cell stress. J. Biol. Chem. 282, 24388–24396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Porter J. R., Burg J. S., Espenshade P. J., Iglesias P. A. (2010) Ergosterol regulates sterol regulatory element binding protein (SREBP) cleavage in fission yeast. J. Biol. Chem. 285, 41051–41061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sehgal A., Lee C. Y., Espenshade P. J. (2007) SREBP controls oxygen-dependent mobilization of retrotransposons in fission yeast. PLoS Genet. 3, e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kearney P. H., Ebert M., Kuret J. (1994) Molecular cloning and sequence analysis of two novel fission yeast casein kinase-1 isoforms. Biochem. Biophys. Res. Commun. 203, 231–236 [DOI] [PubMed] [Google Scholar]
- 38. Akashi M., Tsuchiya Y., Yoshino T., Nishida E. (2002) Control of intracellular dynamics of mammalian period proteins by casein kinase Iϵ (CKIϵ) and CKIδ in cultured cells. Mol. Cell. Biol. 22, 1693–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eide E. J., Woolf M. F., Kang H., Woolf P., Hurst W., Camacho F., Vielhaber E. L., Giovanni A., Virshup D. M. (2005) Control of mammalian circadian rhythm by CKIϵ-regulated proteasome-mediated PER2 degradation. Mol. Cell. Biol. 25, 2795–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hayles J., Nurse P. (1992) Genetics of the fission yeast. Schizosaccharomyces pombe. Annu. Rev. Genet. 26, 373–402 [DOI] [PubMed] [Google Scholar]
- 41. Hoekstra M. F., Dhillon N., Carmel G., DeMaggio A. J., Lindberg R. A., Hunter T., Kuret J. (1994) Budding and fission yeast casein kinase I isoforms have dual-specificity protein kinase activity. Mol. Biol. Cell 5, 877–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Espenshade P. J., Hughes A. L. (2007) Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet. 41, 401–427 [DOI] [PubMed] [Google Scholar]
- 43. Bien C. M., Espenshade P. J. (2010) Sterol regulatory element binding proteins in fungi: hypoxic transcription factors linked to pathogenesis. Eukaryot. Cell 9, 352–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee H., Bien C. M., Hughes A. L., Espenshade P. J., Kwon-Chung K. J., Chang Y. C. (2007) Cobalt chloride, a hypoxia-mimicking agent, targets sterol synthesis in the pathogenic fungus Cryptococcus neoformans. Mol. Microbiol. 65, 1018–1033 [DOI] [PubMed] [Google Scholar]
- 45. Chun C. D., Liu O. W., Madhani H. D. (2007) A link between virulence and homeostatic responses to hypoxia during infection by the human fungal pathogen Cryptococcus neoformans. PLoS Pathog. 3, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Willger S. D., Puttikamonkul S., Kim K. H., Burritt J. B., Grahl N., Metzler L. J., Barbuch R., Bard M., Lawrence C. B., Cramer R. A. (2008) A sterol-regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus. PLoS Pathog. 4, e1000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kanafani Z. A., Perfect J. R. (2008) Resistance to antifungal agents: mechanisms and clinical impact. Clin. Infect. Dis. 46, 120–128 [DOI] [PubMed] [Google Scholar]
- 48. Cegielska A., Gietzen K. F., Rivers A., Virshup D. M. (1998) Autoinhibition of casein kinase Iϵ (CKIϵ) is relieved by protein phosphatases and limited proteolysis. J. Biol. Chem. 273, 1357–1364 [DOI] [PubMed] [Google Scholar]
- 49. Gietzen K. F., Virshup D. M. (1999) Identification of inhibitory autophosphorylation sites in casein kinase Iϵ. J. Biol. Chem. 274, 32063–32070 [DOI] [PubMed] [Google Scholar]
- 50. Swiatek W., Tsai I. C., Klimowski L., Pepler A., Barnette J., Yost H. J., Virshup D. M. (2004) Regulation of casein kinase Iϵ activity by Wnt signaling. J. Biol. Chem. 279, 13011–13017 [DOI] [PubMed] [Google Scholar]
- 51. Cruciat C. M., Dolde C., de Groot R. E., Ohkawara B., Reinhard C., Korswagen H. C., Niehrs C. (2013) RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt/β-catenin signaling. Science 339, 1436–1441 [DOI] [PubMed] [Google Scholar]
- 52. Rumpf C., Cipak L., Dudas A., Benko Z., Pozgajova M., Riedel C. G., Ammerer G., Mechtler K., Gregan J. (2010) Casein kinase 1 is required for efficient removal of Rec8 during meiosis I. Cell Cycle 9, 2657–2662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao X., Feng D., Wang Q., Abdulla A., Xie X. J., Zhou J., Sun Y., Yang E. S., Liu L. P., Vaitheesvaran B., Bridges L., Kurland I. J., Strich R., Ni J. Q., Wang C., Ericsson J., Pessin J. E., Ji J. Y., Yang F. (2012) Regulation of lipogenesis by cyclin-dependent kinase 8–mediated control of SREBP-1. J. Clin. Invest. 122, 2417–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Y., Xu S., Mihaylova M. M., Zheng B., Hou X., Jiang B., Park O., Luo Z., Lefai E., Shyy J. Y., Gao B., Wierzbicki M., Verbeuren T. J., Shaw R. J., Cohen R. A., Zang M. (2011) AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 13, 376–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sundqvist A., Bengoechea-Alonso M. T., Ye X., Lukiyanchuk V., Jin J., Harper J. W., Ericsson J. (2005) Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 1, 379–391 [DOI] [PubMed] [Google Scholar]
- 56. Punga T., Bengoechea-Alonso M. T., Ericsson J. (2006) Phosphorylation and ubiquitination of the transcription factor sterol regulatory element-binding protein-1 in response to DNA binding. J. Biol. Chem. 281, 25278–25286 [DOI] [PubMed] [Google Scholar]
- 57. Chatterjee S., Szustakowski J. D., Nanguneri N. R., Mickanin C., Labow M. A., Nohturfft A., Dev K. K., Sivasankaran R. (2009) Identification of novel genes and pathways regulating SREBP transcriptional activity. PLoS One 4, e5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lamia K. A., Storch K. F., Weitz C. J. (2008) Physiological significance of a peripheral tissue circadian clock. Proc. Natl. Acad. Sci. U.S.A. 105, 15172–15177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rudic R. D., McNamara P., Curtis A. M., Boston R. C., Panda S., Hogenesch J. B., Fitzgerald G. A. (2004) BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol. 2, e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marcheva B., Ramsey K. M., Buhr E. D., Kobayashi Y., Su H., Ko C. H., Ivanova G., Omura C., Mo S., Vitaterna M. H., Lopez J. P., Philipson L. H., Bradfield C. A., Crosby S. D., JeBailey L., Wang X., Takahashi J. S., Bass J. (2010) Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinemia and diabetes. Nature 466, 627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Turek F. W., Joshu C., Kohsaka A., Lin E., Ivanova G., McDearmon E., Laposky A., Losee-Olson S., Easton A., Jensen D. R., Eckel R. H., Takahashi J. S., Bass J. (2005) Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308, 1043–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ralph M. R., Menaker M. (1988) A mutation of the circadian system in golden hamsters. Science 241, 1225–1227 [DOI] [PubMed] [Google Scholar]
- 63. Xu Y., Padiath Q. S., Shapiro R. E., Jones C. R., Wu S. C., Saigoh N., Saigoh K., Ptácek L. J., Fu Y. H. (2005) Functional consequences of a CKIδ mutation causing familial advanced sleep phase syndrome. Nature 434, 640–644 [DOI] [PubMed] [Google Scholar]
- 64. Lowrey P. L., Shimomura K., Antoch M. P., Yamazaki S., Zemenides P. D., Ralph M. R., Menaker M., Takahashi J. S. (2000) Positional syntenic cloning and functional characterization of the mammalian circadian mutation tau. Science 288, 483–492 [DOI] [PMC free article] [PubMed] [Google Scholar]