

Background: Rosiglitazone, a well known PPARγ agonist, stimulates adipocyte differentiation.
Results: In C3H10T1/2 cells, rosiglitazone induces prolyl hydroxylase domain proteins 1, 2, and 3, resulting in the degradation of anti-adipogenic proteins such as GATA-3, KLF-2, and TAZ.
Conclusion: Three isoforms of the prolyl hydroxylase domain protein play a key role in rosiglitazone-induced adipocyte differentiation.
Significance: Novel mechanisms involved in rosiglitazone-induced adipogenesis would provide a better understanding of adipocyte biology.
Keywords: Adipocyte, Adipogenesis, Diabetes, Krüppel-like Factor (KLF), Peroxisome Proliferator-activated Receptor (PPAR), Adipocyte Differentiation, GATA-3, PHD, PPARγ, Rosiglitazone
Abstract
Rosiglitazone, a well known insulin sensitizer, stimulates adipocyte differentiation via the activation of peroxisome proliferator-activated receptor γ (PPARγ). Previous two-dimensional proteomics studies using C3H10T1/2 murine mesenchymal pluripotent stem cells revealed that prolyl hydroxylase domain protein (PHD) levels significantly increased during rosiglitazone-induced adipocyte differentiation (RIAD). In this study, we investigated the functional role played by PHD during RIAD. Three PHD isoforms (PHD1, 2, and 3) were found to be up-regulated in C3H10T1/2 cells during RIAD, whereas PHD knockdown and treatment with PHD inhibitors (dimethyloxalyl glycine or ethyl-3,4-dihydroxybenzoate) blocked RIAD. PHD inhibition was found to be associated with increases in the levels of anti-adipogenic proteins such as GATA-3, KLF-2, and transcriptional coactivator with PDZ binding motif (TAZ), with their reduced ubiquitination, suggesting that PHDs evoke the ubiquitination/proteasomal degradation of anti-adipogenic proteins. On the other hand, MG-132 (a proteasomal inhibitor) prevented the degradation of anti-adipogenic proteins and retarded RIAD. PPARγ antagonists (bisphenol A diglycidyl ether or GW9662) blunted the effects of rosiglitazone on PHD regulation. Furthermore, putative PPARγ binding sites were identified in the promoter region of PHDs by ChIP-PCR, implying that rosiglitazone may induce PHD up-regulation directly by PPARγ activation. Consistent with in vitro results, oral administration of rosiglitazone to ob/ob mice for 2 weeks increased adipose PHD levels and decreased anti-adipogenic protein levels by increasing their ubiquitination. These results suggest that rosiglitazone increases PHD expression in a PPARγ-dependent manner and that this leads to the commitment of anti-adipogenic proteins to the ubiquitination-proteasomal pathway and to the subsequent induction of adipocyte differentiation.
Introduction
Rosiglitazone is a well known peroxisome proliferator-activated receptor γ (PPARγ)3 agonist that increases peripheral insulin sensitivity and, thus, improves glycemic control in type 2 diabetes. However, rosiglitazone induces adipogenesis in cell culture models and increases weight gain in rodents and humans (1). Thus, the dissection of insulin sensitivity and adipogenesis is required to develop agents that retain the insulin-sensitizing effect and minimize the weight gain effect of rosiglitazone, which is, at least in part, due to adipocyte differentiation. Adipocyte differentiation is controlled by coordinated actions of many transcription factors, such as CCAAT enhancer binding proteins (C/EBP) α, β, and γ and PPARγ (2, 3). On the other hand, GATA-binding protein 3 (GATA-3) (4), Krüppel-like factor 2 (KLF-2) (5), and transcriptional coactivator with PDZ binding motif (TAZ) have anti-adipogenic effects because they oppose the effects of PPARγ activation (6).
Prolyl hydroxylase domain (PHD) proteins are known to regulate hypoxia-inducible factor 1α (HIF-1α) by hydroxylating two proline residues (Pro-402 and Pro-564) in its α subunit in response to cellular oxygen availability (7, 8). As a result, prolyl hydroxylated HIF-1α is recognized by von Hippel-Lindau (VHL) protein and subjected to ubiquitination and proteasomal degradation (9–11). To date, three PHD isoforms (PHD1, 2, and 3; also known as EGLN 2, 1, and 3, respectively) have been identified in mammalian cells. These isoforms differ in terms of their mRNA abundances (12), substrate specificities (7), and inducibilities (7), although all are expressed ubiquitously. Originally, PHD was considered a regulator of HIF-1α, but recent reports suggest that it participates in differentiation independently of hypoxic status and in cellular homeostasis. For example, under normoxic conditions, all three PHDs increased throughout adipogenesis, and pharmacological inhibition of PHD activity abrogated adipocyte differentiation and reduced the expressions of all three PHDs (13), and dimethyloxalyl glycine (DMOG, a PHD inhibitor) caused osteoblasts to adopt adipocytic phenotypes (14). Fu et al. (15) reported that PHD3 regulates skeletal muscle differentiation by modulating the stability of myogenin protein, a known key player in myogenic differentiation.
We previously described a new PPARγ agonist, KR-62980, with partial agonistic activity (16). More specifically, KR-62980 increased insulin sensitivity but displayed a weak adipogenic potential relative to rosiglitazone. To elucidate the mechanisms responsible for their different effects in adipocyte differentiation, we performed a two-dimensional proteomics analysis after treating C3H10T1/2 cells with rosiglitazone or KR-62980 and identified PHD as one potential target expressed differentially that increased significantly upon RIAD. In this study, we investigated the functional role played by PHD in RIAD using C3H10T1/2 cells, and modulation of PHD was accomplished by PHD shRNAs and PHD inhibitors.
EXPERIMENTAL PROCEDURES
Materials
DMEM, FBS, penicillin, and streptomycin were obtained from Invitrogen. Rosiglitazone, MG-132, DMOG, ethyl-3,4-dihydroxybenzoate (EDHB), Oil Red O, BADGE, GW9662, and all other chemicals were from Sigma. Antibodies against PHD1, PHD2, and PHD3 were from Novus Biologicals (Littleton, CO). Antibodies against PPARγ, GATA-3, KLF-2, and goat anti-mouse IgG were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against TAZ, ubiquitin, and actin were from Cell Signaling Technology (Beverly, MA).
Animals
C57BL/6J-Lepob leptin-deficient mice (ob/ob mice, 9 weeks old, male) were bred at the Korean Research Institute of Chemical Technology (Taejeon). Animals were housed under specific pathogen-free conditions in an air-conditioned room at 23 ± 2 °C. Food and water were supplied ad libitum. All mice were randomly allocated to control and treatment groups. All animal procedures were approved by the Animal Care and Use Committee at Gachon University. After treatment, adipose tissues were isolated, quickly frozen in liquid nitrogen, and stored at −70 °C until required.
Cell Culture and Adipocyte Differentiation
C3H10T1/2 cells were obtained from the ATCC and maintained in DMEM supplemented with 2 mm l-glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% (v/v) heat-inactivated fetal bovine serum in a humidified 5% CO2 atmosphere at 37 °C. To induce adipocyte differentiation, cells (1 × 106 cells/ml, 12-well plates) were grown to 70–80% confluency. Differentiation was induced 2 days later by adding 10 μm of rosiglitazone in adipogenesis-inducing medium containing 1 μm dexamethasone, 0.5 mm isobutyl-methylxantine, 0.01 mg/ml insulin, and 10% FBS in DMEM. After 72 h, the medium was changed every other day for adipogenesis-inducing medium containing insulin (0.01 mg/ml) in the presence or absence of rosiglitazone. Cultivation was continued for up to 10 days after induction.
Oil Red O Staining
Lipid accumulation was evaluated by staining with Oil Red O as described previously (17). Briefly, cells were washed twice with PBS, fixed with 10% formalin in PBS for 30 min, washed twice with PBS, and stained with filtered Oil Red O solution (120 mg of Oil Red O dissolved in 40 ml of isopropyl alcohol) for 1 h. Lipid accumulation was observed by light phase-contrast microscopy.
Two-dimensional Proteomic Analysis
Cells were lysed in ice-cold lysis buffer (8 m urea containing 10% (v/v) 0.5 m Tris-HCl (pH 7.4), 0.02 m EDTA, 0.05 m DTT, 10% (v/v) glycerol, 6% (v/v) ampholytes (Resolyte (pH 3.5–10, Merck-BDH), 2% (v/v) CHAPS, 0.2 mg/ml RNase, and 0.2 mg/ml DNase). The lysate was clarified by centrifugation at 10,000 × g for 5 min at 4 °C, and the supernatant (80 μg of protein) was applied to 13 cm of immobilized pH gradient gels (Immobiline DryStrip 3–10 NL, Amersham Biosciences). Strips were rehydrated for 12 h at 50 V, followed by focusing for 1 h at 500 V, 1 h at 1000 V, and 10 h at 8000 V on an IPGPhor (Amersham Biosciences). The immobilized pH gradient strips were then equilibrated in a buffer (50 mm Tris-HCl (pH 8.8), 6 m urea, 30% (v/v) glycerol, and 2% (w/v) SDS) containing 1% (w/v) DTT for 30 min, followed by a further 30 min of incubation in the same buffer containing 2.5% (w/v) iodoacetamide in place of the DTT. The equilibrated immobilized pH gradient strips were rinsed gently with distilled water and then applied to a 10% SDS-polyacrylamide gel (18 × 16 cm). The second-dimension separation was performed at 150 V for 5 h. For analytical gels, the proteins were detected by silver staining using the Plus-OneTM silver kit (Amersham Biosciences) according to the protocol of the manufacturer. The stained gels were scanned using a Molecular Dynamics personal densitometer (Amersham Biosciences) at 50-μm resolution to generate 8-bit images. These images were transferred to Phoretix 2DTM analytical software, version 6.01c (Nonlinear Dynamics, Newcastle, UK). All image analyses and comparisons were carried out using this software. Selected spots were cut from stained gels and subjected to in-gel trypsin digestion. Protein identification by MALDI-TOF or electrospray ionization quadrupole TOF tandem mass spectrometry of the MS/MS analysis was performed at the Korea Basic Science Institute (Taejeon, Korea).
PHD Knockdown and Treatment with PHD Inhibitors
The transfection of shRNAs (100 ng of each/well) against PHD1, PHD2, PHD3, or control (Santa Cruz Biotechnology) into C3H10T1/2 cells (5 × 105 cells/well, 70–80% confluent) was accomplished using Lipofectamine 2000 reagent according to the instructions of the manufacturer (Invitrogen). Six hours after transfection, the medium was replaced with DMEM containing 10% FBS, and cells were treated with or without rosiglitazone. PHD inhibitors (1 mm DMOG or 100 μm EDHB, 1 μl/ml in medium) were added to C3H10T1/2 cells at induction and were maintained in medium during medium changes (the medium was changed every other day). C3H10T1/2 cells were first pretreated with inhibitors for 2 h before rosiglitazone was added. Dimethyl sulfoxide was used as a solvent for both inhibitors, and the final concentration of dimethyl sulfoxide present (0.1%) had no effect.
PPARγ Antagonist Treatment
PPARγ antagonists (20 μm BADGE or 10 μm GW9662, 1 μl/ml in media) were added to C3H10T1/2 cells at induction and maintained in medium during medium changes on alternate days. Antagonists were pretreated for 2 h before rosiglitazone was added. Dimethyl sulfoxide was used as a solvent for GW9662 and BADGE.
RT-PCR
Total RNA was isolated from cells using the easy-BLUE total RNA extraction kit (iNtRON Inc., Korea). Reverse transcription of total RNA (1 μg) was performed using AccuPower RT PreMix (Bioneer Inc. Korea). PCR primers for the amplifications of PHD1, 2, and 3; PPARγ; aP2; and GAPDH (the internal standard) were as follows: PHD1, 5′-GAG GAT ACC ACT CCC AAC AGA CC-3′ (forward) and 5′-GAG GAT ACC ACT CCC AAC AGA CC-3′ (reverse); PHD2, 5′-ATG AGC ACA GAA AGC ATG ATC-3′ (forward) and 5′-TAC AGG CTT GTC ACT CGA ATT-3′ (reverse); PHD3, 5′-TTG CTA ACC TGA CAC CCT TTG-3′ (forward) and 5′-CGG TGC AGG TTG AGC ATG TA-3′ (reverse); PPARγ, 5′-TTT TCA AGG GTG CCA GTT TC-3′ (forward) and 5′-AAT CCT TGG CCC TCT GAG AT-3′ (reverse); aP2, 5′-TGG AAG CTT GTC TCC AGT GA-3′ (forward) and 5′-GCT CTT CAC CTT CCT GTC GT-3′ (reverse); and GAPDH, 5′-TTC ACC ACC ATG GAG AAG GC-3′ (forward) and 5′-GGC ATG GAC TGT GGT CAT GA-3′ (reverse). Reverse transcription PCR was conducted using 35 amplification cycles (denaturation at 94 °C for 1 min, annealing at 60 °C for 1 min, and extension at 72 °C for 1 min), followed by a 10-min extension at 72 °C. PCR reaction mixtures were electrophoresed in 1.5% agarose gel and visualized under UV light after Gel Red (Elpis Biotech, Seoul, Korea) staining. Relative abundances of mRNA are expressed versus GAPDH mRNA.
Western Blot Analysis and Immunoprecipitation
Cells were harvested in lysis buffer containing 50 mm HEPES (pH 7.0), 250 mm NaCl, 5 mm EDTA, 0.1% Nonidet P-40, 1 mm phenylmethylsulfonyl fluoride, 0.5 mm dithiothreitol, 5 mm Na fluoride, 0.5 mm sodium orthovanadate, and 5 μg/ml each of leupeptin and aprotinin and incubated for 10 min at 4 °C. Following centrifugation at 12,000 rpm and subsequent denaturation, 50 μg of whole cell lysates was subjected to 8 or 10% SDS-PAGE and then transferred to nitrocellulose membranes (Amersham Biosciences). Blocked membranes were then incubated with anti-PHD1, PHD2, or PHD3 and blotted with secondary antibodies conjugated to HRP. For immunoprecipitation, precleared lysates (100 μg of extracts) were incubated with polyubiquitinated antibody or negative control normal mouse IgG for 2 h at 4 °C, and then protein A/G plus-agarose beads (50 μl) (Santa Cruz Biotechnology) were added. After overnight incubation at 4 °C with constant agitation, immunoprecipitated materials were eluted using SDS-PAGE loading buffer, heated for 5 min, fractionated by SDS-PAGE, and subjected to Western blot analysis. Immunoreactive bands were visualized by enhanced chemiluminescence (Amersham Biosciences Life Science), and band densities were quantified using UN-SCAN-IT Gel 5.1 software (Silk Scientific, Inc., Orem, UT) and normalized versus β-actin. The protein concentrations were determined using Bio-Rad protein assay reagent according to the instructions of the manufacturer.
ChIP-PCR
ChIP assays were performed as described previously (18). Briefly, C3H10T1/2 cells (1 × 107 cells) were cross-linked with 1% formaldehyde for 10 min at 37 °C. Cross-linking was stopped by the addition of glycine to a final concentration of 0.125 m. The cells were washed three times with ice-cold phosphate-buffered saline and kept on ice for 10 min in 25 mm HEPES (pH 7.8), 1.5 mm MgCl2, 10 mm KCl, 0.1% Nonidet P-40, 1 mm dithiothreitol, 0.5 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture (Roche). Nuclei were collected and resuspended in sonication buffer for 60 min on ice and sonicated on ice to an average length of 200 bp using a tissue lyser set at 50% amplitude. After sonication, the chromatin solution (500 μg) was incubated with Dynabeads protein A (Invitrogen) and 5 μg of rabbit anti-PPARγ (Cell Signaling Technology) or 5 μg of rabbit normal IgG (Santa Cruz Biotechnology) at 4 °C overnight. Antibody-bound complexes were obtained, and DNA fragments extricated from these complexes were purified using a QIAquick PCR purification kit (Qiagen, Shanghai, China). The purified ChIPed DNA samples were analyzed by conventional PCR. Primers for PCR were as follows: PHD2, 5′-GTTCCGGTTCGAGACCTCTG-3′ (forward) and 5′-GGGCTGCGTCTCAAGACAAC-3′ (reverse); PHD3, 5′-AGCATCTGGGTGAGCTCCAC-3′ (forward) and 5′-CCAGAGAGGTGACTAGCCCA-3′ (reverse).
In Vivo Study
Ob/ob mice were orally administered rosiglitazone (10 mg/kg/day, n = 10) or vehicle (an equivalent volume of saline, n = 10) for 2 weeks. Tail blood glucose concentrations (Accu-Check; Roche Diagnostics GmbH) and body weights were measured weekly. After 2 weeks of rosiglitazone administration, adipose tissues were removed and immediately frozen and stored in liquid nitrogen until required for assay. For protein analyses, adipose tissues were homogenized and lysed with Prep-sol (iNtRON Inc., Korea).
Statistical Analysis
Results are expressed as mean ± S.E. One-way analysis of variance followed by Tukey's multiple comparison test was used to determine statistical significances. Statistical significance was accepted for p values < 0.05.
RESULTS
PHD Expression Increased during Rosiglitazone-induced Adipocyte Differentiation
To examine the differential expression profiles of proteins between control, KR-62980-treated, and rosiglitazone-treated cells, we carried out two-dimensional gel electrophoresis. Fig. 1 shows the two-dimensional gel images of proteins from treated cells. From gel-to-gel comparison, protein spots whose normalized spot volume changed more than 2-fold in rosiglitazone-treated cells (relative to KR-62980-treated cells) were excised from the gels, in-gel-digested with trypsin, and then their proteins were identified. Of 32 proteins identified, PHD2 showed up-regulation in rosiglitazone-treated cells (Fig. 1C, circle) and was subjected to further study of its role during RIAD.
FIGURE 1.
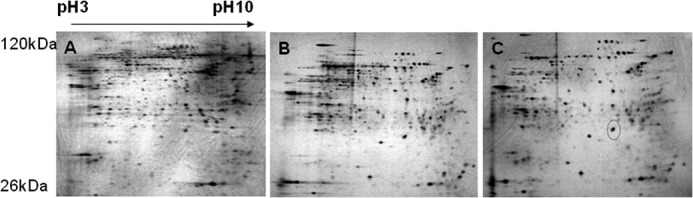
Two-dimensional images and comparative analysis of the expressed protein patterns between control, KR-62980-treated, and rosiglitazone-treated C3H10T1/2 cells. C3H10T1/2 cells were differentiated in the presence of either KR-62980 or rosiglitazone. Lysates from these cells were separated on a two-dimensional gel and then silver-stained for visualization of protein spots. A, control cells. B, KR-62980-treated cells. C, rosiglitazone-treated cells.
To confirm rosiglitazone-induced PHD2 up-regulation observed from two-dimensional proteomic analysis, expression patterns of PHD2 as well as the other isoforms (PHD1 and PHD3) were examined during RIAD. As expected, three PHD isoforms, PHD1, 2, and 3, were induced significantly during RIAD at the mRNA (Figs. 2B and 3B) and protein levels (Figs. 2C and 3C). PHD expression was found to be correlated with the extent of adipocyte differentiation as assessed by Oil Red O staining (Figs. 2A and 3A) and by PPARγ and aP2 mRNA levels (representative markers of adipocyte differentiation), implying that PHD induction is related to the extent of adipocyte differentiation by rosiglitazone.
FIGURE 2.
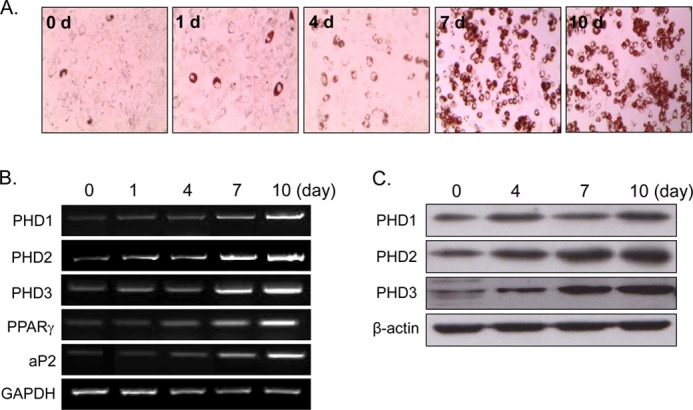
Time-dependent PHD induction by rosiglitazone. C3H10T1/2 cells were treated with rosiglitazone (10 μm) at the indicated times. The extent of adipocyte differentiation was determined by Oil Red O staining (A), RT-PCR (B), and Western blotting (C). Experiments were carried out three times, and representative results are shown. Densitometric analysis was conducted using UN-SCAN-IT Gel version 5.1 software (Silk Scientific).
FIGURE 3.
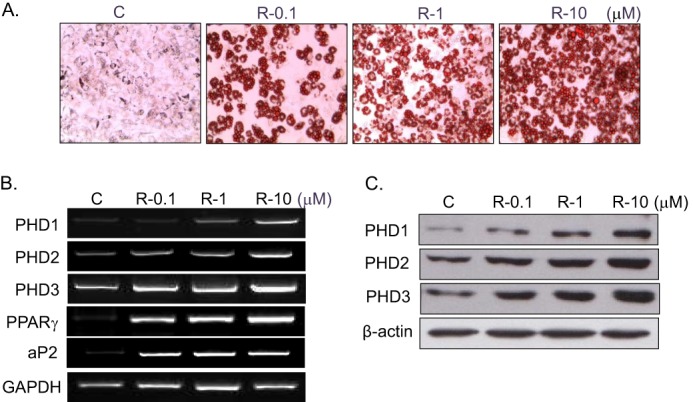
Concentration-dependent PHD induction by rosiglitazone. C3H10T1/2 cells were treated with the indicated concentrations of rosiglitazone for 10 days, and the extent of adipocyte differentiation was determined by Oil Red O staining (A), RT-PCR (B), and Western blotting (C). Experiments were conducted three times, and representative results are shown. Densitometric analysis was conducted using UN-SCAN-IT Gel version 5.1 software (Silk Scientific). C, control; R, rosiglitazone.
PHD Induction by Rosiglitazone Is Mediated by PPARγ Activation
To determine whether induction of three PHD isoforms by rosiglitazone is mediated by PPARγ activation, we examined the effects of PPARγ antagonists on PHD expression. As shown in Fig. 4, BADGE (20 μm) or GW9662 (10 μm) (two well known PPARγ antagonists) prevented increases in PHD mRNA levels (B) and inhibited RIAD (A), indicating that rosiglitazone up-regulated PHD expression in a PPARγ-dependent manner.
FIGURE 4.

Involvement of PPARγ in rosiglitazone-induced PHD up-regulation. C3H10T1/2 cells were treated with BADGE (20 μm) or GW9662 (10 μm) in the presence of rosiglitazone (10 μm) for 10 days. Adipocyte differentiation was determined by Oil Red O staining (A), and the mRNA levels of PHD1, 2, and 3 were analyzed by RT-PCR (B). C, ChIP-PCR was performed using C3H10T1/2 cells treated with either rosiglitazone or vehicle and evaluated by semiquantitative PCR and gel electrophoresis. C, control; R, rosiglitazone; B, BADGE; G, GW9662; M, size marker. Two biological replicates for PPARγ and control IgG were tested. Densitometric analysis was conducted using UN-SCAN-IT Gel version 5.1 software (Silk Scientific).
To further examine whether PPARγ directly regulates the expression of PHDs, we performed a ChIP assay. NURBS, a database for ChIP-Seq-determined nuclear receptor binding sites, revealed several PPARγ binding regions on each PHD gene (19). Consistent with the finding that PHD2 and PHD3 were identified to have PPARγ binding regions on their proximal promoter by genome-wide ChIP-chip, we found the PPARγ binding on PHD2 and PHD3 promoter using ChIP-PCR. As shown in Fig. 4C, PPARγ enrichment over IgG was greater in rosiglitazone-treated differentiated cells than in undifferentiated cells, suggesting that PPARγ may directly regulate the transcription of PHDs via its binding to the promoter regions of each PHD.
PHD Inhibitors Prevented Rosiglitazone-induced Adipocyte Differentiation
To ascertain the involvement of PHD in RIAD, two well known PHD inhibitors, DMOG (1 mm) or EDHB (100 μm), were cotreated with rosiglitazone, and the extent of adipocyte differentiation was monitored by Oil Red O staining. As shown in Fig. 5 (A and C), both inhibitors attenuated RIAD, which was accompanied by reduced mRNA levels of aP2 and PPARγ (B and D), suggesting that PHD plays an important role in RIAD. Consistent with results reported previously (13), the mRNA expressions of PHD isoforms were reduced by PHD inhibitors. Dimethyl sulfoxide (0.1%) had no effect on RIAD.
FIGURE 5.

Effects of PHD inhibitors on rosiglitazone-induced adipocyte differentiation. C3H10T1/2 cells were treated with either DMOG (1 mm) or EDHB (100 μm) in the presence or absence of rosiglitazone (10 μm) for 10 days (the medium was changed every 48 h). The extent of adipocyte differentiation was monitored by Oil Red O staining (A and C), and the mRNA levels of each gene were determined by RT-PCR (B and D). Experiments were conducted three times, and representative results are shown. C, control; R, rosiglitazone.
The Expression of Anti-adipogenic Proteins Was Modulated by Rosiglitazone in the Presence or Absence of PHD Inhibitors
Because PHDs are known to cause the hydroxylation of proline residues of several proteins and, thus, to cause the ubiquitination and degradation of these proteins, we examined the effects of rosiglitazone in the presence or absence of PHD inhibitors on the expression levels of anti-adipogenic proteins; that is, GATA-3, KLF-2, and TAZ. The protein levels of GATA-3, KLF-2, and TAZ were reduced by rosiglitazone concentration-dependently (Fig. 6A), and this was prevented by cotreatment with PHD inhibitors (Fig. 6B).
FIGURE 6.

Effects of rosiglitazone and PHD inhibitors on the expression of anti-adipogenic proteins. C3H10T1/2 cells were treated with the indicated concentrations of rosiglitazone (A) or with DMOG (D; 1 mm) or EDHB (E; 100 μm) (B) in the presence of rosiglitazone (10 μm) for 10 days. The medium was changed every 48 h, and cell extracts were analyzed for the expression of GATA-3, KLF-2, and TAZ by Western blotting. Experiments were carried out three times, and the representative results are shown. C, control; R, rosiglitazone. C, C3H10T1/2 cells were treated with rosiglitazone (10 μm) for 10 days, and whole cell extracts were immunoprecipitated with each PHD isoform followed by immunoblotting with antibodies of GATA-3, KLF-2, and TAZ. Experiments were carried out twice, and representative results are shown.
To confirm whether PHD isoforms indeed bind to anti-adipogenic proteins to induce their prolyl hydroxylation, subsequent to ubiquitination, we carried out immunoprecipitation experiments. As expected, rosiglitazone treatment increased anti-adipogenic proteins coimmunoprecipitated by each PHD antibody (Fig. 6C). These results suggest that rosiglitazone-induced PHDs bind to anti-adipogenic proteins, subsequently leading to their ubiquitination.
Rosiglitazone-induced PHD Up-regulation Leads to Ubiquitination of Anti-adipogenic Proteins
To further confirm that PHD induces the ubiquitination of anti-adipogenic proteins, cell lysates were immunoprecipitated with anti-polyubiquitin antibody, and this was followed by immunoblotting with anti-GATA-3, KLF-2, or TAZ antibodies. The levels of anti-adipogenic proteins in whole cell extracts were decreased by rosiglitazone, but the levels of the polyubiquitinated forms of these proteins were increased (Fig. 7A). Furthermore, these increased levels of the ubiquitinated forms of GATA-3, KLF-2, and TAZ by rosiglitazone were prevented by cotreatment with PHD inhibitors (Fig. 7B). These results indicate that PHDs promote the ubiquitination and, thus, the degradation of anti-adipogenic proteins during RIAD and that PHDs play a key role in PPARγ-mediated adipocyte differentiation by modulating the stabilities of anti-adipogenic proteins.
FIGURE 7.

Effects of rosiglitazone on the ubiquitination of anti-adipogenic proteins. C3H10T1/2 cells were treated with rosiglitazone (R) (10 μm) in the presence or absence of PHD inhibitors for 10 days. Whole cell extracts were immunoprecipitated (IP) with anti-polyubiquitination antibody, and this was followed by Western blotting using anti-GATA-3, anti-KLF-2, or anti-TAZ (A and B). As a control, immunoprecipitation was carried out using mouse IgG antibody (C). Separately, whole cell extracts were analyzed by Western blotting for these anti-adipogenic proteins. D, DMOG; E, EDHB; Ub, ubiquitin; C, control. MG-132 (5 μm) was added in differentiation medium at the end of the 10-day differentiation protocol, incubated for either 6 or 24 h, and then whole cell extracts were analyzed by Western blotting (C). Experiments were carried out twice, and representative results are shown.
To determine whether the effects of PHDs on anti-adipogenic proteins are indeed mediated by proteasomal degradation, cells were cotreated with rosiglitazone and MG-132 (5 μm), a widely used proteasomal inhibitor. MG-132 maintained the levels of anti-adipogenic proteins at the control level (Fig. 7C), further confirming the function of PHDs during the proteasomal degradation of GATA-3, KLF-2, and TAZ.
PHD shRNA Prevented Rosiglitazone-induced Adipocyte Differentiation
To define the PHD isoforms involved in the effect of rosiglitazone, shRNAs specific for each PHD isoform were transfected into C3H10T1/2 cells at days 0 and 4 twice with a 4-day interval. PHD shRNAs efficiently decreased PHD expression (68.5% by PHD1 knockdown, 51.6% by PHD2 knockdown, and 76.1% by PHD3 knockdown) (Fig. 8A). Concomitantly, each shRNA attenuated RIAD to similar extents, as determined by Oil Red O staining (Fig. 8B), suggesting that rosiglitazone induced all three isoforms during adipogenesis and that the three isoforms were involved in RIAD.
FIGURE 8.

Effects of PHD knockdown on rosiglitazone-induced adipocyte differentiation. The PHD1, 2, and 3 isoforms were knocked down by transfecting with isoform-specific shRNAs (sh) at day 0 and day 4 during a 10-day differentiation period. The extent of the knockdown was determined by RT-PCR (A), and adipocyte differentiation was monitored by Oil Red O staining (B). C, control; R, rosiglitazone. C, after knockdown of each PHD expression, immunoprecipitation (IP) of whole cell lysates with anti-polyubiquitination (Ub) antibody, followed by Western blotting (WB) using anti-GATA-3, anti-KLF-2, or anti-TAZ, was carried out. As a control, immunoprecipitation was carried out using mouse IgG antibody (C). Experiments were conducted twice, and representative results are shown. *, p < 0.05; **, p < 0.01 versus controls.
Consistent with the effects of PHD inhibitors on ubiquitination of anti-adipogenic proteins, shRNAs specific for each PHD isoform decreased the ubiquitinated forms of GATA-3, KLF-2, and TAZ when compared with increased levels of their ubiquitination upon control shRNA treatment (Fig. 8C). These results further support the finding that PHDs induce ubiquitination of anti-adipogenic proteins, leading to stimulation of adipocyte differentiation.
PHD Isoform Up-regulation in Adipose Tissues by Rosiglitazone
To examine the involvement of PHD isoforms in RIAD in vivo, we compared PHD 1, 2, and 3 levels in the adipose tissues of ob/ob mice treated with or without rosiglitazone. Oral administration of rosiglitazone (10 mg/kg, 2 weeks, once a day) reduced plasma glucose levels (Fig. 9A). Body weights tended to increase, but not significantly (Fig. 9B). Consistent with our in vitro results, rosiglitazone increased the protein levels of the three PHD isoforms in mesenteric, subcutaneous, and reproductive fat tissues (Fig. 9C) and, concomitantly, reduced the protein levels of GATA-3, KLF-2, and TAZ (Fig. 9D) and increased the levels of their polyubiquitinated forms (Fig. 9E). These results suggest that rosiglitazone induces PHD expressions in adipose tissues in vivo and that these up-regulations are accompanied by the ubiquitination of anti-adipogenic GATA-3, KLF-2, and TAZ.
FIGURE 9.


In vivo effects of rosiglitazone in ob/ob mice. Rosiglitazone (R) (10 mg/kg, once a day) was administered orally to ob/ob mice (9 weeks old, male) for 2 weeks. Plasma glucose concentrations (A) and body weights (B) were determined weekly. Fat tissues were isolated after 2 weeks of administration, and protein expression was determined by Western blotting (C and D) and by immunoprecipitation with anti-polyubiquitination antibody followed by Western blotting (E). V, vehicle; R, rosiglitazone. Experiments were conducted three times, and representative results are shown. Densitometric analysis was conducted using UN-SCAN-IT Gel version 5.1 software (Silk Scientific). *, p < 0.05; **, p < 0.01 versus vehicle controls.
DISCUSSION
This study describes the functional roles played by PHD isoforms during RIAD and shows that these PHD isoforms regulate adipocyte differentiation by controlling the stabilities of anti-adipogenic proteins such as GATA-3, KLF-2, and TAZ. PHD1, 2, and 3 were up-regulated in C3H10T1/2 mesenchymal pluripotent cells treated with rosiglitazone, and these up-regulations were coupled with the ubiquitination of the anti-adipogenic proteins GATA-3, KLF-2, and TAZ, which act to suppress adipocyte differentiation. Furthermore, when rosiglitazone was orally administered to ob/ob mice for 2 weeks, PHD1, 2, and 3 levels were up-regulated, and GATA-3, KLF-2, and TAZ were down-regulated in adipose tissues, thus confirming that these PHD isoforms regulate the stabilities of anti-adipogenic proteins during RIAD.
Adipocyte differentiation is critical for energy and endocrine homeostasis (20), and several transcription factors are strictly controlled to regulate the adipocyte differentiation program. Although it has been shown that C/EBP and PPARγ drive adipocyte differentiation via a positive feedback loop (21), the participation of counteractive transcription factors has been demonstrated. For example, Tong et al. (4) showed that GATA-2 and GATA-3 (GATA transcription factors) suppress adipocyte differentiation during the early stage and that they do so, at least partly, by suppressing PPARγ. That is, they function as negative regulators of the preadipocyte-to-adipocyte transition. TAZ was originally identified as a 14-3-3-interacting protein and was later found to act as an anti-adipogenic regulator by repressing PPARγ-dependent gene transcription (6). Likewise, KLF-2, a zinc finger transcription factor, was reported to inhibit PPARγ expression by directly binding to the promoter region of PPARγ and to act as a negative regulator of adipogenesis (5). Interestingly, the three representative anti-adipogenic proteins examined in this study were found to function by suppressing the transcription of PPARγ.
The PHD family is composed of dioxygenases that utilize oxygen and 2-oxoglutarate as cosubstrates (7). However, despite the considerable amount of work performed on the characterization of PHDs, their biological roles remain unclear. Originally, HIF-1α was the best known substrate for PHD, and studies on this interaction showed that PHDs hydroxylate proline residues in the oxygen-dependent degradation domain of HIF-1α and result in its proteasomal degradation. Accordingly, PHDs play important roles in cellular adaptation to oxygen availability by regulating the stability of HIF-1α (9, 10). On the other hand, recent reports suggest that PHDs have other functions, such as in cellular homeostasis independently of oxygen availability. For example, phenotypic changes from osteoblasts to adipocytes were observed under hypoxic conditions, and PHDs, especially PHD2 and 3, were implicated in the mediation of this response (14). In studies reported by Floyd et al. (13), it was found, using a murine 3T3-L1 adipocyte model, that PHD inhibition prevented adipocyte formation under normoxia and that the relative expression of PHD isoforms differed during the progress of adipocyte differentiation. That is, PHD1 was expressed during early adipogenesis and PHD2 and 3 during late adipogenesis. Furthermore, PHD-mediated adipocyte differentiation appears to be HIF-1α-independent because HIF-1α expression was found to be up-regulated during the early stage of adipogenesis under normoxic conditions. These investigators suggest that HIF-1α activation is associated with insulin-induced adipocyte differentiation via the transcriptions of genes involved in adipogenesis. Similarly, in this study, increased HIF-1α expression was observed in parallel to PHD isoform up-regulation (results not shown), which is in line with previous reports regarding the non HIF-1α-regulated action of PHD isoforms during adipocyte differentiation. In addition, all three PHD isoforms were induced by rosiglitazone, and individual PHD isoform knockdowns by isoform-specific shRNAs impaired RIAD, indicating that each isoform plays a specific role. However, although PHD1, PHD2, and PHD3 were found to have the potential to promote adipocyte differentiation, their relative contributions to adipogenesis under physiological condition have yet to be determined.
Recently, Fu et al. (15) reported a new role for PHD3 (EGLN3) in skeletal muscle differentiation and demonstrated that PHD3 binds to and protects myogenin protein from VHL-mediated degradation and, thus, stimulates skeletal muscle differentiation. On the other hand, the function of PHD3 appears not to be linked with prolyl hydroxylase activity. Later, the same group reported that PHD3 negatively regulates NF-κB, a known suppressor of myogenic differentiation that requires prolyl hydroxylase activity but not HIF-1α (22). In a similar context, adipogenic action of PPARγ-induced three PHD isoforms is dependent on PHD activities because PHD inhibitors blocked the degradation of anti-adipogenic proteins and adipocyte differentiation in this study. It is intriguing how three PHDs interact with GATA-3, KLF-2, and TAZ. All three of these anti-adipogenic proteins share features with zinc finger domains, which have been shown to mediate interactions between proteins and other biomolecules, such as DNA, RNA, and other proteins (23). Further investigations are required to identify the sites responsible for interactions between PHD isoforms (PHD1, 2, and 3) and these anti-adipogenic proteins and to determine whether these reactions are dependent on the nature of the PHD isoform.
PPARγ is expressed mainly in adipocytes (24) and is well known to participate in adipocyte differentiation by regulating the transcriptions of various adipogenic genes (25, 26). Furthermore, rosiglitazone was developed as a synthetic ligand of PPARγ to enhance insulin sensitivity and adipocyte differentiation. In this study, we provide a new mechanistic basis for the action of PPARγ during adipocyte differentiation; namely, that it up-regulates PHDs, and that this is followed by the degradation of anti-adipogenic proteins. PPARγ activation by rosiglitazone increased PHD isoform expression at the mRNA and protein levels, suggesting the transcriptional regulation of PHDs. Furthermore, the PPARγ antagonists BADGE and GW1882 abolished rosiglitazone-induced PHD expression, indicating that PHD induction by rosiglitazone is mediated by PPARγ. Previous reports suggest that there are potential PPARγ binding sites in the promoter region of PHDs on the basis of the PPARγ ChIP sequence database in 3T3-L1 adipocytes (27, 28). In this study, we confirmed direct binding of PPARγ to the proximal promoter of PHD2 and 3 using ChIP-PCR, suggesting that PPARγ may directly regulate transcription of PHDs. On the other hand, NO was found to induce PHD2 and 3 (29), and, interestingly, in this study, NO production was increased by rosiglitazone in parallel with adipocyte differentiation4, leaving the possibility that NO, in part, may induce PHD isoform expression indirectly.
Weight gain is one of the side effects elicited by rosiglitazone, and this is possibly due to its potent adipogenic potential (1). Consistently, rosiglitazone treatment has been reported to increase weight gain in rodent models, which was also observed in this study. Concomitantly, PHD protein levels were increased in various adipose tissues, including mesenteric and reproductive fat tissues in rosiglitazone-treated ob/ob mice, but anti-adipogenic proteins were down-regulated and their ubiquitinated forms up-regulated. These results suggest that in vivo PHD induction in adipose tissues may be, at least in part, responsible for the underlying mechanisms of weight gain initiated by rosiglitazone.
In summary, this study demonstrates a novel role for PHDs in RIAD via the PPARγ-dependent regulation of the stabilities of anti-adipogenic proteins. More importantly, these findings may have physiological relevance to the rosiglitazone-induced weight gain encountered clinically and provide new insight for developmental approaches to more effective PPARγ activators with fewer side effects.
This work was supported by the Korean Government (Ministry of Education and Science Technology (MEST)) National Research Foundation of Korea (NRF) Grant 2011-0013253.
J. Kim, H. J. Kwak, J. Y. Cha, Y. S. Jeong, S. D. Rhee, and H. G. Cheon, unpublished results.
- PPARγ
- peroxisome proliferator-activated receptor γ
- BADGE
- bisphenol A diglycidyl ether
- PHD
- prolyl hydroxylase domain
- DMOG
- dimethyloxalyl glycine
- EDHB
- ethyl-3,4-dihydroxybenzoate
- TAZ
- transcriptional coactivator with PDZ binding motif.
REFERENCES
- 1. Schoonjans K., Staels B., Auwerx J. (1996) The peroxisome proliferator-activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim. Biophys. Acta 1302, 93–109 [DOI] [PubMed] [Google Scholar]
- 2. Rosen E. D., Sarraf P., Troy A. E., Bradwin G., Moore K., Milstone D. S., Spiegelman B. M., Mortensen R. M. (1999) PPARγ is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 4, 611–617 [DOI] [PubMed] [Google Scholar]
- 3. Rosen E. D., Hsu C. H., Wang X., Sakai S., Freeman M. W., Gonzalez F. J., Spiegelman B. M. (2002) C/EBPα induces adipogenesis through PPARγ. A unified pathway. Gene Dev. 16, 22–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tong Q., Dalgin G., Xu H., Ting C. N., Leiden J. M., Hotamisligil G. S. (2000) Function of GATA transcription factors in preadipocyte-adipocyte transition. Science 290, 134–138 [DOI] [PubMed] [Google Scholar]
- 5. Banerjee S. S., Feinberg M. W., Watanabe M., Gray S., Haspel R. L., Denkinger D. J., Kawahara R., Hauner H., Jain M. K. (2003) The Krüppel-like factor KLF-2 inhibits peroxisome proliferator-activated receptor-γ expression and adipogenesis. J. Biol. Chem. 278, 2581–2584 [DOI] [PubMed] [Google Scholar]
- 6. Hong J. H., Hwang E. S., McManus M. T., Amsterdam A., Tian Y., Kalmukova R., Mueller E., Benjamin T., Spiegelman B. M., Sharp P. A., Hopkins N., Yaffe M. B. (2005) TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078 [DOI] [PubMed] [Google Scholar]
- 7. Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 [DOI] [PubMed] [Google Scholar]
- 8. Bruick R. K., McKnight S. L. (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340 [DOI] [PubMed] [Google Scholar]
- 9. Jaakkola P., Mole D. R., Tian Y. M., Wilson M. I., Gielbert J., Gaskell S. J., von Kriegsheim A., Hebestreit H. F., Mukherji M., Schofield C. J., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Targeting of HIFα to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]
- 10. Ivan M., Kondo K., Yang H., Kim W., Valiando J., Ohh M., Salic A., Asara J. M., Lane W. S., Kaelin W. G., Jr., (2001) HIF α targeted for VHL-mediated destruction by proline hydroxylation. Implication for O2 sensing. Science 292, 464–468 [DOI] [PubMed] [Google Scholar]
- 11. Masson N., Willam C., Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) Independent function of two destruction domains in hypoxia-inducible factor chains activated by prolyl hydroxylation. EMBO J. 20, 5197–5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lieb M. E., Menzies K., Moschella M. C., Ni R., Taubman M. B. (2002) Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem. Cell Biol. 80, 421–426 [DOI] [PubMed] [Google Scholar]
- 13. Floyd Z. E., Kilroy G., Wu X., Gimble J. M. (2007) Effects of prolyl hydroxylase inhibitors on adipogenesis and hypoxia inducible factor 1 α levels under normoxic conditions. J. Cell Biochem. 101, 1545–1557 [DOI] [PubMed] [Google Scholar]
- 14. Irwin R., LaPres J. J., Kinser S., McCabe L. R. (2007) Propyl-hydroxylase inhibition and HIF activation in osteoblasts promotes an adipocytic phenotype. J. Cell Biochem. 100, 762–772 [DOI] [PubMed] [Google Scholar]
- 15. Fu J., Menzies K., Freeman R. S., Taubman M. B. (2007) EGLN3 prolyl hydroxylase regulates skeletal muscle differentiation and myogenin protein stability. J. Biol. Chem. 282, 12410–12418 [DOI] [PubMed] [Google Scholar]
- 16. Kim K. R., Lee J. H., Kim S. J., Rhee S. D., Jung W. H., Yang S. D., Kim S. S., Ahn J. H., Cheon H. G. (2006) KR-62980. A novel peroxisome proliferator-activated receptor gamma agonist with weak adipogenic effects. Biochem. Pharmacol. 72, 446–454 [DOI] [PubMed] [Google Scholar]
- 17. Yagi K., Kondo D., Okazaki Y., Kano K. (2004) A novel preadipocyte cell line established from mouse adult mature adipocytes. Biochem. Biophys. Res. Commun. 321, 967–974 [DOI] [PubMed] [Google Scholar]
- 18. Cha J. Y., Repa J. J. (2007) The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 282, 743–751 [DOI] [PubMed] [Google Scholar]
- 19. Fang Y., Liu H. X., Zhang N., Guo G. L., Wan Y. J., Fang J. (2013) NURBS. A database of experimental and predicted nuclear receptor binding sites of mouse. Bioinformatics 29, 295–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spiegelman B. M., Flier J. S. (1996) Adipogenesis and obesity. Rounding out the big picture. Cell 87, 377–389 [DOI] [PubMed] [Google Scholar]
- 21. Rosen E. D., Spiegelman B. M. (2000) Molecular regulation of adipogenesis. Annu. Rev. Cell Dev. Biol. 16, 145–171 [DOI] [PubMed] [Google Scholar]
- 22. Fu J., Taubman M. B. (2010) Prolyl hydroxylase EGLN3 regulates skeletal myoblast differentiation through an NF-κB-dependent pathway. J. Biol. Chem. 285, 8927–8935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matthews J. M., Sunde M. (2002) Zinc fingers. Folds for many occasions. IUBMB Life 54, 351–355 [DOI] [PubMed] [Google Scholar]
- 24. Vidal-Puig A. J., Considine R. V., Jimenez-Liñan M., Werman A., Pories W. J., Caro J. F., Flier JS. (1997) Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J. Clin. Invest. 99, 2416–2422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chawla A., Schwarz E. J., Dimaculangan D. D., Lazar M. A. (1994) Peroxisome proliferator-activated receptor (PPAR)γ. Adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 135, 798–800 [DOI] [PubMed] [Google Scholar]
- 26. Tontonoz P., Hu E., Spiegelman B. M. (1994) Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 79, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 27. Lefterova M. I., Zhang Y., Steger D. J., Schupp M., Schug J., Cristancho A., Feng D., Zhuo D., Stoeckert C. J., Jr., Liu X. S., Lazar M. A. (2008) PPARγ and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Gene Dev. 22, 2941–2952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lefterova M. I., Steger D. J., Zhuo D., Qatanani M., Mullican S. E., Tuteja G., Manduchi E., Grant G. R., Lazar M. A. (2010) Cell-specific determinants of peroxisome proliferator-activated receptor γ function in adipocytes and macrophages. Mol. Cell Biol. 30, 2078–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berchner-Pfannschmidt U., Tug S., Trinidad B., Oehme F., Yamac H., Wotzlaw C., Flamme I., Fandrey J. (2008) Nuclear oxygen sensing. Induction of endogenous prolyl-hydroxylase 2 activity by hypoxia and nitric oxide. J. Biol. Chem. 283, 31745–31753 [DOI] [PubMed] [Google Scholar]