

Background: Large Hsp70s are structurally similar to conventional Hsp70s but functionally less well understood.
Results: The endoplasmic reticulum cognate Grp170 binds directly to substrates in vivo, which is modulated by structural elements characteristic for large Hsp70s.
Conclusion: Grp170 serves as a nucleotide exchange factor and a chaperone.
Significance: In addition to BiP another Hsp70 superfamily member fulfills ER chaperone functions.
Keywords: Endoplasmic Reticulum (ER), ER Quality Control, Glycoprotein, Molecular Cell Biology, Molecular Chaperone, Protein Folding
Abstract
The Hsp70 superfamily is a ubiquitous chaperone class that includes conventional and large Hsp70s. BiP is the only conventional Hsp70 in the endoplasmic reticulum (ER) whose functions include: assisting protein folding, targeting misfolded proteins for degradation, and regulating the transducers of the unfolded protein response. The ER also possesses a single large Hsp70, the glucose-regulated protein of 170 kDa (Grp170). Like BiP it is an essential protein, but its cellular functions are not well understood. Here we show that Grp170 can bind directly to a variety of incompletely folded protein substrates in the ER, and as expected for a bona fide chaperone, it does not interact with folded secretory proteins. Our data demonstrate that Grp170 and BiP associate with similar molecular forms of two substrate proteins, but while BiP is released from unfolded substrates in the presence of ATP, Grp170 remains bound. In comparison to conventional Hsp70s, the large Hsp70s possess two unique structural features: an extended C-terminal α-helical domain and an unstructured loop in the putative substrate binding domain with an unknown function. We find that in the absence of the α-helical domain the interaction of Grp170 with substrates is reduced. In striking contrast, deletion of the unstructured loop results in increased binding to substrates, suggesting the presence of unique intramolecular mechanisms of control for the chaperone functions of large Hsp70s.
Introduction
The endoplasmic reticulum (ER)2 is a dedicated protein folding compartment where nascent polypeptide chains fold, acquire further modifications like glycosylation and disulfide bonds and often assemble with other subunits before traversing further along the secretory pathway. These processes are both assisted and monitored by molecular chaperones (1, 2). Two major chaperone classes of the ER are the lectins calnexin and calreticulin and the Hsp70 family member BiP (3). In the case of BiP, binding to incompletely folded proteins is regulated by its bound nucleotide, ATP or ADP, and by various ER-localized DnaJ family members (ERdjs), which can deliver substrates to BiP and connect it to the ER functions of protein synthesis, folding or degradation (4, 5). ERdjs stimulate hydrolysis of the BiP-bound ATP to ADP and thereby increase BiP's affinity for the substrate. Ultimately, ADP must be exchanged back to ATP so that substrates can be released and either fold or be degraded, a process controlled by nucleotide exchange factors (NEFs) (2, 6).
One of the two NEFs in the mammalian ER is Grp170, also known as oxygen-regulated protein of 150 kDa (ORP150) (7–9), which is a member of the large Hsp70 family. Homologs have been identified in the ER of all eukaryotic organisms examined (10), and cytosolic orthologs also exist in these organisms (11, 12). Large Hsp70s have a high degree of homology to conventional Hsp70s, as both possess an N-terminal nucleotide binding domain (NBD) followed by a β-sheet domain, which acts as the substrate binding domain (SBD) in conventional Hsp70s, and an α-helical domain at the C terminus. The increased size of large Hsp70s is due to the insertion of an acidic, unstructured loop in their β-sheet domain and an extended unstructured C terminus following the α-helical domain (11, 12). In conventional Hsp70s, the α-helical domain serves as a lid for the SBD, whereas structural studies on Sse1p, the yeast cytosolic ortholog of Grp170, show instead that its α-helical domain reaches out to embrace the NBD of Hsp70 (13, 14).
In addition to functioning as a NEF, both cytosolic and ER large Hsp70s have been shown to prevent heat-induced aggregation of luciferase in vitro (13, 15–18), but not much is understood about their ability to bind substrates in vivo. Grp170 co-immunoprecipitates with immunoglobulin (Ig) heavy and light chains (19, 20) and several other proteins (21–23). However, it has remained unclear whether the association of Grp170 with these substrates is direct or occurs via its association with BiP, since both proteins are part of a multi-chaperone complex in the ER (20), and BiP was present in the immunoprecipitated material in these studies (19, 21, 22).
To more fully understand potential substrate binding by the large Hsp70s in vivo, we investigated the binding of Grp170 to a variety of substrates synthesized in the ER of mammalian cells. We demonstrate that Grp170 directly binds unfolded Ig subunits and to a lesser extent to nascent secreted substrates, suggesting that it acts as a bona fide chaperone in vivo. Grp170 remains bound to its substrates in the presence of ATP, while BiP is released from its substrates under these conditions, arguing for independent binding. Grp170 and BiP exhibit similar substrate binding patterns with various oxidation intermediates of the non-secreted light chain (NS-1 LC) and with two glycoforms of the T cell receptor β-chain (TCRβ). Finally, we show that both domains unique to large Hsp70s, the extended α-helical domain and the unstructured loop in the β-sheet domain, modulate substrate binding to Grp170. Together our data reveal that Grp170 directly interacts with unfolded protein substrates in cells, but the regulation of its substrate binding function is strikingly different than for conventional Hsp70s.
EXPERIMENTAL PROCEDURES
Cell Lines and Antibodies
COS-1 cells, an African green monkey kidney fibroblast cell line, were cultured in DMEM (Cellgro) supplemented with 10% FBS, 2 mm l-glutamine, and 100 units/ml penicillin-streptomycin at 37 °C in 3% CO2, and P3U.1 cells, a mouse plasmacytoma line, were cultured in RPMI 1640 (Cellgro) supplemented with 15% FBS, 2 mm l-glutamine, 100 units/ml penicillin-streptomycin and 55 nm β-mercaptoethanol at 37 °C in 5% CO2. A polyclonal anti-rodent BiP antiserum was used (24). The monoclonal anti-HA antibody (12CA5) producing cell line was kindly provided by Dr. Al Reynolds (Vanderbilt University). The anti-Grp170 antiserum was produced by Rockland Immunochemicals Inc., by immunizing rabbits with the peptides STGQKRPLKNDEL and SAGQKRPSKNDEL corresponding to the human and mouse Grp170 C terminus, respectively, along with the purified NBD (aa 36–427) of human Grp170. All other antibodies were obtained commercially: goat anti-mouse κ LC (1050–01) and goat anti-mouse λ LC (1060–01) (SouthernBiotech); rabbit anti-FLAG (F7425) (Sigma-Aldrich); HRP-conjugated goat anti-rabbit (sc-2054), and HRP-conjugated donkey anti-goat (sc-2020) (Santa Cruz Biotechnology); anti-TCRβ (TCR1151) (Thermo Fisher Scientific).
Expression Vectors and Generation of Grp170 Mutants
The pSVL expression vectors encoding the truncated γHC with a double HA-tag, mHCHA (25), NS-1 LC (26), murine λ LC (26), λCL (27), and TCRβ (28) have been described, as have the pMT vectors expressing wild-type BiP and the BiPT37G mutant (29, 30). A human Grp170 cDNA clone in the pcDNA3.1(+) vector was a generous gift of Dr. Osamu Hori (Kanazawa University, Japan), into which we introduced a Kozak sequence (GCCACC) before the initiating methionine to increase expression. Where indicated, a FLAG-tag (DYKDDDDK) flanked by a GS-linker at the 5′- and 3′-site was inserted before the KNDEL ER-retention sequence of Grp170 (Grp170wtFL) to distinguish between endogenous and transfected Grp170. The resulting construct was used as a template for generating Grp170 mutants via the restriction-free mutagenesis method (31). To obtain the Grp170 domain mutant lacking the predicted C-terminal α-helical domain (Grp170ΔC-termFL), amino acids 712–994 were deleted. The unstructured loop-deficient Grp170 mutant (Grp170ΔloopFL) was generated by replacing amino acids 591–696 with a GS-linker to connect the flanking β-sheets. A double mutant lacking both the α-helical domain and the unstructured loop (Grp170ΔC-term, ΔloopFL) was produced by using the Grp170ΔloopFL construct as a template and deleting the C-terminal α-helical domain as described above. All constructs were sequenced for verification.
Transfections
COS-1 cells were plated 24 h prior to transfection, which was performed using GeneCellin (BioCellChallenge, France) according to the manufacturer's protocol. For the analysis of Grp170 binding to Ig-substrates (Figs. 1 and 6), 0.4 μg of Grp170, 0.6 μg of BiP and 2.5 μg of indicated substrates were used for a p60 dish. For the analysis of Grp170 binding to NS-1 LC in the presence of BiPWT and BiPT37G (Fig. 4), the amount of Grp170 cDNA transfected was reduced to 0.02 μg to prevent accumulation of an unglycosylated form of Grp170. Grp170-TCRβ interaction experiments were carried out in p100 dishes transfected with 0.06 μg Grp170, 1.8 μg BiP and 7.5 μg TCRβ. To determine interactions between Grp170 mutants and BiP (Fig. 5, C and D), 0.4 μg of the indicated Grp170 construct and 1.5 μg of BiP were transfected in a p60 dish of COS-1 cells.
FIGURE 1.
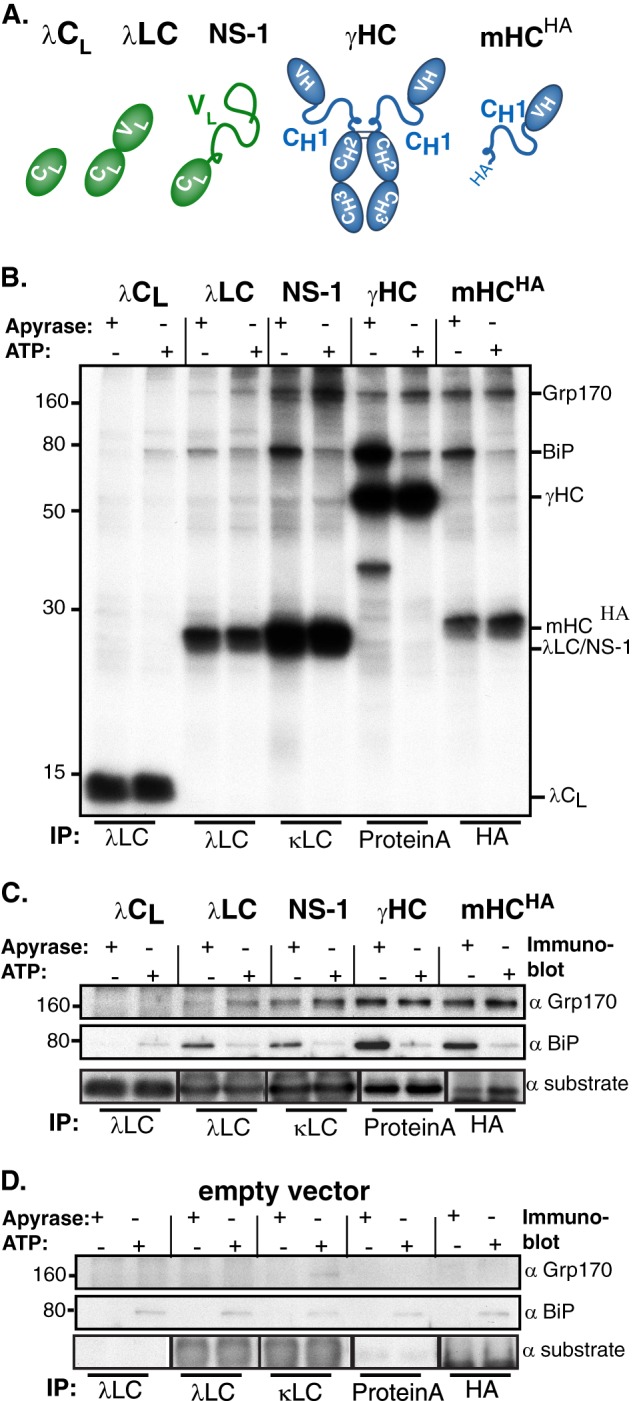
Grp170 directly binds to Ig substrates in vivo. A, schematic of substrates used to analyze substrate binding properties of Grp170 in vivo. Color-filled ovals represent folded Ig domains, and lines indicate unfolded regions. B, COS-1 cells were transfected with these Ig substrates along with Grp170 and BiP. Cells were pulse-labeled with [35S]cysteine/methionine for 1 h and chased for 1 h to allow maturation of the chaperones before lysing either in the presence of ATP or apyrase. Lysates were immunoprecipitated with the indicated reagents and separated by 12% SDS-PAGE followed by autoradiography. C, lysates from cells transfected as in B were immunoprecipitated with substrate specific antibodies, separated by SDS-PAGE and transferred to membranes that were blotted with either anti-Grp170, anti-BiP or anti-substrate antisera followed by species-specific secondary reagents. D, COS-1 cells were transfected and analyzed as in C, except an empty vector was transfected instead of ones encoding the substrates.
FIGURE 6.

Grp170's unstructured loop and C-terminal α-helical domain modulate substrate binding. COS-1 cells were transfected with FLAG-tagged Grp170 constructs, BiP and the indicated Ig proteins. Following a 1 h pulse-label with [35S]cysteine/methionine and 1 h chase, cells were lysed in the presence of ATP, and the cell lysate was equally divided for immunoprecipitation with substrate-specific antisera or FLAG-tag as indicated. Proteins were separated on 10% SDS-gels, followed by autoradiography. The signals of Grp170 constructs bound to NS-1 and mHCHA were quantified and corrected for the Grp170 and substrate expression levels. The value obtained for Grp170wtFL was set to 1 (NS-1 LC: n = 7 ± S.E., *, p ≤ 0.007; mHCHA: n = 3 ± S.E., **, p ≤ 0.02).
FIGURE 4.
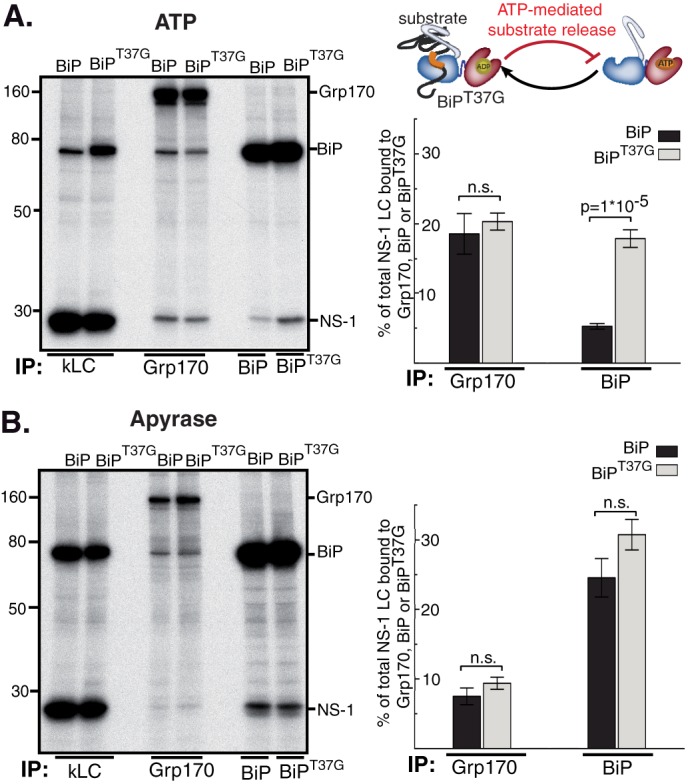
Release from BiP is not required for substrate binding to Grp170. COS-1 cells were transfected with NS-1 LC, Grp170, and BiPWT or BiPT37G as indicated. Subsequently cells were pulse-labeled with [35S]cysteine/methionine for 0.5 h, chased for 1 h and lysed in the presence of ATP (A) or apyrase (B). The lysate was equally divided and immunoprecipitated with the indicated immune reagents. For quantitative analysis the signal of NS-1 LC bound to Grp170, BiP or BiPT37G was divided by the value for NS-1 LC obtained with κLC immunoprecipitations (n = 4 ± S.E., wild-type BiP, and n = 8 ± S.E., BiPT37G; n.s.: not significant).
FIGURE 5.

Construction and characterization of Grp170 domain deletion mutants. A, schematic of the structural organization of Grp170 and the domain deletion mutants (blue: NBD, magenta: linker, green: β-sheet domain and unstructured loop insertion, orange: α-helical domain). The structure of human Grp170 (shown in ribbon) was modeled using Yasara Structure (www.yasara.org) based on the crystal structures of its cytosolic yeast orthologue Sse1p (13, 14, 40) and used to design FLAG-tagged Grp170 whole domain deletion mutants, which are numbered. B, COS-1 cells were transfected with empty pSVL vector, BiP, and the indicated Grp170 constructs. After a 1 h pulse-label with [35S]cysteine/methionine and a 1 h chase, cells were lysed in the presence of ATP. After centrifugation, samples were divided into Nonidet P-40 soluble and Nonidet P-40 insoluble fraction and immunoprecipitated with antiserum against the FLAG-tag. The eluted protein from cell lysates was divided and left undigested or treated with Endo H. Samples were analyzed by 10% SDS-PAGE, followed by autoradiography. The numbers above each group correspond to the deletion mutants shown in A. C and D, COS-1 cells were transfected with BiP and the indicated Grp170 constructs. After labeling with [35S]cysteine/methionine for 5 h and a chase period of 16 h, cells were lysed in the presence of apyrase (C) or ATP (D). Cell lysates were divided equally and immunoprecipitated with an antiserum against either the FLAG-tag (FL), BiP, or protein A (PrA) only. The proteins were separated on 10% SDS-PAGE, followed by autoradiography. Deletion mutants are indicated by the number above each group.
Metabolic Labeling Experiments
Twenty-four h post-transfection, cells were pre-incubated in complete DMEM labeling media (Cellgro) supplemented with 10% dialyzed FBS for 30 min, and pulse-labeled with 100 μCi/p60 or 300 μCi/p100 of EasyTagTM EXPRESS35S Protein Labeling Mix (Perkin Elmer) as indicated. To analyze chaperone association with substrates, the cells were washed and chased in complete media supplemented with 2 mm unlabeled Cys and Met for 1 h to allow chaperones to mature properly and enter an active pool. Cells were lysed in 1 ml of Nonidet P-40 lysis buffer (50 mm Tris/HCl, pH 7.5, 150 mm NaCl, 0.5% Nonidet P40 substitute, 0.5% sodium deoxycholate, 0.1 mm PMSF, 1× Roche complete protease inhibitor tablets w/o EDTA) supplemented as indicated with either with 10 units/ml of apyrase (Sigma-Aldrich) or with 2 mm Mg-ATP (Sigma-Aldrich) and 25 mm KCl (Fisher Scientific). After clearing the lysate at 20,000 × g for 15 min at 4 °C, the supernatant was divided and immunoprecipitated with the indicated antibodies overnight. Immune complexes were isolated with CaptivATM PriMAB Protein A agarose slurry, washed with Nonidet P-40 washing buffer (50 mm Tris/HCl, pH 7.5, 400 mm NaCl, 0.5% Nonidet P40 substitute, 0.5% sodium deoxycholate), eluted with 2x reducing Laemmli buffer and analyzed by SDS-PAGE. The resulting gels were incubated in Amplify (GE Healthcare, Pittsburgh, PA) supplemented with 3% glycerol for 30 min at room temperature before they were dried. To determine the kinetics of NS-1 LC binding to Grp170 and BiP, P3U.1 cells were split the day prior to the experiment, and 15 × 10∧6 cells were pulse-labeled with 1 mCi of EasyTagTM EXPRESS35S Protein Labeling Mix in 10 ml RPMI labeling media (Cellgro) that was supplemented with 15% dialyzed FBS and chased for the indicated times. Cells were lysed in 1.5 ml Nonidet P-40 buffer in the presence of apyrase by rotating at 4 °C for 1 h. 150 μl of the clarified lysate were immunoprecipitated with anti-mouse κ LC, 200 μl with anti-rodent BiP and 1100 μl with anti-Grp170. Further steps were carried out as described above.
To determine the solubility of the Grp170 single domain mutants, cells were lysed with Nonidet P-40 lysis buffer supplemented with ATP. After centrifugation, the Nonidet P-40 insoluble pellets were dissolved in 50 μl of Tris-SDS-buffer (50 mm Tris/HCl, pH 8.0, 2% SDS) and incubated at 95 °C for 20 min with frequent agitation. Subsequently, 950 μl of Nonidet P-40 lysis buffer with ATP was added to the solubilized pellets, and proteins were analyzed via immunoprecipitation as described above. Endo H (NEB) digestions were carried out according to the manufacturer's instructions.
Immunoprecipitation Coupled to Western Blot Experiments
To examine the molecular forms of NS-1 LC that Grp170 and BiP interact with, 2 × 106 P3U.1 murine plasmacytoma cells, which naturally synthesize the NS-1 LC, were used. 10 mm DTT was added to one half of the culture 30 min before lysis to reduce the LC while the other half remained untreated. In both cases, cells were washed with PBS supplemented with 20 mm NEM and lysed in the presence of apyrase or ATP as indicated with Nonidet P-40 lysis buffer supplemented with 20 mm NEM. Lysates were pre-cleared with CaptivATM PriMAB protein A-agarose slurry for 30 min at 4 °C, and immunoprecipitations were carried out with the indicated immune reagents as described above. After separating proteins on 13% non-reducing SDS-PAGE gels and transferring them to a PVDF membrane, proteins were detected by blotting with the indicated antiserum following a standard protocol using Gelatin Buffer (0.1% gelatin, 15 mm Tris/HCl, pH 7.5, 130 mm NaCl, 1 mm EDTA, 0.1% Triton X-100, 0.002% NaN3). Primary antibodies were used at a dilution of 1:1000, while secondary species-specific HRP-conjugated antibodies were diluted 1:10,000.
Quantification and Analysis of the Data
To quantify metabolically labeled proteins, a Storm 860 phosphorimager scanner (GE Healthcare, Pittsburgh, PA) was used, and the signals were analyzed with the ImageQuant TL software (GE Healthcare). For the analysis of the percentage of NS-1 LC bound to Grp170, BiP or BiPT37G, signal of NS-1 co-immunoprecipitating with the various chaperones was divided by the signal obtained by immunoprecipitating with anti-κ LC antibody. Association of Grp170 domain deletion mutants with mHCHA or NS-1 LC was calculated as follows in Equation 1.
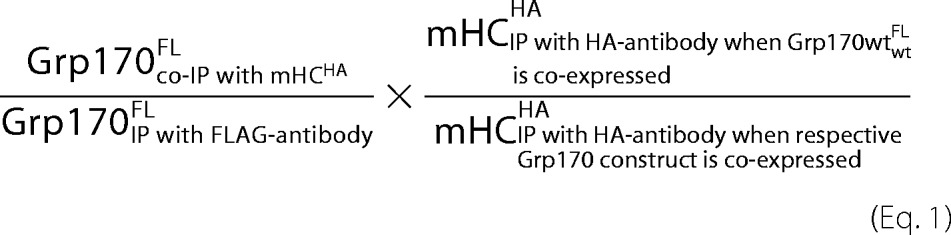 |
Only the signal of the fully glycosylated form of Grp170 was included in the analysis, and the value of calculations in the presence of Grp170wtFL was set to 1. The binding of Grp170 single domain deletion mutants to NS-1 LC was calculated analogously to mHCHA. p values associated with all comparisons were derived from paired two-tailed Student's t-tests. Results are shown as means ± S.E.
Structural Modeling of Grp170
The homology models of human Grp170 was based on the crystal structure of Sse1p bound to Hsp70 NBD (Protein Data Bank code: 3D2F) and built with Yasara Structure (www.yasara.org) as previously described (32).
RESULTS
Grp170 Directly Binds to Unfolded Ig Substrates in Vivo
To study Grp170's substrate binding characteristics in vivo, we chose several Ig proteins with well-defined folding characteristics. These included the well-folded constant domain of the Ig lambda light chain (λ CL) (27), a secreted lambda light chain (λ LC) that transiently interacts with BiP during its maturation (27), and three substrates that possess unfolded domains: the NS-1 LC, whose VL domain does not fold (26); an Ig gamma heavy chain (γHC) in which the first constant domain (CH1) remains unfolded in the absence of its partner, the LC (33, 34); and a truncated, HA-tagged version of the γHC that is devoid of the second and third constant domain (mHCHA) (25) (Fig. 1A).
COS-1 cells were co-transfected with BiP, Grp170 and one of the various Ig constructs and metabolically labeled 24 h later. As the binding of BiP to substrates is stabilized in the absence of ATP, whereas ATP addition releases substrates (1, 5, 30), cell lysates were prepared with either Mg-ATP or with apyrase to hydrolyze ATP. Subsequently samples were immunoprecipitated with substrate-specific immune reagents (Fig. 1B). As expected, in the presence of apyrase a band migrating at the size of BiP co-precipitated with all of the Ig proteins, except the well-folded λCL domain, and was released from these substrates by ATP (Fig. 1B). We observed that in the presence of apyrase an additional band migrating at the expected size of Grp170 similarly bound to the substrates with permanently unfolded domains: NS-1 LC, γHC, and mHCHA. Only trace amounts of this band were detected with the secretion-competent λLC, whereas no binding was observed to the well-folded λCL domain (Fig. 1B). The identity of the co-precipitating bands as BiP and Grp170 was confirmed using immunoprecipitation coupled with Western blotting (Fig. 1C). Nonspecific binding of the different substrate-targeted antibodies to Grp170 or BiP was excluded by transfecting cells with BiP, Grp170, and an empty vector instead of substrates (Fig. 1D). Since Grp170 and BiP bound to the same substrates, and because Grp170 and BiP form a complex in vivo (20), the binding of Grp170 to these substrates in the presence of apyrase could be due to its interactions with BiP. However, when we released BiP by adding ATP, no decrease in Grp170 association with any of these substrates was detected (Fig. 1, B and C). This demonstrates for the first time that Grp170 binds substrates independent of the presence of BiP in vivo.
Grp170 and BiP Bind to the Same Molecular Species of Different Substrates
The fact that BiP and Grp170 bind the same Ig substrates (Fig. 1, B and C), led us to ask if they recognized different molecular intermediates of the various substrates. NS-1 LC exists in two different oxidization states in the cell; one in which only the CL domain has formed its internal disulfide bridge (ox1) and one where the VL and CL domains are both oxidized (ox2) (26, 35). To analyze the interaction of BiP and Grp170 with this substrate in a native setting, we used the myeloma cell-line P3U.1, which naturally produces the NS-1 LC (26) and performed immunoprecipitation-coupled Western blotting assays. As previously reported (26, 35), BiP co-immunoprecipitated only the ox1 form of the NS-1 LC under non-reducing conditions (Fig. 2A). Reduction of the NS-1 LC disulfide bonds in cells with DTT resulted in a slight reduction in the LCs mobility (ox0 form), as expected, and dramatically enhanced co-precipitation with BiP (Fig. 2A). Of note, Grp170 also only bound to the partially oxidized (ox1) species of NS-1 LC in non-treated cells and also showed increased binding to the fully reduced form (ox0) when cells were treated with DTT (Fig. 2A). Thus, in terms of oxidation status, our data show that BiP and Grp170 recognize the same molecular form of the NS-1 LC in a native setting and both exhibit increased binding to the fully reduced ox0 form.
FIGURE 2.
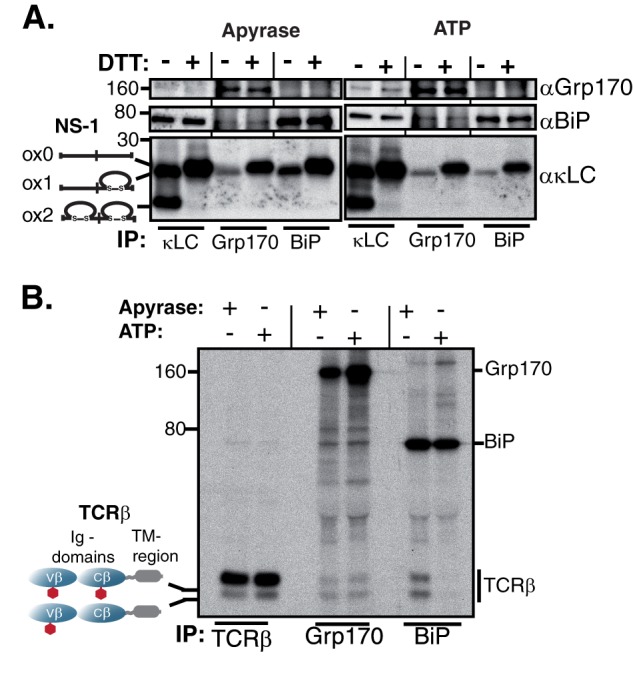
Grp170 and BiP bind to the same molecular species of Ig substrates. A, unlabeled P3U.1 murine myeloma cells were lysed either in the presence of apyrase or ATP, immunoprecipitated with the indicated reagents and analyzed under non-reducing conditions. As BiP is ∼10-fold more abundant than Grp170 in the cell (46), five times more lysate was used for Grp170 immunoprecipitations than for κLC or BiP. Isolated proteins were separated on 13% SDS-PAGE gels and transferred for blotting with the indicated reagents. B, COS-1 cells were transfected with vectors encoding TCRβ, Grp170, and BiP, pulse-labeled with [35S]cysteine/methionine for 0.5 h, chased for 1 h and lysed either in the presence of apyrase or ATP. Interactions between TCRβ, Grp170, and BiP were analyzed via immunoprecipitation with the indicated antisera and separation on 10% SDS gels. Four times less lysate was used for the TCRβ immunoprecipitation than for Grp170 or BiP to make both species of TCRβ visible. The mobility of the two TCRβ glycoforms is indicated.
In addition to disulfide bond formation, N-linked glycosylation is another typical modification occurring on proteins synthesized in the ER. Many ER proteins are glycosylated on asparagine residues within Asn-X-Ser/Thr sequons (1, 3). It has been reported that the T cell receptor β-chain (TCRβ) populates two isoforms that vary in their degree of glycosylation and which are readily distinguished by SDS-PAGE (28, 36). Although the majority of TCRβ is glycosylated at two sites co-translationally, a species can be detected that is only glycosylated on a single site (28). COS-1 cells were co-transfected with vectors encoding TCRβ, BiP, and Grp170. Metabolic labeling experiments revealed that BiP bound a significantly smaller fraction of the fully glycosylated form than of the less glycosylated form (Fig. 2B). When Grp170 association with TCRβ was examined, we found that although it bound to less of the TCRβ than BiP, its pattern of binding to the two glycosylation species was very similar to that of BiP (Fig. 2B). Thus, our data show for two different substrates and two different types of ER-specific protein modifications that Grp170 and BiP bind similar molecular species in vivo.
Release from BiP Is Not Required for Substrate Binding to Grp170
Our data revealed that Grp170 and BiP associated with the same substrates (Fig. 1, B and C) and to the same molecular forms of two different substrates (Fig. 2). As the NEF function of Grp170 could induce BiP release from substrates leading to a subsequent transfer to Grp170, we wished to determine if Grp170 acted downstream of BiP. To address this, we performed a pulse-chase experiment using the P3U.1 cells so that interactions between endogenous proteins in a native setting could be examined. Consistent with a previous report (37), the NS-1 LC could be co-immunoprecipitated with BiP immediately after labeling, and the amount bound to BiP decreased over time as the LC was degraded (Fig. 3, left panel). When the binding of NS-1 LC to Grp170 was examined, no association was detected initially, but in contrast to its binding to BiP, we observed a slight increase of NS-1 LC binding to Grp170 over time (Fig. 3, right panel).
FIGURE 3.
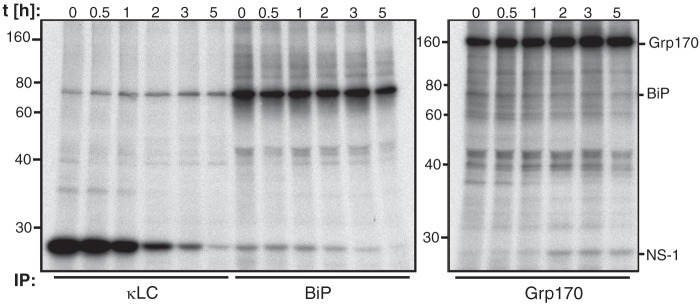
Grp170 and BiP exhibit different binding kinetics to NS-1 LC. P3U.1 murine plasmacytoma cells were pulse-labeled with [35S]cysteine/methionine for 30 min followed by the indicated chase times. After lysing in the presence of apyrase, the clarified lysate was divided for immunoprecipitation with the indicated antisera. Due to different expression levels of Grp170 and BiP (46), five times more lysate was used for Grp170 immunoprecipitations. Proteins were separated by 10% SDS-PAGE and visualized by autoradiography.
To better understand what appeared to be a temporal relationship between the binding of these two chaperones to the NS-1 LC, we asked if BiP must be released from the substrate for Grp170 to bind. To do so, we expressed a BiP “trap” mutant that is defective in ATP-induced substrate release (BiPT37G) (38) (Fig. 4A) and examined the effect on Grp170 binding to NS-1 LC. If Grp170 binding to its substrates would depend on direct substrate transfer from BiP, then the amount of NS-1 LC bound to Grp170 in the presence of BiPT37G should be reduced. As expected, significantly more NS-1 LC remained bound to BiPT37G after the addition of ATP as compared with wild-type BiP, whereas the association of NS-1 LC with Grp170 under these conditions was not significantly affected (Fig. 4A). When cell extracts were prepared in the presence of apyrase, the percentage of NS-1 LC associated with BiPT37G was slightly increased (Fig. 4B). Although the increased binding to BiPT37G was not dramatic, there was no indication that this resulted in a decrease in the amount of NS-1 LC bound to Grp170 (Fig. 4B), suggesting that Grp170 binding to a substrate does not depend on its release from BiP.
Grp170's Characteristic Structural Elements Modulate Substrate Binding
The fact that ATP did not have as profound of an effect on substrate release from Grp170 as has been established for the conventional Hsp70 BiP (30, 39), led us to explore how substrate binding to Grp170 might be regulated. All large Hsp70s possess unique structural features, which set them apart from conventional Hsp70s: an unstructured loop insertion within the β-sheet domain and an extended α-helical domain at the C terminus (Fig. 5A). Although uncharacterized in this regard, we hypothesized that these structural elements might play a role in substrate binding in vivo. Thus, we modeled Grp170 based on the crystal structures of its cytosolic orthologue Sse1p (13, 14, 40) and constructed a number of domain deletions mutants (Fig. 5A) with a FLAG-tag for specific isolation. We found that all mutants, except for a double mutant of Grp170 in which both the unstructured loop insertion and extended α-helical domain were deleted (Grp170ΔC-term, Δloop), where the structural integrity was likely perturbed, remained soluble (Fig. 5B) and none were secreted (data not shown). Consistent with nine predicted glycosylation sites, all domain deletion mutants were glycosylated (Fig. 5B), suggesting that glycosylation was occurring throughout the protein. This finding is in contrast to a previous study on mouse Grp170 expressed in insect cells where only the NBD was glycosylated (17). We next determined the effect of the domain deletions on the ability of Grp170 to interact with BiP, since structural data obtained for the yeast large Hsp70s argued that the α-helical domain at the C terminus reaches out to embrace the NBD of its partner Hsp70 when acting as a NEF (7, 13, 14). A very minor unglycosylated species was detected for all mutants (Fig. 5B), which was less stable than the fully glycosylated form. As we were concerned that this unglycosylated form might interact with BiP as an unfolded substrate instead of a NEF, we performed metabolic labeling experiments with an extended chase period of 16 h to allow the unglycosylated species to be degraded (Fig. 5, C and D). The association of the two deletion mutants with BiP was determined both in the presence of apyrase (C) and ATP (D). Under these conditions, both Grp170 deletion mutants bound to BiP as well as the full-length protein and this association was not disrupted by the presence of ATP (Fig. 5D) arguing that neither the extended α-helical domain nor the unstructured loop insertion were essential for the BiP-Grp170 interaction in vivo and that none of these associations represented a chaperone:client interaction. Similar results were obtained when interactions were examined by Western blotting (data not shown).
To assess the effect of the domain deletions on substrate binding, COS-1 cells were co-transfected with the indicated Grp170 constructs along with BiP and the various Ig substrates (Fig. 6). Cells were lysed in the presence of ATP to release BiP. All detected interactions were specific, as none of the antibodies used to immunoprecipitate substrates bound any of the Grp170 constructs (data not shown). Similar to data obtained with wild-type Grp170 (Fig. 1, B and C and Fig. 6), neither of the Grp170 mutants bound to the well-folded λCL domain (Fig. 6). Interestingly, deletion of the α-helical domain at the C terminus of Grp170 (Grp170 ΔC-term) appeared to decrease its binding to mHCHA, NS-1 LC and λ LC compared with wild-type Grp170 (Fig. 6). A quantitative analysis, where feasible due to signal intensities, confirmed a significant reduction in the binding of Grp170ΔC-term to mHCHA and NS-1 LC (Fig. 6) relative to wild-type Grp170. In striking contrast, absence of the unstructured loop visibly increased binding of Grp170Δloop to these same three substrates (Fig. 6). Quantitative analyses showed that the binding of Grp170Δloop to mHCHA and NS-1 LC was ∼2.5 fold greater than for Grp170WT (Fig. 6). To rule out any influence from slight variations in the expression levels of the Grp170 mutants, the binding values were normalized to the amount of Grp170 present (see experimental procedures for details). These results demonstrate an important role for both of the large Hsp70-specific domains in substrate binding in vivo: the α-helical domain at the C terminus increases substrate binding, whereas the unstructured loop negatively regulates Grp170 binding to substrates.
DISCUSSION
Identification and Characterization of Direct Grp170:Substrate Interactions in Vivo
Our understanding of the ability of large Hsp70 family members to bind to substrates is based on a combination of in vitro studies with purified proteins (13, 15–18, 41, 42) and co-immunoprecipitation with the multi-chaperone complexes formed in vivo (19–23, 43). The contribution of the various domains and regulation of substrate binding to large Hsp70s has remained poorly understood. In the case of experiments where Grp170 was co-immunoprecipitated from cell lysates, BiP was also present (19–22), thus leaving it ambiguous as to whether Grp170 bound to substrates directly or only via BiP. The release of BiP from isolated complexes by ATP allowed us to demonstrate that Grp170 directly binds to several partially folded or unfolded ER substrates in vivo, extending in vitro studies with purified recombinant mammalian (17) and yeast (15) Grp170, as well as other cytosolic large Hsp70s (13, 16, 18, 44), which showed that all of them prevented the aggregation of denatured luciferase.
A crystal structure of Sse1p in complex with the NBD of Hsp70 revealed that the C-terminal α-helical domain provides a major interaction site with the Hsp70 NBD, which is important for Sse1p's nucleotide exchange activity (13, 14). Cross-linking studies confirmed a similar interaction between ER resident large and conventional Hsp70s (7). In our studies, we found that deletion of the α-helical domain of Grp170 did not disrupt Grp170′s interaction with BiP in vivo (Fig. 5, C and D), but did significantly reduce its interaction with substrates (Fig. 6). These data together with an in vitro study demonstrating that a purified protein construct corresponding to only this C-terminal region of Grp170 prevented luciferase aggregation (17), suggests a more direct role for this domain in substrate interaction. Thus, it is possible that the α-helical domain at the C terminus exists in different conformations to support the two distinct roles of large Hsp70s as NEFs or chaperones or that this domain has additional functions in the ER orthologue.
Regulation of Substrate Binding by Grp170
The binding and release of conventional Hsp70s to substrates is regulated by whether ATP or ADP is present in the NBD (1, 5). Indeed, when we prepared cell lysates with ATP, the BiP:substrate complexes were dissociated as expected. However, the Grp170:Ig substrate complexes were not. This is compatible with data showing that recombinant yeast Grp170, Lhs1p, could suppress the aggregation of denatured luciferase regardless of whether it was in the apo-form or had ADP or ATP present in its NBD (15). That study also demonstrated that unlike Hsp70s, the ADP- versus ATP-bound form of Lhs1p did not give distinct protease protection patterns, which in the case of Hsp70s reflects changes in the interaction between the NBD and SBD that affect substrate binding (15), although there may be some species-specific differences (41). Consistent with these in vitro studies, we found that Grp170 remained bound to the Ig substrates when ATP was added. Together these data reveal differences in the regulation of substrate binding to the large and conventional Hsp70s.
In contrast to the α-helical domain, the function of the acidic unstructured loop insertion in the β-sheet domain has remained completely unclear. Deletion of this loop in yeast Sse1p diminished its ability to suppress luciferase aggregation in vitro (13). Conversely, deletion of this loop from human Hsp110 increased its ability to maintain luciferase in a soluble form but decreased its ability to refold it in the presence of reticulocyte lysates which contain conventional Hsp70s (16). The latter data might suggest enhanced binding of Hsp110 to luciferase but reduced release, which is compatible with our finding that deletion of this domain from Grp170 resulted in strongly enhanced binding to all of the Ig substrates we analyzed in this study. It is conceivable that the unstructured loop occludes the SBD (e.g. when Grp170 is functioning as a NEF for BiP), and that the removal of this region exposes the SBD thereby increasing its affinity for substrates. Alternatively, it is possible that the unstructured loop is critical for substrate release, as the mechanism for substrate release remains unknown for all large Hsp70 family members. Further structural studies with a large Hsp70 bound to either a peptide or a substrate or identification of potential co-factors for these large chaperones will be required to address these possibilities.
Why Have Two Hsp70-type Proteins That Bind Similar Molecular Forms of the Same Substrates?
An intriguing finding of our study was that both BiP and Grp170 not only bound the same substrates but within the limitations of our study even appeared to interact with the same molecular forms of these proteins. Since BiP is much more abundant than Grp170 in a cell (45, 46), questions arise as to why both proteins are present and essential, and why the large Hsp70s like Grp170 possess the dual ability to bind to substrates as well as to have NEF activity. Although definitive answers to these questions will require further studies, it is interesting to speculate on possible reasons. First, it is conceivable that Grp170's substrate binding activity becomes much more important under conditions where BiP levels become limiting, as could occur during certain cellular differentiation stages or during ER stress. In the ER, where BiP is the only ER Hsp70 family member, this is a possibility, but in the cytosol, which contains multiple constitutive and inducible Hsp70s and in some cases also possesses several large Hsp70s, this possibility is less appealing. Alternatively, it is possible that Grp170 and BiP bind to different sites on the same substrates or do not have completely overlapping substrates, as suggested by in vitro studies with large Hsp70s (42, 43, 47). Finally, the binding of Grp170 to proteins might signal a different fate for the substrate than when it is associated with BiP. In support of the latter possibility, a recent study demonstrated that Lhs1p was required for the degradation of the α-subunit of the ENaC channel, while BiP was dispensable (48). In this regard it is interesting to note that we observed different binding kinetics for BiP and Grp170 with the NS-1 LC, with Grp170 binding later than BiP. More studies are required to determine if this is a general activity of the large Hsp70s, and our work now provides a set of substrates to address these questions.
In conclusion, we have demonstrated that mammalian Grp170, the only ER member of the large Hsp70 family, can directly bind to a variety of incompletely folded protein substrates in vivo establishing it as a chaperone. We found that Grp170 shows a pattern of binding to these substrates that is similar to BiP, including their folded state and the effects of post-translational modifications. The regulation of its binding to substrates, both in terms of dependence on nucleotide and intramolecular contributions, was strikingly distinct from that of conventional Hsp70s. Based on the high degree of structural conservation between large Hsp70 family members in the cytosol and ER, our data also provide new insights into the regulation of chaperone functions for other large Hsp70s.
Acknowledgments
We thank Drs. Johannes Buchner, Matthias J. Feige, Tanja Mittag, and Stephen White for helpful discussions and comments on the manuscript. We thank Isaac Estrada for generating the Grp170 antibody.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM54068 (to L. M. H.), the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital, and a Boehringer Ingelheim Fonds Ph.D. fellowship (to J. B.).
- ER
- endoplasmic reticulum
- BiP
- immunoglobulin heavy chain-binding protein
- γHC
- gamma heavy chain
- Grp170
- glucose-regulated protein of 170 kDa
- Hsp70
- heat-shock protein of 70 kDa
- mHC
- mini heavy chain
- NBD
- nucleotide-binding domain
- NEF
- nucleotide exchange factor
- NS-1 LC
- non-secreted light chain
- ORP150
- oxygen-regulated protein of 150 kDa
- SBD
- substrate-binding domain
- TCRβ
- T-cell receptor beta.
REFERENCES
- 1. Braakman I., Bulleid N. J. (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 2. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 3. Hebert D. N., Molinari M. (2012) Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem. Sci 37, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Otero J. H., Lizák B., Hendershot L. M. (2010) Life and death of a BiP substrate. Semin Cell Dev. Biol. 21, 472–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kampinga H. H., Craig E. A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bukau B., Weissman J., Horwich A. (2006) Molecular chaperones and protein quality control. Cell 125, 443–451 [DOI] [PubMed] [Google Scholar]
- 7. Andréasson C., Rampelt H., Fiaux J., Druffel-Augustin S., Bukau B. (2010) The endoplasmic reticulum Grp170 acts as a nucleotide exchange factor of Hsp70 via a mechanism similar to that of the cytosolic Hsp110. J. Biol. Chem. 285, 12445–12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Steel G. J., Fullerton D. M., Tyson J. R., Stirling C. J. (2004) Coordinated activation of Hsp70 chaperones. Science 303, 98–101 [DOI] [PubMed] [Google Scholar]
- 9. Weitzmann A., Volkmer J., Zimmermann R. (2006) The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett. 580, 5237–5240 [DOI] [PubMed] [Google Scholar]
- 10. Craven A. R., Tyson J. R., Stirling C. J. (1997) A novel subfamily of Hsp70s in the endoplasmic reticulum. Trends Cell Biol. 7, 277–282 [DOI] [PubMed] [Google Scholar]
- 11. Easton D. P., Kaneko Y., Subjeck J. R. (2000) The hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones 5, 276–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shaner L., Morano K. A. (2007) All in the family: atypical Hsp70 chaperones are conserved modulators of Hsp70 activity. Cell Stress Chaperones 12, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Polier S., Dragovic Z., Hartl F. U., Bracher A. (2008) Structural basis for the cooperation of Hsp70 and Hsp110 chaperones in protein folding. Cell 133, 1068–1079 [DOI] [PubMed] [Google Scholar]
- 14. Schuermann J. P., Jiang J., Cuellar J., Llorca O., Wang L., Gimenez L. E., Jin S., Taylor A. B., Demeler B., Morano K. A., Hart P. J., Valpuesta J. M., Lafer E. M., Sousa R. (2008) Structure of the Hsp110:Hsc70 nucleotide exchange machine. Mol. Cell 31, 232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Keyzer J., Steel G. J., Hale S. J., Humphries D., Stirling C. J. (2009) Nucleotide binding by Lhs1p is essential for its nucleotide exchange activity and for function in vivo. J. Biol. Chem. 284, 31564–31571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Oh H. J., Easton D., Murawski M., Kaneko Y., Subjeck J. R. (1999) The chaperoning activity of hsp110. Identification of functional domains by use of targeted deletions. J. Biol. Chem. 274, 15712–15718 [DOI] [PubMed] [Google Scholar]
- 17. Park J., Easton D. P., Chen X., MacDonald I. J., Wang X. Y., Subjeck J. R. (2003) The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry 42, 14893–14902 [DOI] [PubMed] [Google Scholar]
- 18. Polier S., Hartl F. U., Bracher A. (2010) Interaction of the Hsp110 molecular chaperones from S. cerevisiae with substrate protein. J. Mol. Biol. 401, 696–707 [DOI] [PubMed] [Google Scholar]
- 19. Lin H. Y., Masso-Welch P., Di Y. P., Cai J. W., Shen J. W., Subjeck J. R. (1993) The 170-kDa glucose-regulated stress protein is an endoplasmic reticulum protein that binds immunoglobulin. Mol. Biol. Cell 4, 1109–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meunier L., Usherwood Y. K., Chung K. T., Hendershot L. M. (2002) A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 13, 4456–4469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bando Y., Ogawa S., Yamauchi A., Kuwabara K., Ozawa K., Hori O., Yanagi H., Tamatani M., Tohyama M. (2000) 150-kDa oxygen-regulated protein (ORP150) functions as a novel molecular chaperone in MDCK cells. Am. J. Physiol. Cell Physiol. 278, C1172–C1182 [DOI] [PubMed] [Google Scholar]
- 22. Schmidt B. Z., Perlmutter D. H. (2005) Grp78, Grp94, and Grp170 interact with alpha1-antitrypsin mutants that are retained in the endoplasmic reticulum. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G444–G455 [DOI] [PubMed] [Google Scholar]
- 23. Ozawa K., Tsukamoto Y., Hori O., Kitao Y., Yanagi H., Stern D. M., Ogawa S. (2001) Regulation of tumor angiogenesis by oxygen-regulated protein 150, an inducible endoplasmic reticulum chaperone. Cancer Res. 61, 4206–4213 [PubMed] [Google Scholar]
- 24. Hendershot L. M., Wei J. Y., Gaut J. R., Lawson B., Freiden P. J., Murti K. G. (1995) In vivo expression of mammalian BiP ATPase mutants causes disruption of the endoplasmic reticulum. Mol. Biol. Cell 6, 283–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee Y. K., Brewer J. W., Hellman R., Hendershot L. M. (1999) BiP and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Mol. Biol. Cell 10, 2209–2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Skowronek M. H., Hendershot L. M., Haas I. G. (1998) The variable domain of nonassembled Ig light chains determines both their half-life and binding to the chaperone BiP. Proc. Natl. Acad. Sci. U.S.A. 95, 1574–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hellman R., Vanhove M., Lejeune A., Stevens F. J., Hendershot L. M. (1999) The in vivo association of BiP with newly synthesized proteins is dependent on the rate and stability of folding and not simply on the presence of sequences that can bind to BiP. J. Cell Biol. 144, 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feige M. J., Hendershot L. M. (2013) Quality control of integral membrane proteins by assembly-dependent membrane integration. Mol. Cell 51, 297–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaut J. R., Hendershot L. M. (1993) Mutations within the nucleotide binding site of immunoglobulin-binding protein inhibit ATPase activity and interfere with release of immunoglobulin heavy chain. J. Biol. Chem. 268, 7248–7255 [PubMed] [Google Scholar]
- 30. Hendershot L., Wei J., Gaut J., Melnick J., Aviel S., Argon Y. (1996) Inhibition of immunoglobulin folding and secretion by dominant negative BiP ATPase mutants. Proc. Natl. Acad. Sci. U.S.A. 93, 5269–5274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van den Ent F., Löwe J. (2006) RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67, 67–74 [DOI] [PubMed] [Google Scholar]
- 32. Howes J., Shimizu Y., Feige M. J., Hendershot L. M. (2012) C-terminal mutations destabilize SIL1/BAP and can cause Marinesco-Sjogren syndrome. J. Biol. Chem. 287, 8552–8560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feige M. J., Groscurth S., Marcinowski M., Shimizu Y., Kessler H., Hendershot L. M., Buchner J. (2009) An unfolded CH1 domain controls the assembly and secretion of IgG antibodies. Mol. Cell 34, 569–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hendershot L., Bole D., Köhler G., Kearney J. F. (1987) Assembly and secretion of heavy chains that do not associate posttranslationally with immunoglobulin heavy chain-binding protein. J. Cell Biol. 104, 761–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Okuda-Shimizu Y., Hendershot L. M. (2007) Characterization of an ERAD pathway for nonglycosylated BiP substrates, which require Herp. Mol Cell 28, 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee S. J. (1998) Endoplasmic reticulum retention and degradation of T cell antigen receptor beta chain. Exp. Mol. Med. 30, 159–164 [DOI] [PubMed] [Google Scholar]
- 37. Knittler M. R., Haas I. G. (1992) Interaction of BiP with newly synthesized immunoglobulin light chain molecules: cycles of sequential binding and release. EMBO J. 11, 1573–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wei J., Gaut J. R., Hendershot L. M. (1995) In vitro dissociation of BiP-peptide complexes requires a conformational change in BiP after ATP binding but does not require ATP hydrolysis. J. Biol. Chem. 270, 26677–26682 [DOI] [PubMed] [Google Scholar]
- 39. Vanhove M., Usherwood Y. K., Hendershot L. M. (2001) Unassembled Ig heavy chains do not cycle from BiP in vivo but require light chains to trigger their release. Immunity 15, 105–114 [DOI] [PubMed] [Google Scholar]
- 40. Liu Q., Hendrickson W. A. (2007) Insights into Hsp70 chaperone activity from a crystal structure of the yeast Hsp110 Sse1. Cell 131, 106–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Raviol H., Bukau B., Mayer M. P. (2006) Human and yeast Hsp110 chaperones exhibit functional differences. FEBS Lett. 580, 168–174 [DOI] [PubMed] [Google Scholar]
- 42. Xu X., Sarbeng E. B., Vorvis C., Kumar D. P., Zhou L., Liu Q. (2012) Unique peptide substrate binding properties of 110-kDa heat-shock protein (Hsp110) determine its distinct chaperone activity. J. Biol. Chem. 287, 5661–5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spee P., Subjeck J., Neefjes J. (1999) Identification of novel peptide binding proteins in the endoplasmic reticulum: ERp72, calnexin, and grp170. Biochemistry 38, 10559–10566 [DOI] [PubMed] [Google Scholar]
- 44. Goeckeler J. L., Stephens A., Lee P., Caplan A. J., Brodsky J. L. (2002) Overexpression of yeast Hsp110 homolog Sse1p suppresses ydj1–151 thermosensitivity and restores Hsp90-dependent activity. Mol. Biol. Cell 13, 2760–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Finka A., Goloubinoff P. (2013) Proteomic data from human cell cultures refine mechanisms of chaperone-mediated protein homeostasis. Cell Stress Chaperones 18, 591–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Weitzmann A., Baldes C., Dudek J., Zimmermann R. (2007) The heat shock protein 70 molecular chaperone network in the pancreatic endoplasmic reticulum - a quantitative approach. FEBS J 274, 5175–5187 [DOI] [PubMed] [Google Scholar]
- 47. Goeckeler J. L., Petruso A. P., Aguirre J., Clement C. C., Chiosis G., Brodsky J. L. (2008) The yeast Hsp110, Sse1p, exhibits high-affinity peptide binding. FEBS Lett. 582, 2393–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Buck T. M., Plavchak L., Roy A., Donnelly B. F., Kashlan O. B., Kleyman T. R., Subramanya A. R., Brodsky J. L. (2013) The Lhs1/GRP170 chaperones facilitate the endoplasmic reticulum associated degradation of the epithelial sodium channel. J. Biol. Chem. 288, 18366–18380 [DOI] [PMC free article] [PubMed] [Google Scholar]