

Background: Phytochromes are photochromic bili-proteins vital to microbial and plant photoperception.
Results: NMR spectroscopy generated three-dimensional structures of the photosensing module from a cyanobacterial variant in the dark and photoactivated states.
Conclusion: Photoconversion involves thioether bond rupture, bilin isomerization and sliding, and increased protein disorder.
Significance: Combined with crystallographic models, these paired NMR structures provide an unprecedented view into photoconversion of a phytochrome-type photoreceptor.
Keywords: Cyanobacteria, NMR, Phototransduction, Plant, Protein Structure, Bilin, Cyanobacteriochrome, Photoconversion, Photoreceptor, Phytochrome
Abstract
The phytochrome superfamily of photoreceptors exploits reversible light-driven changes in the bilin chromophore to initiate a variety of signaling cascades. The nature of these alterations and how they impact the protein moiety remain poorly resolved and might include several species-specific routes. Here, we provide a detailed picture of photoconversion for the photosensing cGMP phosphodiesterase/adenylyl cyclase/FhlA (GAF) domain from Thermosynechococcus elongatus (Te) PixJ, a member of the cyanobacteriochrome clade. Solution NMR structures of the blue light-absorbing dark state Pb and green light-absorbing photoactivated state Pg, combined with paired crystallographic models, revealed that the bilin and GAF domain dynamically transition via breakage of the C10/Cys-494 thioether bond, opposite rotations of the A and D pyrrole rings, sliding of the bilin in the GAF pocket, and the appearance of an extended region of disorder that includes Cys-494. Changes in GAF domain backbone dynamics were also observed that are likely important for inter-domain signal propagation. Taken together, photoconversion of T. elongatus PixJ from Pb to Pg involves complex structural changes within the GAF domain pocket that transduce light into a mechanical signal, many aspects of which should be relevant to others within the extended phytochrome superfamily.
Introduction
Biological light perception is mediated by a collection of photoreceptors that initiate various signaling cascades upon photoactivation. One influential set in plants and microorganisms is the phytochromes, a superfamily of bilin (or open-chain tetrapyrrole)-containing chromoproteins that employ photoreversible conformational changes in the bilin to monitor the ambient light environment (1, 2). The bilin is cradled within a signature cGMP phosphodiesterase/adenylyl cyclase/FhlA (GAF)3 domain through numerous electrostatic and van der Waals interactions and fixed by a cysteine-based thioether linkage involving the C3 side chain on pyrrole ring A (see Fig. 2).
FIGURE 2.

Schematic diagram showing intermolecular NOE partners between the TePixJ(GAF) polypeptide (magenta) and the isotopically labeled 15N- or 13C-PVB as Pb (top, blue) and Pg (bottom, green). Cys-494 and Cys-522 that participate in the thioether linkages between the chromophore and polypeptide are located by the red arrowheads. The C-terminal His6 tag was included in the sequence. The locations of the predicted β-strands and α-helices are shown above the sequence. The proposed structures of bound PVB and carbon atom assignments for each state are included for reference. HN represents the pyrrole nitrogen protons (A-D rings are identified in superscript).
Phytochromes assume two photointerconvertible states, typically a dark Pr state that absorbs red light, and a metastable Pfr state that absorbs far red light and is generated only upon red light illumination. These unique spectral properties are created by a number of conserved chromophore/GAF domain contacts, including an extensive hydrogen-bonding network involving a centrally positioned “pyrrole water” and the four pyrrole ring nitrogens (3–8). It has long been assumed that the Pr → Pfr photocycle is triggered by a Z to E isomerization of the bilin at the C15=C16 double bond that flips the D pyrrole ring (see Refs. 9 and 10), but structural and biochemical studies indicate that other routes might also exist for some phytochrome subtypes, including light-driven rotation of the A pyrrole ring (11) and light-induced changes in bilin protonation (12). These light-driven alterations in the chromophore then induce conformational changes within the protein moiety that ultimately allow phytochromes to act as long-lived photoswitches. Numerous signaling outputs are possible; the most common initiates two component-type phosphorelays (1, 2).
In addition to the canonical red/far red light-absorbing phytochromes, photosynthetic bacteria are populated with variants with peak absorptions that span near UV light and much of the visible spectrum despite incorporating the same or similar bilin (13). This diversity is particularly widespread in cyanobacteria, where a unique phytochrome subclade, called cyanobacteriochromes (CBCRs), assembles with phycocyanobilin (PCB) to acquire reversible end states ranging from violet/blue to green/red (e.g. Refs. 12 and 14–16). CBCRs possess two key properties helpful to investigations on phytochrome photochemistry. In many cases the GAF domain alone is sufficient for their unique absorption and photoconversion, and for at least some, photoexcitation of the dark state leads to near complete conversion to the activated state (14, 15, 17). These collective properties are not typical among more canonical phytochromes for which additional domains are often photochemically essential (i.e. Per/Arndt/Sim (PAS) and PHY domains) and where ground state contamination is unavoidable due to spectral overlap (2). Expanding studies on CBCRs revealed that multiple photocycles are possible, including the use of a second thioether link to spectrally tune light absorption (15–18).
One CBCR that has attracted considerable interest is PixJ from the thermotolerant cyanobacterium Thermosynechococcus elongatus (Te), which photointerconverts between a blue light-absorbing dark state Pb (428 nm maximum) and a green light-absorbing photoactivated state Pg (529 nm maximum). Its hypsochromically shifted spectra are generated by an isomerase activity intrinsic to the GAF domain that transposes the C4=C5 double bond within the bound PCB to the C2-C3 position, thus generating the bilin variant phycoviolobilin (PVB) (19, 20). In addition to the conventional linkage involving the C31 carbon and Cys-522, the Pb state of TePixJ links PVB via a second thioether bond between the C10 carbon and Cys-494 (15, 20, 21). The end result is a bisected bilin π-conjugation system that yields a blue light-absorbing system involving the C and D pyrrole rings and a UV-absorbing system derived from the B ring.
Comparisons between recent x-ray crystallographic structures of Pb and Pg also implied that blue-light irradiation drives D-ring rotation via Z- to E-isomerization of the C15=C16 double bond and concomitant rupture of the C10/Cys-494 linkage to change the geometry of C10 from tetrahedral to planar (21, 22). However, interpretation of the paired crystal models is challenged by potential constraints imposed upon TePixJ(GAF) in the crystalline state, especially with regard to a non-native disulfide bridge between GAF domains within the asymmetric unit of the Pg crystal (22), and a general lack of information regarding the dynamics of the GAF domain.
To overcome these challenges, we determined the solution structures of TePixJ(GAF) for both the Pb and Pg states using high resolution NMR spectroscopy. By comparing our NMR models to those previously generated by x-ray crystallography (21, 22), a more complete picture of photoconversion for a CBCR GAF domain is now possible. It shows that transformation between Pb and Pg includes Z to E photoisomerization about the C15=C16 double bond, a photolabile thioether linkage, sliding of the bilin within the GAF domain pocket concomitant with extensive changes in chromophore/protein contacts, and multiple effects on protein disorder, which collectively should be relevant to how TePixJ signals to downstream effectors. The paired dark (Pb) and photoactivated state (Pg) structures shown here likely reveal common themes for phytochrome photochemistry as well as emphasize the striking diversity within the superfamily.
EXPERIMENTAL PROCEDURES
TePixJ(GAF) Protein Expression and Purification
The TePixJ(GAF-C555A) fragment (residues 430–593) bearing an N-terminal Met and a C-terminal His6 tag was assembled recombinantly with PCB as described (15, 21). The protein moiety was labeled with 15N and 13C by adding 15NH4Cl and [13C]glycerol and an excess of unlabeled bilin precursor α-aminolevulinic acid to the culture medium. PVB was labeled by adding [U-13C,15N]-, U-13C]-, [1,2-13C]-, [3-13C]-, [4-13C]-, or α-[5-13C]aminolevulinic acid to the medium along with unlabeled NH4Cl and glycerol.
NMR Data Collection
NMR assignments and three-dimensional structures of the Pb and Pg states were generated by multidimensional, multinuclear NMR analyses. TePixJ(GAF) samples (between 0.7 and 1.3 mm) used for backbone, side chain, nuclear Overhauser effect (NOE), and backbone dynamics experiments contained [U-13C,15N]TePixJ(GAF) with unlabeled PVB in 10 mm deuterated Tris-HCl (pH 8.0), 0.03% NaN3, and 7% D2O. Similar samples of 0.3–0.8 mm [U-13C,15N]Te-PixJ GAF supplemented with 15 mg/ml filamentous pf1 phage (ASLA Biotech, Riga, Latvia) were used to record 1DNH and 1DCαHα residual dipolar couplings (RDCs). NMR experiments were recorded in Shigemi microcells at 25 °C on 600, 800, and 900 MHz Varian INOVA and 600 and 700 MHz Bruker AVANCE spectrometers equipped with cryogenic probes. NMR spectra of the Pg state were collected under continuous 430-nm irradiation using a fiber optic cable passing through the plunger of the Shigemi microcell.
Backbone assignments were obtained from standard three-dimensional CBCA(CO)NH, HNCACB, and HNCO experiments. Side-chain assignments were derived primarily from two-dimensional 13C-heteronuclear single-quantum coherence (HSQC), 15N-HSQC, (HB)CB(CGCD)HD and (HB)CB(CGCDCE)HE, and three-dimensional C(CO)NH, H(CCO)NH, HBHA(CO)NH, and HCCH-TOCSY data. Distance constraints were obtained from three-dimensional 15N-edited NOESY (tmix = 150 ms) and three-dimensional 13C-edited NOESY (aliphatic and aromatic, tmix = 150 ms) experiments. Backbone dynamics were extracted from 15N T1, 15N T2, and 1H,15N NOE data acquired on a 600-MHz Varian INOVA spectrometer. NH and CαHα couplings were measured from a three-dimensional HNCO antiphase 1H-coupled in the 15N dimension (23) and a three-dimensional HCA(CO)N antiphase 1H-coupled in the 13Cα dimension, respectively.
Resonance Assignments and Secondary Structure Calculations
Data were processed with the NMRPipe package (24). For the respective Pb and Pg states, 91 and 84% of the backbone resonances were assigned manually using the PIPP/STAPP software (25). The TALOS+ program (26) was used to provide 147 and 136 pairs of ϕ/ψ backbone torsion angle restraints for the Pb and Pg states, respectively, and to identify the secondary structural elements, which were confirmed by local NOEs. The protein and chromophore NOE statistics for Pb and Pg are presented in Tables 1 and 2, respectively. Hydrogen bond restraints were inferred initially for α-helices and later for β-strands when the level of structural refinement allowed their unambiguous alignment within the β-sheet. Distance restraints of 1.9 and 2.9 Å per involved pair of residues were used to represent hydrogen bonds for HN-O and N-O, respectively (27).
TABLE 1.
Structural statistics for the solution structure of TePixJ GAF domain as Pb
| Restraints and structural statistics | r.m.s.d. |
|
|---|---|---|
| 〈Sa〉a | Lowest energy | |
| Experimental 1H-1H distance restraints | ||
| GAF domain (Å) (2847) | ||
| Intraresidue (1017) | 0.091 ± 0.003 | 0.090 |
| Sequential |i − j| = 1 (772) | 0.063 ± 0.004 | 0.058 |
| Short range 1 < |i − j| ≤ 5 (575) | 0.123 ± 0.015 | 0.125 |
| Long range |i − j| > 5 (483) | 0.242 ± 0.026 | 0.239 |
| PVB (Å) (163) | ||
| Intraresidue (67) | 0.295 ± 0.003 | 0.295 |
| Sequential |i − j| = 1 (26) | 0.031 ± 0.022 | 0.040 |
| Short range 1 < |i − j| ≤ 5 (38) | 0.420 ± 0.019 | 0.392 |
| Long range |i − j| > 5 (32) | 0.436 ± 0.091 | 0.603 |
| Hydrogen bonds (Å) (136) | 0.095 ± 0.007 | 0.087 |
| Predicted dihedral restraints (°) (294) | ||
| ϕ (147) / ψ (147) | 2.02 ± 0.50 | 1.95 |
| Residual dipolar couplings (Hz) (217) | ||
| 1DNH Pf1 (128) Rb = 18.2% | 1.43 ± 0.03 | 1.44 |
| 1DCαHα Pf1 (89) Rc = 31.2% | 2.50 ± 0.04 | 2.56 |
| Deviations from idealized covalent geometry | ||
| Bonds (Å) (2738) | 0.0014 ± 0.0001 | 0.0014 |
| Angles (°) (4946) | 0.398 ± 0.001 | 0.402 |
| Impropers (°) (1498) | 0.85 ± 0.02 | 0.86 |
| Coordinate precisiond | ||
| Backbone (N, Cα, C') | 0.20 ± 0.04 | 0.16 |
| All non-hydrogen atoms | 0.62 ± 0.04 | 0.58 |
a 〈Sa〉 represents the 10 lowest-energy structures. For 〈Sa〉, each value represents the mean± S.D. with the number of restraints used to calculate these values shown in parentheses.
b R factor is defined as in XPLOR-NIH (28).
c R and r.m.s.d. of CαHα RDCs are scaled to NH.
d Average r.m.s.d. of the 10 lowest energy structures from the mean coordinates for structured residues 12–50, 72–83 and 94–148.
TABLE 2.
Structural statistics for the solution structure of TePixJ GAF domain as Pg
| Restraints and structural statistics | r.m.s.d. |
|
|---|---|---|
| 〈Sa〉a | Lowest energy | |
| Experimental 1H-1H distance restraints | ||
| GAF domain (Å) (2444) | ||
| Intraresidue (879) | 0.182 ± 0.004 | 0.182 |
| Sequential |i − j| = 1 (599) | 0.083 ± 0.004 | 0.079 |
| Short range 1 < |i − j| ≤ 5 (468) | 0.116 ± 0.010 | 0.119 |
| Long range |i − j| > 5 (498) | 0.113 ± 0.008 | 0.111 |
| PVB (Å) (62) | ||
| Intraresidue (22) | 0.486 ± 0.030 | 0.471 |
| Sequential |i − j| = 1 (3) | 0.0 ± 0.0 | 0.0 |
| Short range 1 < |i − j| ≤ 5 (9) | 0.130 ± 0.022 | 0.134 |
| Long range |i − j| > 5 (28) | 0.152 ± 0.048 | 0.115 |
| Hydrogen bonds (Å) (140) | 0.082 ± 0.005 | 0.083 |
| Predicted dihedral restraints (°) (272) | ||
| ϕ (136)/ψ (136) | 2.16 ± 0.08 | 2.19 |
| Residual dipolar couplings (Hz) (201) | ||
| 1DNH Pf1 (109) Rb = 10.2% | 1.30 ± 0.04 | 1.44 |
| 1DCαHα Pf1 (92) Rc = 23.2% | 3.04 ± 0.06 | 3.00 |
| Deviations from idealized covalent geometry | ||
| Bonds (Å) (2742) | 0.0014 ± 0.0001 | 0.0014 |
| Angles (°) (4954) | 0.373 ± 0.002 | 0.370 |
| Impropers (°) (1496) | 0.79 ± 0.01 | 0.78 |
| Coordinate precisiond | ||
| Backbone (N, Cα, C') | 0.33 ± 0.12 | 0.29 |
| All non-hydrogen atoms | 0.73 ± 0.10 | 0.63 |
a 〈Sa〉 represents the 10 lowest-energy structures. For 〈Sa〉, each value represents the mean ± S.D. with the number of restraints used to calculate these values shown in parentheses.
b R factor is defined as in XPLOR-NIH (28).
c R and r.m.s.d. of CαHα RDCs are scaled to NH.
d Average r.m.s.d. of the 10 lowest energy structures from the mean coordinates for structured residues 12–50, 72–83, and 94–148.
Three-dimensional Structure Calculations and Refinements
15N T2 measurements with 1.2 mm [U-13C,15N]Te-PixJ(GAF) (see below) yielded average values around 55 ms for the rigid part of the molecule, indicating that the protein behaved as a monomer. In support, the line widths of 1H,15N HSQC cross-peaks were independent of protein concentration from 0.3 to 1.3 mm. Peak intensities in three-dimensional NOESY spectra were assigned using the PIPP/STAPP package and converted into a continuous distribution of 2847 and 2444 approximate interproton distance restraints for Pb and Pg, respectively, with a uniform 40% distance error applied to take into account spin diffusion. In addition, 163 and 62 chromophore distance restraints (Pb and Pg, respectively) were extracted from NOESY spectra of the labeled chromophore/unlabeled protein samples. Structure calculations and refinements made use of the torsion angle molecular dynamics and the internal variable dynamics modules of XPLOR-NIH (28). The Dundee PRODRG2 Server (29) was used to generate the PVB topology and parameters. A separate structure calculation run (100 structures) was used to identify and generously constrain side chain dihedral angles if these were consistent with a unique rotameric state in >90% of the structures. The magnitude and rhombicity of the alignment tensors and the agreement with NMR and x-ray structures are presented in Table 3. PyMol (Delano Scientific, LLC) and VMD-XPLOR (30) were used to prepare the structure figures.
TABLE 3.
Agreement of the TePixJ(GAF) NMR and x-ray crystallographic structures with experimental RDCs
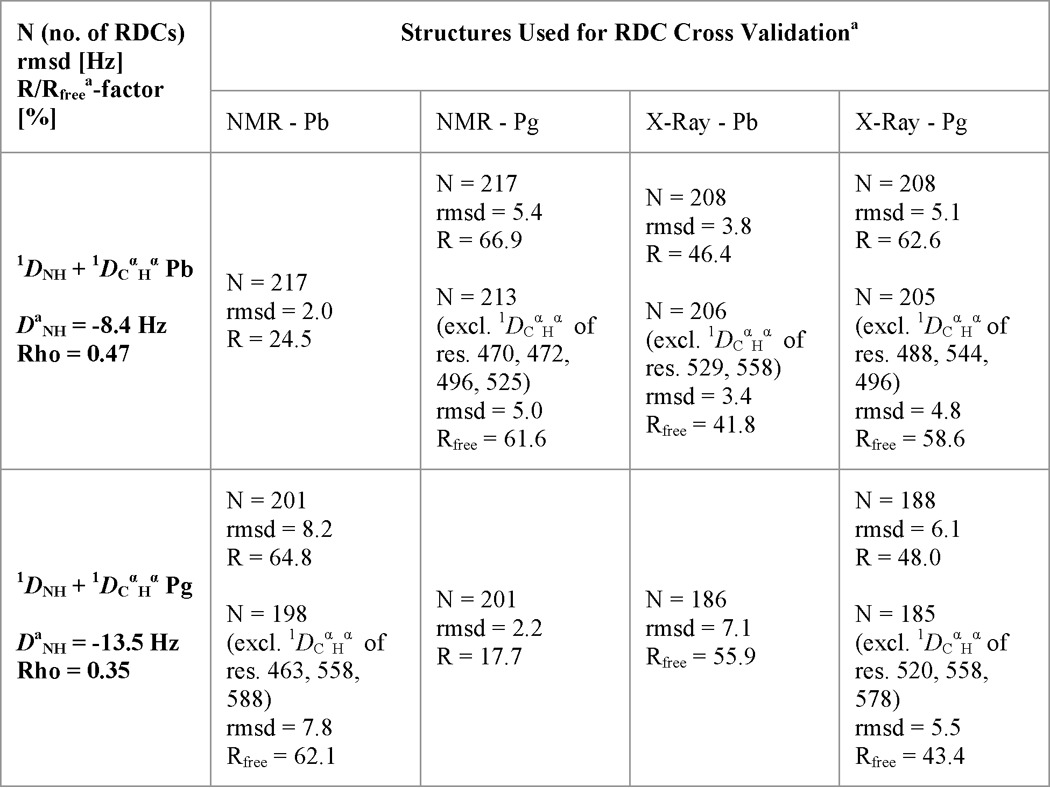
a The lowest energy NMR structure was used for Pb. For Pg, the lowest energy structure with no NOE violations was used.
b Rfree defines how well experimental RDCs fit a structure when they were not used as structural restraints.
RESULTS AND DISCUSSION
Solution NMR Analysis of TePixJ(GAF)
Here, we examined a TePixJ(GAF) fragment encompassing residues 430–591 and bearing the Cys-555 to Ala mutation, which was assembled recombinantly with PCB. Previous studies showed that this preparation is highly stable, fully photochromic, and amenable to photostate-specific structural characterization by NMR spectroscopy after isotopic labeling of either the bilin or protein moieties (15, 21). To counteract Pg → Pb thermal reversion (21), the Pg samples were irradiated continuously with low fluence rate blue light during data collection. Pg retention and the absence of photoconversion intermediates were ensured by periodic inspection of 1H,15N HSQC spectra collected during and after irradiation.
By using an assortment of two- and three-dimensional NMR spectroscopic methods, including measurement of RDCs, we solved the solution structures of TePixJ(GAF) in its Pb and Pg states (PDB ID codes 2M7U and 2M7V, respectively). Statistical support for the two structures is found in Tables 1 and 2, and agreement of the structures with experimental RDCs is shown in Table 3. Like other phytochrome-type GAF domains (2), TePixJ(GAF) consists of an arched six-stranded β-sheet (β1-β6) in a 3-2-1-6-5-4 β-strand sequence, which together with α-helices α2 and α4 create the bilin binding pocket, and two anti-parallel α-helices (α1 and α5) that extend to other domains and might promote dimerization (Fig. 1, A–C). With the exception of local changes near strand β3 in the Pg state (see below), the respective protein folds of the Pb and Pg states retained high similarity to each other and to the pair deduced by x-ray crystallography (PDB ID codes 4GLQ and 3VV4, respectively (21, 22)) (Fig. 1, B–D). Superposition of the main chain atoms in the Pb structures determined by NMR and x-ray crystallography gave a root mean square deviation (r.m.s.d.) of 1.4 Å, whereas superposition of the Pg structures gave an r.m.s.d. of 1.3 Å when the lowest energy NMR structures were used. When the Pb and Pg structures generated by the same method were compared, r.m.s.d. values of 1.2 and 0.7 Å were obtained from the pair deduced by NMR and x-ray crystallography, respectively.
FIGURE 1.

Three-dimensional structures of the GAF domain from TePixJ in the Pb and Pg states. A, ribbon diagrams of the five lowest energy conformers for Pb (blue) and Pg (green) as determined by solution NMR spectroscopy. The relatively disordered N and C termini were truncated for clarity. B, superposition of the Pb crystal structure (gray) with its lowest energy solution structure (blue). C, superposition of the lowest energy solution structures for Pg (blue) and Pg (green). D, superposition of the crystal structures for Pb (gray) and Pg (pink). The positions of Cys-494 and Cys-522 that form thioether linkages to PVB are indicated. Cyan, pyrrole nitrogens; red, oxygens; orange and yellow, side-chain carbon and sulfur atoms of Cys-494 and Cys-522, respectively. The α-helices and β-strands are labeled as are the N terminus (N) and C terminus (C).
Despite their overall similarity, close comparison of the Pb and Pg structures generated by x-ray crystallography identified several significant differences in backbone positions (Fig. 1D). These were confirmed in the NMR structures (Fig. 1C) and supported by fitting of the Pb and Pg state experimental RDCs to the NMR and x-ray crystallographic models (Table 3). For example, the Rfree factor of Pb RDCs was lower when fitted to the x-ray Pb structure than when fitted to the NMR or x-ray structures of Pg, whereas no significant differences in Rfree were observed when the NMR and crystallographic Pg models were compared and vice versa. Taken together, it appears that photoconversion of TePixJ, like that seen for more canonical phytochromes (10, 11), does not appear to globally alter the GAF domain but acts to more subtly reposition existing structural elements. The RDC cross-validations also implied that the NMR and x-ray crystallographic structures are near equivalent in accuracy.
All other characterized phytochromes assemble as homodimers, with studies on several indicating a head-to-head orientation (2). Prior crystal structures of TePixJ(GAF) gave conflicting results, with one crystalline form packing as a head-to-tail dimer and the other two packing as head-to-head dimers, both via helices α1 and α5 (21, 22). The crystal structure of Pg also included a surprising Cys-494–Cys-494 disulfide bridge between separate GAF domains within the asymmetric unit (22). In contrast, our solution models of TePixJ(GAF) were monomeric based on several NMR parameters, including line widths of 1H,15N HSQC cross-peaks at various concentrations, RDCs, and 15N transverse relaxation (T2) measurements (data not shown and see Fig. 7). Even though the full-length photoreceptor is likely a dimer, we concluded that the disulfide bridge and the associated solvent projection of residues 493–496 in the Pb model are crystallization artifacts, and that the contacts between sister GAF domains are insufficient to maintain the dimer by themselves even at high protein concentrations.
FIGURE 7.

Mobility predictions for TePix(GAF) assembled with PVB in the Pb (blue) and Pg states (green). Plots show the experimental 15N backbone amide relaxation parameters versus the residue number determined by NMR spectroscopy at 600 Mhz. Data include longitudinal relaxation time (T1), transverse relaxation time (T2), and the steady-state 1H,15N NOEs. The positions of the α-helices and β-strands are indicated above the panels. The magenta open circles indicate non-proline, non-termini residues for which the amides could not be assigned, and the black open circles indicate residues that were excluded from the amide dynamics analysis due to signal overlap or low signal to noise. Arrows locate the positions of Cys-494 and Cys-522 that form thioether bonds with the bilin. het, heteronuclear.
Bilin Movements during Photoconversion
The transition from Pb to Pg induced several distinct structural changes in the chromophore detectable by NMR. For the bilin as Pb, 83-13C and 81-15N NOE-derived distance constraints allowed relatively precise determination of its conformation and orientation within the GAF domain cavity (Fig. 2). As in the crystal model (21), the Pb chromophore was identified as a rubinoid derivative of PVB that has sp3 hybridization at the C10 position and a Z configuration at the C15=C16 double bond (Fig. 3). In fact, the diagnostic C4—C5 single bond for PVB was easily detected in the 13C,1H HSQC spectra of 13C,15N-labeled chromophore/unlabeled protein samples by the methylene signal at the C5 position (5.6 ppm/38.1 ppm). The composite five lowest energy models also revealed a substantially contorted chromophore, with the planes of the A and D rings oriented by 46° and 124° out of plane relative the B and C-ring planes, respectively, and the B and C rings oriented out of plane from each other by 34° (Fig. 3E). Cys-494 was modeled in close proximity to the bridge between the B and C rings by numerous NOE contacts (Fig. 2), which supported a second thioether linkage in Pb involving C10 and this cysteine (21).
FIGURE 3.

Conformational rearrangements of PVB during TePixJ(GAF) photoconversion. A, proposed chemical structures of PVB bound to the GAF domain in the Pb and Pg states. The A-D pyrrole rings and the various pyrrole carbon atoms are labeled. Carbons highlighted in blue and green indicate the residues interrogated by NMR experiments for Pb and Pg, respectively. B, two-dimensional 13C,1H HSQC spectrum of TePixJ(GAF) with 13C incorporated into PVB carbons 21, 32, 71, 82, 122, 131, 171, and 182 as Pb (blue) and Pg (green). C, two-dimensional NOE spectrum of the sample in B showing the 1Hx/1Hy cross-peaks for Pb and Pg. Significant chemical shift changes for the 131, 171, and 182 carbons are highlighted. D, top and side views of the 10 lowest energy structures of PVB in the Pb (blue) and Pg (green) states as determined by solution NMR spectroscopy. E, top and side views of the lowest energy structures of PVB shown in panel D in the Pb (blue) and Pg (green) states. Light-induced rotation at the C5 and C15 bridges and the D ring flip are highlighted (the asterisk marks C17 carbonyl). Cyan, pyrrole nitrogens; red, oxygens; orange and yellow, side-chain carbon and sulfur atoms of Cys-494 and Cys-522, respectively.
Assignment of the 1H,15N HSQC signals for rings A and D based on multiple isotope labeling patterns detected major and minor conformers of Pb (data not shown). For ring D, the 1H,15N signals for both conformers correlated with a unique 13C19 carbonyl signal at 176 ppm, indicative of similar configurations for the two. However, very different 1H,15N and 13C1 carbonyl signals were obtained for ring A, indicating that multiple conformations exist for this ring as Pb. Given our observations of greater A ring plasticity in prior crystallographic analyses of TePixJ(GAF) as Pb (21) and in NMR studies on a more canonical phytochrome (SyBCph1; Ref. 11), it is possible that A ring mobility is an intrinsic feature of phytochromes in the dark state.
After photoconversion to Pg, all 1H,15N chromophore signals became vanishingly weak, most likely due to solvent exchange, and the chromophore appeared more mobile as evidenced by the resolution of substantially fewer chromophore 13C,1H- and 1H,15N NOE constraints (62 and 0, respectively), even after increasing the number of spectral scans to the practical limit (Figs. 2 and 3C). Depleted NOE constraints were especially evident for the A-C pyrrole rings. For the A ring, this ambiguity generated a varied ensemble of acceptable positions in the lowest energy solution structures, which is likely reflective of the true solution state(s) of the photoreceptor (Fig. 3D). Comparisons of the crystal structures for TePixJ(GAF) supported the proposed break of the C10/Cys-494 link followed by reformation of the C10=C11 double bond (21, 22). For our NMR studies, the lack of NOE signals unambiguously assigned to C4-H, C10-H, and C15-H prevented direct detection of this break (15). However, the break resulting in the Pg state was evident by the loss of all NOE contacts between Cys-494 and PVB and migration of Cys-494 away from C10 (Figs. 1A and 2). Retention of PVB as opposed to its possible re-isomerization back to PCB after photoconversion (16) was supported by the easily detected C5 methylene signal in Pg (1H = 4.2 ppm, 13C = 38.1 ppm) and by its NOE contact to the chromophore C32 methyl. Taken together, we conclude that photoconversion of TePixJ to Pg restores PVB, thus generating a bathochromically shifted π-conjugated system that now encompasses the B, C, and D rings (Fig. 3A).
Our NMR analyses implied that the D pyrrole ring is more rigid than the A-C rings in the Pg state and fully supported photoisomerization about the C15=C16 bridge to generate the 15E configuration (Fig. 3, D and E). Photostate-dependent changes in the unambiguously assigned NOE contacts between ring D and the protein (Fig. 2) supported an averaged 148° flip embodied in our five lowest energy structures, which was consistent with large displacements of the methyl signals at the 13C1 and 17C1 positions (Fig. 3B). By comparison, superposition of the Pb and Pg crystal structures estimated a 165° D-ring flip (21, 22). This 15Z to E isomerization is in agreement with the 15E photoproduct of PVB when bound to cyanobacterial photosynthetic antennae protein α-phycoerythrocyanin (31) and with the photoconversion mechanism proposed for a canonical phytochrome assembled with PCB (9). Photoconversion in solution also induced an ∼71° swivel of the A ring based on the average of the five lowest energy NMR structures (Fig. 3, D and E). The significance of the A ring movement is unclear, but its rotational freedom might optimize photoinduced sliding of PVB upon rotation of the D ring while still retaining the 3C1 thioether linkage (see below).
Photoinduced Changes in the Chromophore Pocket
After Pb → Pg photoconversion, structural changes in PVB drive numerous alterations in chromophore/protein contacts. Both the solution and crystal structures show extensive displacement of the bilin that is likely induced by migration of the flipped D-ring (21, 22). This migration can best be seen by the alignment of rings C and D with individual strands of the β-sheet (Fig. 4). In Pb, the D ring straddles the grooves between the side chains of the β1-β6 and β5-β6 strand pairs, whereas in Pg it slides into the β1-β2 groove and the space between β2 and the displaced β3 strand. In Pb, the C-ring methyl and propionate groups sits atop the side chain ridges formed by strands β5 and β4, respectively, and slide into the grooves between the β1-β6 and β5-β6 strand pairs in Pg. Notably, Yang et al. (32) detected similar sliding of the bilin within the GAF pocket of the Pseudomonas aeruginosa bathyphytochrome BphP from comparison of its Pfr structure with that for the Q188L mutant, which appears to generate a Pr/Pfr-mixed state. These observations suggest that bilin sliding is a general mechanism for both CBCRs and more canonical phytochromes.
FIGURE 4.
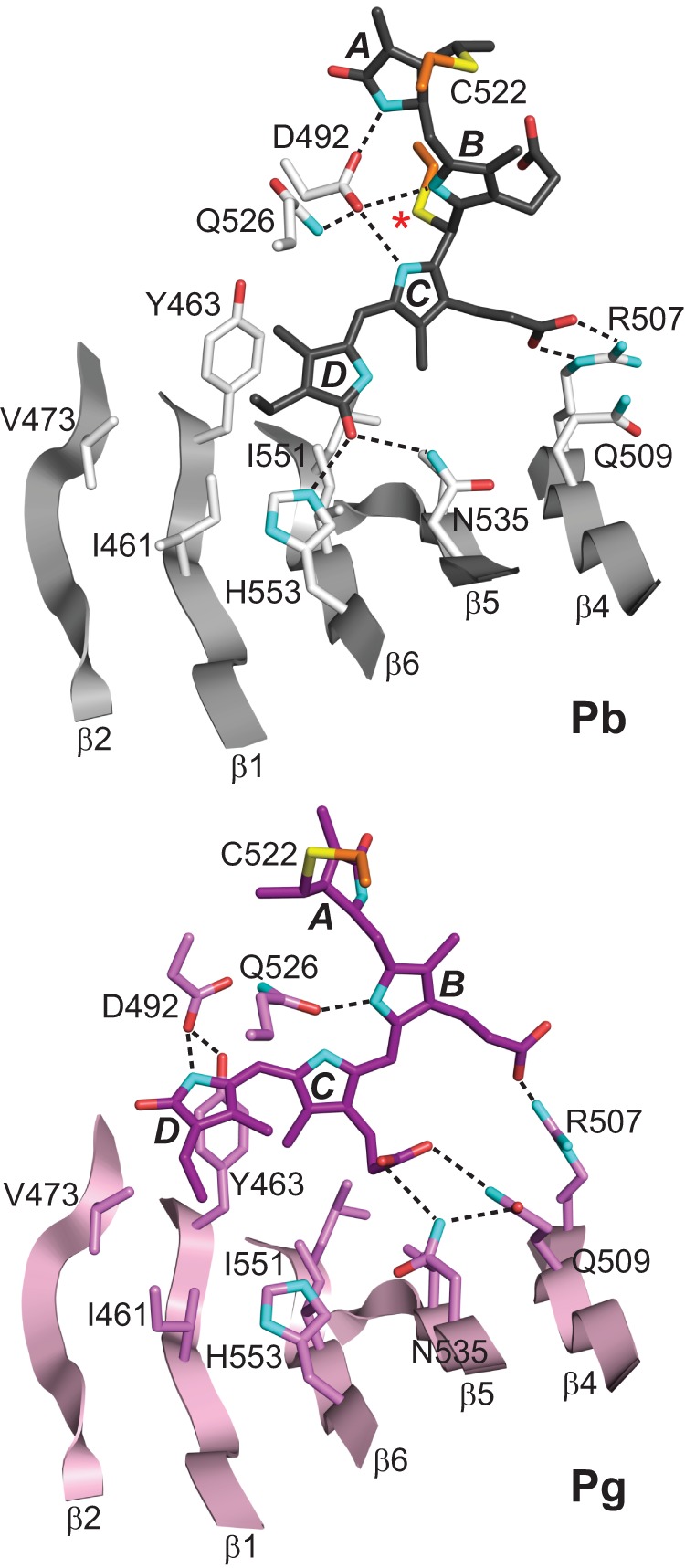
Repositioning of PVB in the TePixJ(GAF) pocket during photoconversion. The three-dimensional structures of the Pb (gray) and Pg states (pink) were determined by x-ray crystallography (21, 22). The relevant β-strands and amino acids are shown along with key hydrogen bond contacts (dashed lines). Cys-494 is located by an asterisk. Cyan, nitrogens; red, oxygens; orange and yellow, side-chain carbon and sulfur atoms of Cys-494 and Cys-522, respectively.
As with other structurally defined phytochromes, TePixJ includes an aspartate (Asp-492) nestled within the crescent configuration of the A-C rings. In canonical phytochromes, the main-chain carbonyl of this aspartate interacts with all three pyrrole nitrogens through the pyrrole water intermediary (3–8), but in the crystallographic structure of TePixJ as Pb, the carboxylate group of Asp-492 is modeled to bind directly to the pyrrole nitrogens (Figs. 4 and 5; Ref. 21). Rotation of ring D during photoconversion dissolves the hydrogen bonds between its carbonyl and Asn-535 and His-553 to now allow a new contact between its pyrrole nitrogen and the carboxylate group of Asp-492, which is repositioned toward the D ring. This new contact apparently serves as an island of stability in Pg given that our NMR analyses showed increased mobility in the residues flanking Asp-492 (see below).
FIGURE 5.
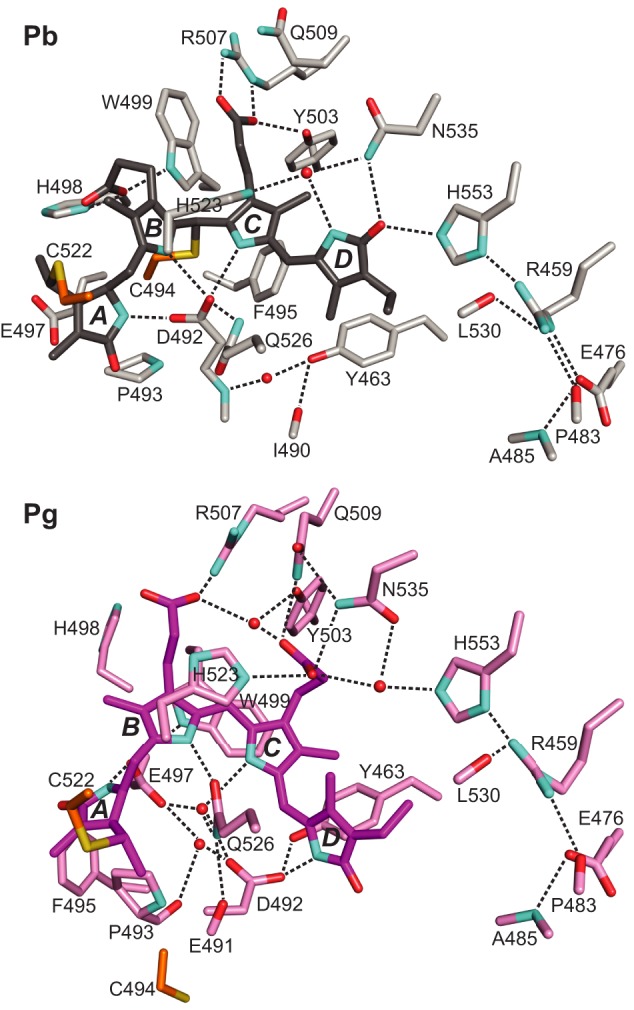
Changes in bilin/protein contacts during Pb → Pg photoconversion of TePixJ(GAF). PVB is colored in gray and purple for the Pb (top) and Pg states (bottom), respectively. The A-D pyrrole rings and key amino acid contacts are labeled. Predicted hydrogen bonds are indicated by the dashed lines. Shown are the x-ray crystallographic structures (21, 22). Cyan, nitrogens; red, oxygens; orange and yellow, side-chain carbon and sulfur atoms of Cys-494 and Cys-522, respectively.
The combined movements upon photoconversion break direct contact of Asp-492 with the A-C rings. However, crystallographic data showed that the hydrogen-bond lattice is maintained indirectly upon entry of two water molecules into the void (22). The presence of these waters might explain in part the loss of bilin 1H,15N NOE signals upon photoconversion that could arise from the new solvent exchange properties of the Pg state (Fig. 2). In the crystallographic structures, the positions of the waters are further stabilized by their hydrogen bonding with the carboxylate of Glu-497, the carbamoyl side chain of Gln-526, and the main chain carbonyl of Pro-493 (Fig. 5). Although Gln-526 moves little during photoconversion, bilin sliding allows its carbamoyl to hydrogen bond with the B-ring nitrogen. Given their central positions, it is not surprising that most Asp-492 and Gln-526 substitutions in TePixJ have abnormal blue/green photochemistry (15, 21). The Q526H mutant in particular fails to form Pg, likely due to repulsion of the histidine imidazole group by the PVB pyrrole nitrogens. Collectively, the NMR and crystallographic data also indicate that the A ring is loosely held in the GAF cavity with the ring rotating and becoming more mobile as Pg (Fig. 3; Refs. 21 and 22).
Both His-523 and Trp-499 show subtle but potentially important movements during photoconversion. In the Pb crystal structure (21), the imidazole ring of His-523 participates in an unusual cation-π interaction with ring B, whereas Trp-499 is displaced away from the chromophore and fixed via a hydrogen bond between its indole NH and the B-ring propionate (Fig. 5). Upon rupture of the Cys-494 linker and sliding of PVB, both amino acids assume a closer and more parallel orientation relative to the bilin, suggesting a role for this displacement in fine-tuning Pg absorption by π-stacking interactions (Ref. 22 and Fig. 5). Our NMR data support these movements and show Trp-499 to be relatively mobile in Pg as evidenced by the uncertainty in the indole ring position. The mobility of the Trp-499 indole ring might play a role in Pg → Pb photoconversion and thermal reversion, as the position of this side chain in Pg should impede access of the bilin C10 for nucleophilic attack by the Cys-494 thiol.
Finally, NOE-derived interatomic distances lend additional credence to the complex set of interaction changes between the bilin propionates and several surrounding amino acids (His-498, Trp-499, Arg-507, Gln-509, Tyr-503, and Asn-535) that might lock the bilin in place after sliding. Surprisingly, the string of solvent-excluded interactions extending across much of the GAF domain β-sheet (including His-553, Leu-530, Arg-459, Glu-476, Pro-483, and Ala-485) were only subtly affected by photoconversion in the ensemble of crystallographic and NMR structures after the electrostatic contact between the D ring and His-553 was broken by the 15Z to E isomerization (Fig. 5).
Photostate-dependent Changes in GAF Domain Architecture
Comparisons of the Pb and Pg solution structures identified a number of changes in the GAF domain upon photoconversion. Prominent main chain differences were seen at residues 485–506 and 512–522. In Pg, the region spanning residues 493–501 exhibits intermediate conformational exchange, resulting in complete broadening of most of its NMR signals. In fact, no Pg NOEs were detected in this region, resulting in a less structured NMR bundle (Fig. 1A). Residues 512–522 include Cys-522, which provides the thioether link with the C3 ethylidene side chain of ring A. Comparisons between the Pb and Pg crystal models suggested that this stretch undergoes a conformational change during photoconversion (Fig. 1D; Refs. 21 and 22), a possibility implied by the observations that the L519S mutant has a highly unstable Pg state (21). Supported by several assigned NOE contacts (Fig. 2), residues 512–522 in our paired NMR models exhibited increased flexibility among the lowest energy structures and a downward displacement of helix α4 and proximal loop bundles toward the chromophore (Figs. 1C and 6, A and B).
FIGURE 6.
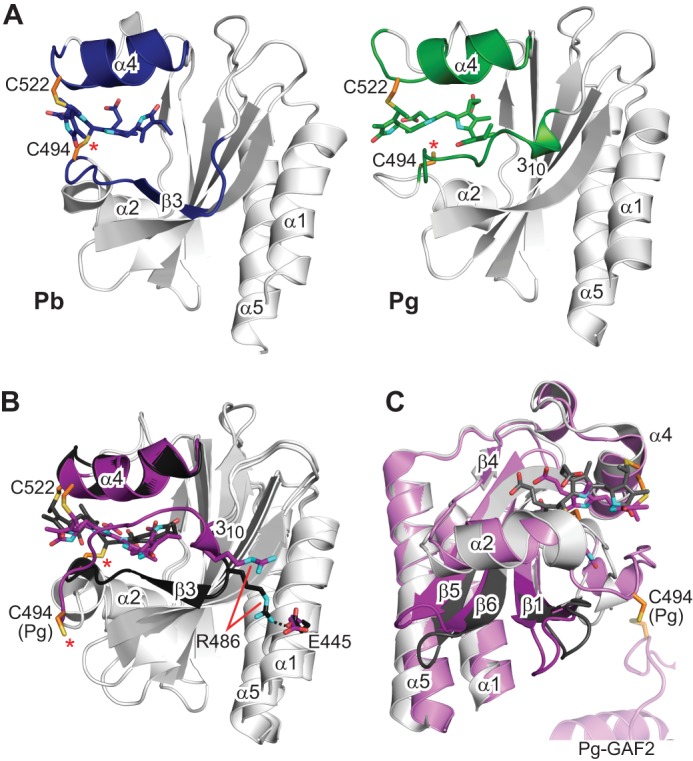
Large light-driven structural changes in TePixJ(GAF) influence the positions of helices α1 and α5. A, strand β3 is well defined in the solution NMR structure of Pb, but it dissolves into a dynamic loop region in Pg that is bounded by a 310-helix and helix α2. The position of the 310-helix draws this entire loop region closer to helix α4. B, the same structural perturbations are identified by comparison of the crystal structures of Pb and Pg (21, 22). Additionally, Pb → Pg photoconversion is coincident with rupture of the Arg-486–Glu-445 salt bridge, which may serve to loosen constraints on the helix α1 position. C, the alternative view provided by the paired crystallographic structures illustrates the sweeping movements of the loops between strands β1-β2 and β5-β6 and correlated motions of helices α1 and α5. The ectopic cystine disulfide found in the Pg crystal structure is included in C, with a portion of the second GAF domain from the asymmetric unit. Cys-494 is located by an asterisk. Cyan, nitrogens; red, oxygens; orange and yellow, side-chain carbon and sulfur atoms of Cys-494 and Cys-522, respectively.
In Pb, residues 485–506 encompass strand β3, helix α2, and a type II β-turn motif formed by residues 485–488 that connects strand β3 with a conserved loop motif (Figs. 1, A and C, and 6A). Strikingly, overlay of the solution models revealed that upon Pb → Pg photoconversion the β3 strand (residues 488–490) becomes largely unstructured, and in agreement with the paired crystal structures, part of the β-turn (residues 485–488) converts to a 310-helix (Fig. 6A). This unusual change is likely driven by collision of the repositioned D ring with Ile-490 and is accompanied by disposition of the entire region toward helix α4 (Fig. 6, A and B). Importantly, visualization of these changes by both NMR and x-ray crystallography verified that they were not generated by the unexpected disulfide bridge in the Pg crystal structure (22) nor by artifacts in crystal packing, the latter of which have been observed previously in other crystal-derived structures (e.g. Refs. 33 and 34).
The net effect of these alterations to strand β3 is to pinch together the newly formed 310-helix in Pg and helix α4 by as much as 6 Å. Notably, 310-helix formation is accompanied by breakage of a potential salt-bridge between Arg-486 and Glu-445 (Fig. 6B). Loss of this interaction in Pg could release helix α1, with the acquired freedom then facilitating signal transmission to other domains. Interestingly, pinching of a comparable region was detected by NMR for the GAF domain of SyBCph1 during its Pr → Pfr photoconversion (11), suggesting that this structural rearrangement might be common among phytochromes.
A conspicuous feature of the GAF domain β-sheet is a prominent ridge that bisects the sheet perpendicular to the β-strands. It is formed by a kink in the β-sheet that begins with the Pb-state-dependent β-turn motif at residues 485–488 (Figs. 1 and 6A) and is accentuated by a β-bulge within strand β2 created by an unpaired residue (Val-474). The ridge is propagated through Val-462 of strand β1 and Leu-550 of strand β6, which is adjacent to the potentially disruptive Pro-539 of strand β5. Comparison of the Pb and Pg crystal structures revealed distinct bending of the sheet at this ridge in a photostate-dependent manner, which was most prominent for residues 462–474 of strands β1-β2 and residues 539–550 of strands β5-β6 relative to helices α1 and α5 (Figs. 1 and 6C). Surprisingly, the remainder of the β-sheet (except for strand β3) exhibits little photostate-dependent movement. Although still evident in the paired NMR structures (Fig. 1), the magnitude of the β-sheet displacement was less, possibly due to limited NOE signals available to precisely position helices α1 and α5.
Photoconversion Effects on Backbone Dynamics
With respect to the 15N backbone dynamics of the GAF domain, our NMR analyses revealed subtle photostate-dependent changes in longitudinal (T1) and transverse (T2) relaxation times, and steady-state 1H,15N NOE measurements. Despite the greater overall rigidity of Pb as compared with Pg, several residues in Pb had unusually dynamic features (Fig. 7). Leu-530 near ring D exhibited exchange dynamics on the μs-ms time scale (shorter T2 values than average without an increase in T1 (Fig. 7)), and residues 556–561, which form a loop between strand β6 and helix α5, and Val-532, which is proximal to the D pyrrole ring, displayed dynamics on the ps-ns time scale (T1 and T2 larger and heteronuclear NOE smaller than average; Fig. 7).
Photoconversion to Pg annulled these dynamic features and introduced new ones to the NMR models. These included residues involved in photo-induced movement of the β-sheet (Fig. 7) such as 471–474 and 493–500, which showed slow conformational dynamics, and Asp-466, which exhibited dynamics on the ps-ns time scale. Several residues near the chromophore showed conformational dynamics in the Pg state, including μs-ms motions for Ile-490 and Leu-527, and ps-ns motions for Gln-488 and Val-489. Strikingly, these Pb and Pg dynamic features occur at opposite sides of Arg-459, an almost invariant residue among phytochromes and part of the buried hydrogen-bonding network connecting the bilin to the protein (Fig. 5). For Pb, both the crystalline and NMR-derived structures showed that Arg-459 is well anchored through electrostatic interactions with the Glu-476 carboxylate, the main-chain carbonyls of Pro-483 and Leu-530, and the His-553 imidazole. The non-degenerate, well-dispersed NηHη signals for Arg-459 as Pb, which are consistent with a salt bridge, disappeared in Pg, thus suggesting increased conformational freedom (data not shown). In support of these observations, we previously demonstrated that Arg-459 is required to stabilize the Pb state but is not required for photoconversion or maintenance of the Pg state (15).
Effects of Photoconversion on Signal Transmission
The differences between the paired Pb and Pg structures of TePixJ(GAF) provide clues as to how light energy is transduced into a mechanical signal. Presumably TePixJ signaling is initiated by photo-induced structural changes in the bilin, which then reverberate through the GAF domain, the up and/or downstream histidine kinase/adenylate cyclase/methyl accepting/phosphatase (HAMP) domains, and ultimately to the methyl-accepting chemotaxis protein signaling (MCP) domain. In this context, light-triggered repositioning the GAF domain relative to the HAMP domain(s) might be facilitated through effects on the connecting GAF domain helices α1 and α5. Consequently, it is easy to imagine that any motion affecting these helices is crucial to signal transduction within the TePixJ dimer.
One possible avenue is via displacement of the β-sheet enabled by the distinct fault propagated by the β-bulge in strand β2 and release of strand β3. Given that helices α1 and α5 are each physically coupled to a pair of β-strands (β1 and β2, and β5 and β6, respectively), displacement of the β sheet would directly impinge on the mobility and positioning of these helices. Pg formation also stabilizes the loop between strand β6 and helix α5, which potentially reorients the helix with respect to rest of the GAF domain. Melting and structural reorganization of strand β3/β-turn into a flexible region/310-helix in addition to affecting the β1 and β2 strands appears to rupture a salt bridge between Arg-486 (just above strand β3) and Glu-445 within helix α1, which would then add mobility to this helical extension. Interestingly, such a photoinduced release of a rigid domain is reminiscent of the flavin-containing light/oxygen/voltage (LOV) photoreceptors. Here, blue light-induced cleavage of a cysteine-based chromophore linkage within a structurally related PAS domain derepresses a downstream kinase effector domain by relaxing a critical interdomain contact (35).
Such rearrangements in TePixJ could affect the GAF domain dimerization contact, loosen constraints on the flanking HAMP domains imposed by the GAF domain as Pb, and/or reorient the HAMP domains relative to the GAF domain via movement or axial rotation of the α1/α5 helical bundles. With respect to the latter possibility, it has been proposed that HAMP domain-containing kinases, in particular, couple ligand binding by the sensory domain to rotation of the HAMP domain helical bundles via a gear box-type mechanism (36). Notably, similar rearrangements could underpin signaling by phytochromes in general. For the red/far red light-absorbing PAS-less phytochrome SyBCph1, we previously showed that upon photoexcitation several bilin-mediated structural changes focus on the GAF domain helical bundles involved in dimerization (11). Clearly, additional insights of the motions underpinning TePixJ signaling will now require more detailed models of photoconversion that include the output HAMP and MCP domains.
Conclusions
The paired solution NMR structures presented here of the CBCR TePixJ GAF domain in the dark and photoactivated states combined with the recently published crystal structures of this same blue-green light-absorbing photosensing module (21, 22) now provide unprecedented atomic details of phytochrome photoconversion. The solution structures confirmed the thioether linkage of the C10 carbon in PVB that underpins the distorted V-like structure of the bilin as Pb, demonstrated blue-light induced breakage of this bond concomitant with Z to E isomerization of the C15=C16 double bond and rotation of the D ring during Pg formation, and supported extensive migration of the bilin within the GAF domain pocket. The NMR structure of Pg also resolved the conformation of the region surrounding Cys-494, which was complicated by the non-native disulfide bridge in the x-ray crystallographic model. In addition, NMR relaxation studies revealed photostate-dependent changes in local dynamics. Key to these events are a number of universally conserved amino acids known to be critical for the photochemistry of TePixJ and others within the phytochrome superfamily (e.g. Asp-492, His-523, Gln-526, Asn-535, Tyr-463, and His-553; Refs. 11, 15, 21, and 37).
Ultimately, these combined changes substantially reorganize a β-strand/β-turn section adjacent to the bilin and cause general bending of the β-sheet along a prominent central ridge. Given the proximity of these conformational changes to the helices potentially involved in TePixJ dimerization, we propose that they reorient the adjacent region(s) responsible for signal transmission by modifying intra- and interdomain contacts within the dimer. Collectively, these paired Pb/Pg structures likely reveal commonalities in light perception within the phytochrome superfamily and provide informative scaffolds to modify signaling by CBCRs and their use as bilin-based fluorophores (15).
Acknowledgments
We thank Dr. Mario Rivera for isotopically labeled α-aminolevulinic acid, Joseph M. Walker for help with sample preparation, and Dr. W. Milo Westler for technical advice.
This work was supported by National Institutes of Health Grants P41RR02301 and P41GM66326 (to the National Magnetic Resonance Facility at Madison, University of Wisconsin and to J. L. M.) and by United States National Science Foundation Grants MCB-07191530 and MCB-1329956 (to R. D. V.).
The atomic coordinates and structure factors (codes 2M7U and 2M7V) have been deposited in the Protein Data Bank (http://wwpdb.org/).
NMR data have been deposited in the BioMagResBank (access codes 19213 and 19214) for Pb and Pg, respectively.
- GAF
- cGMP phosphodiesterase/adenylyl cyclase/FhlA
- Pr
- red light-absorbing form
- Pfr
- far red light-absorbing form
- CBCR
- cyanobacteriochrome
- PCB
- phycocyanobilin
- PVB
- phycoviolobilin
- Pb
- blue light-absorbing form
- Pg
- green light-absorbing form
- Te
- T. elongatus
- HSQC
- heteronuclear single-quantum coherence
- RDC
- residual dipole couplings
- r.m.s.d.
- root mean square deviation
- HAMP
- histidine kinase/adenylate cyclase/methyl-accepting/phosphatase
- PAS
- Per/Arndt/Sim.
REFERENCES
- 1. Auldridge M. E., Forest K. T. (2011) Bacterial phytochromes. More than meets the light. Crit. Rev. Biochem. Mol. Biol. 46, 67–88 [DOI] [PubMed] [Google Scholar]
- 2. Vierstra R. D., Zhang J. (2011) Phytochrome signaling. Solving the Gordian knot with microbial relatives. Trends Plant Sci. 16, 417–426 [DOI] [PubMed] [Google Scholar]
- 3. Wagner J. R., Brunzelle J. S., Forest K. T., Vierstra R. D. (2005) A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 438, 325–331 [DOI] [PubMed] [Google Scholar]
- 4. Wagner J. R., Zhang J., Brunzelle J. S., Vierstra R. D., Forest K. T. (2007) High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J. Biol. Chem. 282, 12298–12309 [DOI] [PubMed] [Google Scholar]
- 5. Cornilescu G., Ulijasz A. T., Cornilescu C. C., Markley J. L., Vierstra R. D. (2008) Solution structure of a cyanobacterial phytochrome GAF domain in the red-light-absorbing ground state. J. Mol. Biol. 383, 403–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Essen L. O., Mailliet J., Hughes J. (2008) The structure of a complete phytochrome sensory module in the Pr ground state. Proc. Natl. Acad. Sci. U.S.A. 105, 14709–14714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang X., Kuk J., Moffat K. (2008) Crystal structure of Pseudomonas aeruginosa bacteriophytochrome. Photoconversion and signal transduction. Proc. Natl. Acad. Sci. U.S.A. 105, 14715–14720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang X., Stojkovic E. A., Kuk J., Moffat K. (2007) Crystal structure of the chromophore binding domain of an unusual bacteriophytochrome, RpBphP3, reveals residues that modulate photoconversion. Proc. Natl. Acad. Sci. U.S.A. 104, 12571–12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song C., Psakis G., Lang C., Mailliet J., Gärtner W., Hughes J., Matysik J. (2011) Two ground state isoforms and a chromophore D-ring photoflip triggering extensive intramolecular changes in a canonical phytochrome. Proc. Natl. Acad. Sci. U.S.A. 108, 3842–3847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang X., Ren Z., Kuk J., Moffat K. (2011) Temperature-scan cryocrystallography reveals reaction intermediates in bacteriophytochrome. Nature 479, 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ulijasz A. T., Cornilescu G., Cornilescu C. C., Zhang J., Rivera M., Markley J. L., Vierstra R. D. (2010) Structural basis for the photoconversion of a phytochrome to the activated Pfr form. Nature 463, 250–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hirose Y., Rockwell N. C., Nishiyama K., Narikawa R., Ukaji Y., Inomata K., Lagarias J. C., Ikeuchi M. (2013) Green/red cyanobacteriochromes regulate complementary chromatic acclimation via a protochromic photocycle. Proc. Natl. Acad. Sci. U.S.A. 110, 4974–4979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rockwell N. C., Ohlendorf R., Möglich A. (2013) Cyanobacteriochromes in full color and three dimensions. Proc. Natl. Acad. Sci. U.S.A. 110, 806–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshihara S., Shimada T., Matsuoka D., Zikihara K., Kohchi T., Tokutomi S. (2006) Reconstitution of blue-green reversible photoconversion of a cyanobacterial photoreceptor, PixJ1, in phycocyanobilin-producing Escherichia coli. Biochemistry 45, 3775–3784 [DOI] [PubMed] [Google Scholar]
- 15. Ulijasz A. T., Cornilescu G., von Stetten D., Cornilescu C., Velazquez Escobar F., Zhang J., Stankey R. J., Rivera M., Hildebrandt P., Vierstra R. D. (2009) Cyanochromes are blue/green light photoreversible photoreceptors defined by a stable double cysteine linkage to a phycoviolobilin-type chromophore. J. Biol. Chem. 284, 29757–29772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rockwell N. C., Martin S. S., Gulevich A. G., Lagarias J. C. (2012) Phycoviolobilin formation and spectral tuning in the DXCF cyanobacteriochrome subfamily. Biochemistry 51, 1449–1463 [DOI] [PubMed] [Google Scholar]
- 17. Rockwell N. C., Martin S. S., Feoktistova K., Lagarias J. C. (2011) Diverse two-cysteine photocycles in phytochromes and cyanobacteriochromes. Proc. Natl. Acad. Sci. U.S.A. 108, 11854–11859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen Y., Zhang J., Luo J., Tu J. M., Zeng X. L., Xie J., Zhou M., Zhao J. Q., Scheer H., Zhao K. H. (2012) Photophysical diversity of two novel cyanobacteriochromes with phycocyanobilin chromophores. Photochemistry and dark reversion kinetics. FEBS J. 279, 40–54 [DOI] [PubMed] [Google Scholar]
- 19. Ishizuka T., Kamiya A., Suzuki H., Narikawa R., Noguchi T., Kohchi T., Inomata K., Ikeuchi M. (2011) The cyanobacteriochrome, TePixJ, isomerizes its own chromophore by converting phycocyanobilin to phycoviolobilin. Biochemistry 50, 953–961 [DOI] [PubMed] [Google Scholar]
- 20. Rockwell N. C., Martin S. S., Lagarias J. C. (2012) Mechanistic insight into the photosensory versatility of DXCF cyanobacteriochromes. Biochemistry 51, 3576–3585 [DOI] [PubMed] [Google Scholar]
- 21. Burgie E. S., Walker J. M., Phillips G. N., Jr., Vierstra R. D. (2013) A photo-labile thioether linkage to phycoviolobilin provides the foundation for the blue/green photocycles in DXCF-cyanobacteriochromes. Structure 21, 88–97 [DOI] [PubMed] [Google Scholar]
- 22. Narikawa R., Ishizuka T., Muraki N., Shiba T., Kurisu G., Ikeuchi M. (2013) Structures of cyanobacteriochromes from phototaxis regulators AnPixJ and TePixJ reveal general and specific photoconversion mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 918–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cornilescu G., Bax A. (2000) Measurement of proton, nitrogen, and carbonyl chemical shielding anisotropies in a protein dissolved in a dilute liquid crystalline phase. J. Am. Chem. Soc. 122, 10143–10154 [Google Scholar]
- 24. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPIPE. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 25. Garrett D. S., Powers R., Gronenborn A. M., Clore G. M. (1991) A common sense approach to peak picking two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 95, 214–220 [DOI] [PubMed] [Google Scholar]
- 26. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+. A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, pp. 180–182, Wiley Interscience, New York, NY [Google Scholar]
- 28. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The XPLOR-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 29. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG. A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 30. Schwieters C. D., Clore G. M. (2001) The VMD-XPLOR visualization package for NMR structure refinement. J. Magn. Reson. 149, 239–244 [DOI] [PubMed] [Google Scholar]
- 31. Schmidt M., Patel A., Zhao Y., Reuter W. (2007) Structural basis for the photochemistry of α-phycoerythrocyanin. Biochemistry 46, 416–4123 [DOI] [PubMed] [Google Scholar]
- 32. Yang X., Kuk J., Moffat K. (2009) Conformational differences between the Pfr and Pr states of Pseudomonas aeruginosa bacteriophytochrome. Proc. Natl. Acad. Sci. U.S.A. 106, 15639–15644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chou J. J., Li S., Klee C. B., Bax A. (2001) Solution structure of Ca2+-calmodulin reveals flexible hand-like properties of its domains. Nat. Struct. Biol. 8, 990–997 [DOI] [PubMed] [Google Scholar]
- 34. Goto N. K., Skrynnikov N. R., Dahlquist F. W., Kay L. E. (2001) What is the average conformation of bacteriophage T4 lysozyme in solution? A domain orientation study using dipolar couplings measured by solution NMR. J. Mol. Biol. 308, 745–764 [DOI] [PubMed] [Google Scholar]
- 35. Harper S. M., Neil L. C., Gardner K. H. (2003) Structural basis of a phototropin light switch. Science 301, 1541–1544 [DOI] [PubMed] [Google Scholar]
- 36. Ferris H. U., Dunin-Horkawicz S., Hornig N., Hulko M., Martin J., Schultz J. E., Zeth K., Lupas A. N., Coles M. (2012) Mechanism of regulation of receptor histidine kinases. Structure 20, 56–66 [DOI] [PubMed] [Google Scholar]
- 37. Wagner J. R., Zhang J., von Stetten D., Günther M., Murgida D. H., Mroginski M. A., Walker J. M., Forest K. T., Hildebrandt P., Vierstra R. D. (2008) Mutational analysis of Deinococcus radiodurans bacteriophytochrome reveals key amino acids necessary for the photochromicity and proton exchange cycle of phytochromes. J. Biol. Chem. 283, 12212–12226 [DOI] [PMC free article] [PubMed] [Google Scholar]