

Background: S-nitrosylation inhibits mediators of the immune response including the transcription factor NF-κB (p50/p65).
Results: Exogenous and endogenous inhibitors of thioredoxin prevent p65 denitrosylation and downstream activation of NF-κB.
Conclusion: Thioredoxin activates inflammatory signaling through targeted denitrosylation of NF-κB.
Significance: Mechanisms that augment protein S-nitrosylation can be utilized to suppress the immune response.
Keywords: Lung Injury, NF-κB, Nitric Oxide, S-nitrosylation, Thioredoxin
Abstract
S-nitrosylation of nuclear factor κB (NF-κB) on the p65 subunit of the p50/p65 heterodimer inhibits NF-κB DNA binding activity. We have recently shown that p65 is constitutively S-nitrosylated in the lung and that LPS-induced injury elicits a decrease in SNO-p65 levels concomitant with NF-κB activation in the respiratory epithelium and initiation of the inflammatory response. Here, we demonstrate that TNFα-mediated activation of NF-κB in the respiratory epithelium similarly induces p65 denitrosylation. This process is mediated by the denitrosylase thioredoxin (Trx), which becomes activated upon cytokine-induced degradation of thioredoxin-interacting protein (Txnip). Similarly, inhibition of Trx activity in the lung attenuates LPS-induced SNO-p65 denitrosylation, NF-κB activation, and airway inflammation, supporting a pathophysiological role for this mechanism in lung injury. These data thus link stimulus-coupled activation of NF-κB to a specific, protein-targeted denitrosylation mechanism and further highlight the importance of S-nitrosylation in the regulation of the immune response.
Introduction
S-nitrosylation has emerged as a preeminent mechanism by which nitric oxide (NO) modulates cellular immunity, with this post-translational modification shown to regulate numerous proteins in a wide range of immune response pathways. The proinflammatory transcription factor NF-κB2 is the prototypic example, as both subunits of the p50/p65-activating heterodimer are modified by S-nitrosylation at a conserved, redox-sensitive cysteine located in the Rel protein DNA binding domain (1, 2). S-nitrosylation of this reactive thiol results in disruption of p50/p65 DNA binding thereby attenuating antecedent transcription of κB-dependent immune response genes. Importantly, we have previously shown that S-nitrosylation of the NF-κB heterodimer is the central mechanism by which inducible nitric-oxide synthase (NOS2) deactivates NF-κB and inhibits the continued transcription of NOS2 in cytokine-stimulated respiratory epithelial cells and macrophages (1, 3). These findings thus delineate a feedback loop by which S-nitrosylation controls NO production and prevents cellular nitrosative stress in immune-activated cells.
In the lung, we have shown that NF-κB p65 is constitutively S-nitrosylated and that LPS exposure, in a model of acute lung injury (ALI), induces a rapid decrease in S-nitrosylated p65 (SNO-p65) (3). Denitrosylation of SNO-p65 occurs contiguous with NF-κB activation in the respiratory epithelium and initiation of the airway inflammatory response. Augmentation of SNO-p65 levels in the lung by inhalation of ethyl nitrite prevents NF-κB activation and inhibits lung inflammation/injury, highlighting the physiological significance of p65 denitrosylation and suggesting that S-nitrosothiol (SNO)-based therapies may have therapeutic potential in the treatment of inflammatory lung disease (4). However, the mechanism(s) responsible for cytokine-induced p65 denitrosylation remains unexplored and enigmatic.
The oxidoreductase thioredoxin (Trx) is a well known regulator of NF-κB activity. Trx serves to facilitate NF-κB-dependent transcription through direct interaction with the p50/p65 heterodimer in the nucleus reducing the conserved, redox-sensitive cysteine in the Rel DNA binding domain (5, 6). Oxidation of this cysteine, similar to S-nitrosylation, has been shown to preclude p50/p65 DNA binding and inhibit NF-κB activation (6). In addition to its classic role as a disulfide reductase, Trx has also been shown to function in protein denitrosylation (7). The “denitrosylase” and oxidoreductase activities both utilize the Trx active site cysteines for reduction of the targeted protein thiol. We therefore hypothesized that Trx might function in the cytokine-induced denitrosylation and activation of NF-κB within respiratory epithelial cells and in our model of ALI.
Recently, Trx denitrosylase activity in cytokine-activated, NOS2-expressing macrophages was shown to be regulated by the thioredoxin-interacting protein Txnip (8). A NO-dependent decrease in Txnip transcription resulted in an increase in SNO-protein metabolism, preventing the development of cellular nitrosative stress. These studies also suggested there to be a NO-independent, acute decrease in cellular Txnip levels not explained by attenuated transcription. We now show that cytokine-induced denitrosylation of p65 in the respiratory epithelium is mediated by Trx, which is activated predominantly by accelerated ubiquitination and proteasomal degradation of Txnip, and that this mechanism facilitates NF-κB activation and the resultant inflammatory response in a model of ALI (3, 4).
EXPERIMENTAL PROCEDURES
Reagents
All materials were purchased from Sigma unless otherwise indicated. All antibodies were from Santa Cruz Biotechnology except Txnip rabbit polyclonal (Invitrogen), Txnip mouse monoclonal (MBL International), and ubiquitin (Cell Signaling).
Cell Culture
A549 (CCL-185) cells were grown in F12K medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cultures were maintained in 95% air, 5% CO2 at 37 °C. Cells were infected with the Txnip lentivirus expression or empty vector for 36 h prior to cytokine stimulation with TNFα. Whole cell, cytoplasmic, and nuclear lysates were prepared as previously described (1). Protein concentration of cell extracts was determined by BCA (Pierce).
Txnip Lentivirus Construction
Mouse Txnip cDNA was subcloned into the 5′-EcoRI and 3′-XhoI sites of pCDH lentiviral expression (System Biosciences). Txnip and control lentiviruses were produced by transfecting HEK-293T cells with a mixture of pCDH, PAX2, and VSVG plasmids using Lipofectamine (Invitrogen). Cell culture supernatants were harvested 4–5 days later, cleared by centrifugation at 2000 rpm for 5 min, and filtered at 0.45 μm.
Immunoprecipitation (IP)
A549 cells were lysed in IP buffer (0.1% Nonidet P-40, 150 mm NaCI, 10 mm Na2HPO4, 2 mm EDTA), and NF-κB p65, Trx1, and ubiquitin IPs were performed by incubation with primary antibody or nonimmunized rabbit IgG (Cell Signaling) for 2 h at 4 °C, followed by addition of protein G-agarose and overnight incubation at 4 °C. Beads were washed extensively with IP buffer, and proteins were eluted by boiling in Lamelli buffer and separated by SDS-PAGE followed by Western blotting.
Mouse Model of ALI
All procedures were approved by the Duke University Institutional Animal Care and Use Committee. 6–8-week-old, male wild-type C57BL6/J mice were exposed to aerosolized LPS (0111:B4 Escherichia coli LPS, 4 μg/m3) or phosphate-buffered saline (PBS) for 2.5 h as previously described (4).
Mouse lung lavage, tissue extraction, and homogenate preparation were performed as previously described (4). Cell counting of pooled bronchoalveolar lavage fluid (BALF) was performed with a hemocytometer and cell differentials determined on stained cytospin preparations. BALF was centrifuged at 1500 × g for 10 min to collect cells, and the protein concentration of the cell-free supernatant was determined by BCA assay. ELISAs were used to quantify KC (Peprotech) and IL-6 (EBioscience) in the BALF.
Protein SNO Detection
Total cell SNOs were quantified in protein lysates using photolysis-chemiluminescence as previously described (4). The biotin switch assay was used to quantify SNO-p65 as previously described (1). Samples were diluted in HENS buffer (250 mm Hepes, 1 mm EDTA, 0.1 mm neocuproine, 2.5% SDS, pH 7.7) and free thiols blocked by the addition of 3 mm S-methylmethane thiosulfonate. Proteins were then precipitated in cold acetone, washed extensively, and resuspended in HENS buffer. SNOs were reduced by 50 mm sodium ascorbate and labeled with 50 μg/ml biotin-HPDP (Pierce). Biotinylated proteins were precipitated by overnight incubation with Neutravidin-agarose (Pierce). The biotinylated proteins were analyzed by immunoblotting.
NF-κB Activity Assay
Nuclear protein binding to a consensus NF-κB oligonucleotide was determined by ELISA (TransAm p65, Active Motif).
S-nitrosoglutathione Reductase (GSNOR) Activity
Protein was diluted to 1 mg/ml in PBS containing 100 μm DTPA and 500 μm EDTA, and the absorbance at 340 nm was measured over time in the presence of 100 μm reduced nicotinamide adenine dinucleotide (NADH) and 100 μm S-nitrosoglutathione (GSNO). NADH-dependent GSNOR activity was defined as the difference in absorbance in the presence or absence of GSNO. Relative GSNOR activity was calculated from the slope of the absorbance over time.
Thioredoxin Reductase (TrxR) Activity
Protein was diluted to 1 mg/ml in PBS containing 100 μm DTPA and 500 μm EDTA, followed by the addition of 200 μm NADPH and 5 mm 5.5′-dithiobis-(2-nitro benzoic acid) (DTNB). TrxR activity was calculated as the rate of thionitrobenzoate anion (TNB) formation at 412 nm.
Real-time PCR
Total RNA was extracted from A549 cells (RNeasy Mini Kit, Qiagen, Valencia, CA). A total of 1 μg of RNA was used for reverse transcription using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-Time PCR was performed in a Bio-Rad iCycler using SYBR Green reagent (Bio-Rad) under standard conditions. Primer sequences were as following: actin forward, 5′-TCA AGA TCA TTG CTC CTC CTG-3′ and reverse, 5′-CTG CTT GCT GAT CCA CAT CTG-3′; Txnip forward, 5′-GAG TGT GGG TCC ACC TTA GC-3′ and reverse, 5′-TGT ATC ACA ACA TGG GCG CT-3′. Relative quantification was performed using comparative threshold cycle CT analysis. Gene expression changes were expressed as -fold change using actin as internal control.
Data Analysis
Data are expressed as mean ± S.E. Significant differences between groups were identified by Student's t test, or two-way analysis of variance. The ratios of SNO-p65 to total-p65 Western blot densitometry were calculated and log-transformed to satisfy normality. Analysis of variance was performed to examine the effect of blot and chemical on treatment group. Results are presented as estimated marginal means and are adjusted for multiple comparisons (Tukey-Kramer contrast; R statistical Analysis freeware, version 2.15.0).
RESULTS
TNFα-induced Denitrosylation of NF-κB p65 in the Respiratory Epithelium
We have previously shown that S-nitrosylation of p65 inhibits NF-κB activity in the respiratory epithelium and that LPS exposure in a mouse model of ALI induces a rapid decrease in SNO-p65 levels in the lung in conjunction with NF-κB activation in the respiratory epithelium (4). These data suggest that S-nitrosylation of p65 is a critical mechanism for the maintenance of NF-κB in a quiescent state in the respiratory epithelium. To determine whether p65 denitrosylation is a prerequisite for cytokine-induced NF-κB activation, we quantified S-nitrosylation of p65 in A549 respiratory epithelial cells after 1-h stimulation with TNFα, which robustly activates NF-κB in these cells and is secreted at a high concentration into the mouse airway in response to LPS (9). By biotin switch assay, we found significant basal S-nitrosylation of p65 in A549 cells and observed a >50% reduction in SNO-p65 levels following TNFα treatment (Fig. 1, A and B). Interestingly, we found no significant change in total cellular SNOs after TNFα stimulation (Fig. 1C), suggesting that the decrease in SNO-p65 was not the result of a generalized increase in cell SNO metabolism. In addition, TNFα stimulation did not alter the activity of GSNOR, the enzyme that is primarily responsible for regulating intracellular GSNO levels (Fig. 1D). Collectively, these data indicate that the mechanism for TNFα-induced SNO-p65 denitrosylation is protein-specific and suggest a well coordinated, targeted process.
FIGURE 1.
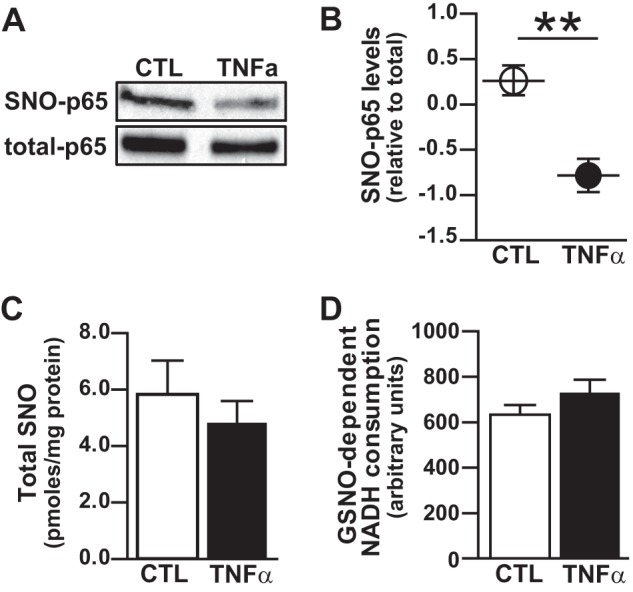
Cytokine-induced denitrosylation of NF-κB p65 in the respiratory epithelium. A549 cells were stimulated with TNFα (10 ng/ml) for 1 h followed by cell lysate preparation. A, S-nitrosylated p65 (SNO-p65) as determined by biotin switch assay. B, predicted marginal means of the ratio of SNO-p65/total-p65 Western blot densitometry values, n = 5, mean ± S.E. (error bars); **, p < 0.01. C, total SNO in cell lysate as quantified by photolysis-chemiluminescence, n = 4, mean ± S.E. D, GSNOR activity in cell lysates as quantified by SNO-dependent NADH consumption, n = 5, mean ± S.E.
TNFα-induced Denitrosylation and Activation of NF-κB Is Trx-dependent
Recent work has established that the oxidoreductase Trx can function as a SNO-protein denitrosylase. For example, in the lymphocyte, Trx mediates Fas-induced denitrosylation and activation of caspase-3 (7). Given the well known role of Trx in regulating NF-κB activity by controlling the redox status of the p50/p65 heterodimer (6, 10), we hypothesized that Trx might similarly serve to mediate TNFα-induced SNO-p65 denitrosylation and NF-κB activation in the respiratory epithelium. Pretreatment of A549 cells with auranofin, an organogold compound that inhibits TrxR thereby blocking redox cycling of Trx, prevented TNFα-induced denitrosylation of SNO-p65 in conjunction with marked attenuation in NF-κB (p50/p65) DNA binding and the NF-κB-dependent transcription of cytokine IL-8 (Fig. 2, A–D). Moreover, auranofin treatment did not affect TNFα-induced IκBα degradation or p65 nuclear translocation (Fig. 2D) indicating that S-nitrosylation of p65 is an inhibitory mechanism distinct from the classically described NF-κB signal transduction pathway, where IκBα phosphorylation, ubiquitination, and proteasomal degradation are the crucial regulators of NF-κB activation.
FIGURE 2.
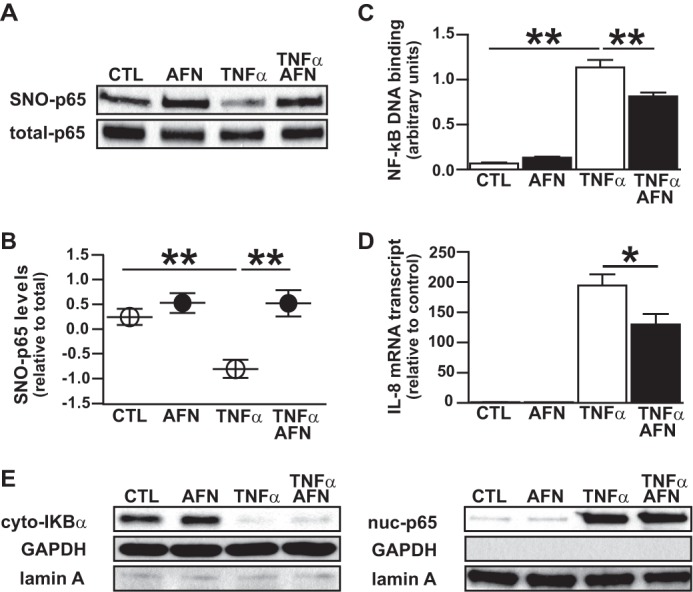
Cytokine-induced denitrosylation and activation of NF-κB p65 is thioredoxin-dependent. A549 cells were treated with 5 μm auranofin (AFN) followed by TNFα stimulation (1 h) and cell lysate preparation. A, SNO-p65 as determined by biotin switch assay. B, predicted marginal means of the ratio of SNO-p65/total-p65 Western blot densitometry values, n = 5, mean ± S.E. (error bars). **, p < 0.01. C, NF-κB p65 oligonucleotide binding as quantified in nuclear lysates using TransAm assay, n = 5, mean ± S.E. **, p < 0.01. D, IL-8 mRNA as quantified by RT-PCR, n = 5. *, p < 0.05. E, IκBα and p65 immunoblots prepared from cytoplasmic and nuclear lysates. GAPDH and lamin A were used as cytoplasm and nuclear protein markers, respectively.
Trx Mediates LPS-induced Airway Inflammation via NF-κB Denitrosylation
We have previously demonstrated that SNO-p65 denitrosylation in the lung occurs in conjunction with NF-κB activation in the respiratory epithelium and is an essential step that initiates the airway inflammatory response to aerosolized LPS (4). To determine whether Trx activity regulates p65 denitrosylation and NF-κB activation in this model of ALI, we inhibited TrxR activity in the mouse lung via intraperitoneal injection of the TrxR inhibitor aurothioglucose prior to LPS exposure. Similar to our findings in A549 cells, inhibition of TrxR activity prevented LPS-induced SNO-p65 denitrosylation and attenuated NF-κB DNA binding in the lung (Fig. 3, A–C). The decrease in NF-κB activation by aurothioglucose resulted in diminished airway expression of the proinflammatory cytokines KC and IL-6 and inhibited the influx of inflammatory cells (i.e. neutrophils) into the airway (Fig. 3, D and E). These findings indicate that Trx-mediated denitrosylation of SNO-p65 is a critical signaling event that is required for NF-κB activation in the lung and is an important mechanism by which the pulmonary immune response is regulated.
FIGURE 3.
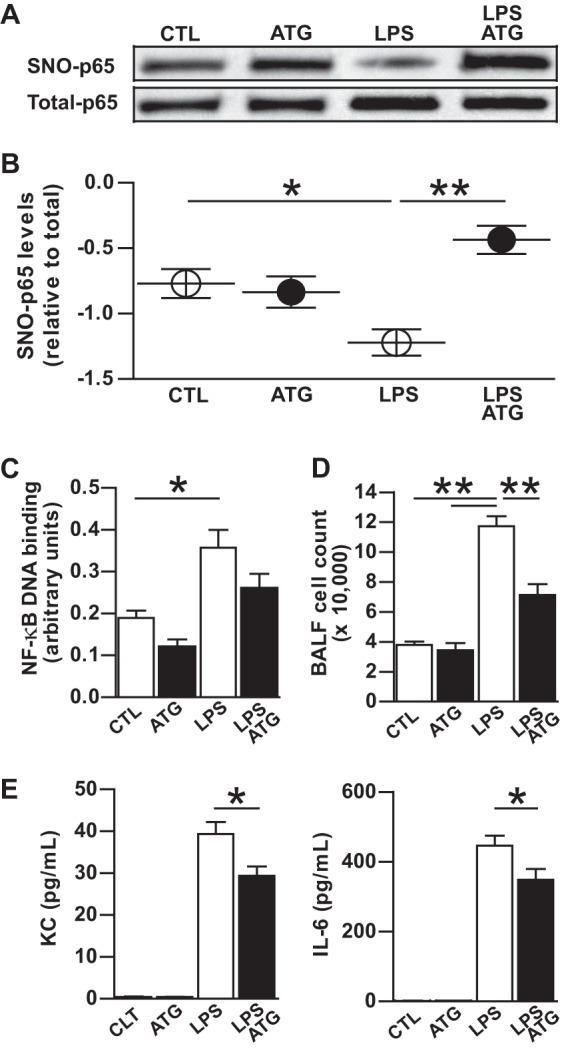
Thioredoxin mediates denitrosylation and activation of NF-κB p65 in LPS-induced lung injury. Mice were administered a 625-μg dose intraperitoneal injection of aurothioglucose (ATG) 24 h prior to treatment with aerosolized saline or LPS. Lung tissues were analyzed 1 h after treatment. A, SNO-p65 as determined in lung tissue homogenate by biotin switch assay. B, predicted marginal means of the ratio of SNO-p65/total-p65 Western blot densitometry values, n = 5, mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01. C, NF-κB p65 oligonucleotide binding as quantified in nuclear lysates prepared from lung homogenate, n = 5; mean ± S.E. *, p < 0.05. D, total WBC in BALF, n = 5, mean ± S.E. **, p < 0.01. E, BALF KC and IL-6 levels as quantified by ELISA, n = 5, mean ± S.E. *, p < 0.05.
Mechanism by Which Trx1 Regulates NF-κB Denitrosylation
The NF-κB p50/p65 heterodimer has previously been shown to undergo protein interaction with Trx1 in both the cytoplasm and nucleus (10). As such, alteration in this interaction is a potential mechanism by which denitrosylation of SNO-p65 could be regulated. To determine whether cytokine stimulation enhances NF-κB-Trx1 interaction in the respiratory epithelium, we analyzed p65 and Trx1 immunoprecipitates prepared from A549 cell lysates. Trx1 co-immunoprecipitated with p65 at base line, and this interaction was confirmed in the reverse (Trx1) IP (Fig. 4A). Interestingly, we observed a slight decrease in Trx1-p65 interaction after TNFα stimulation with no change in either Trx1 or p65 expression. Similarly, we saw no differences in TrxR protein expression or activity in A549 cells following TNFα stimulation (Fig. 4, B and C). These data indicate that neither enhanced TrxR activity nor Trx1 interaction with the NF-κB p50/p65 heterodimer is the mechanism by which TNFα-induced, Trx-mediated denitrosylation of NF-κB p65 is regulated. However, TNFα stimulation did induce a marked decrease in cellular levels of the Trx-inhibiting protein Txnip (Fig. 4C) delineating a potential Trx-activating mechanism. Furthermore, we found that Txnip co-immunoprecipitated with Trx1 but not p65 at base line, suggesting that the interaction of Txnip and p65 with Trx1 is exclusive (Fig. 4D).
FIGURE 4.
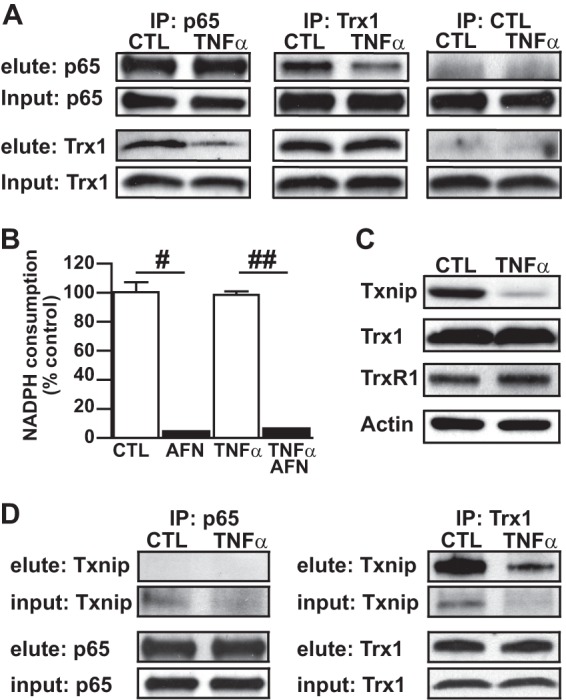
Cytokine-induced activation of thioredoxin. A549 cells were stimulated with TNFα (10 ng/ml) for 1 h followed by cell lysate preparation. A, co-precipitation of p65 with Trx1. p65 and Trx1 immunoprecipitates (IP) were prepared from whole cell lysate and p65 and Trx1 immunoblots from proteins eluted from the neutravidin-agarose bead conjugates. B, TrxR activity quantified in whole cell lysate using a NADPH consumption assay, n = 5, mean ± S.E. (error bars). #, p < 0.001; ##, p < 0.0001. C, immunoblots for Txnip, Trx1, and TrxR1 prepared from whole cell lysates. D, Txnip immunoblots prepared from p65 and Trx1 IP eluate.
Txnip is a constitutively expressed protein that forms a mixed disulfide with the active site Cys32 of Trx, thereby inhibiting its oxidoreductase activity (11). Txnip has a short half-life due to robust degradation via the ubiquitin-proteasome system (12). Recently, NO was shown to down-regulate Txnip transcription in cytokine-stimulated macrophages, thereby providing a feed forward mechanism for activation of protein denitrosylation under settings of high NO output (8). To determine whether ubiquitination of Txnip is increased in TNFα-stimulated cells, we captured ubiquitinated proteins by IP and immunoblotted the eluate for Txnip. A relative increase in Txnip ubiquitination was seen after TNFα stimulation (Fig. 5A), particularly in light of the decrease in total cell Txnip expression (Fig. 5A, Txnip input). To further probe the importance of proteasomal degradation in the observed decrease in Txnip after cytokine stimulation, A549 cells were treated with the proteasome inhibitor MG132 prior to TNFα. Pretreatment with MG132 fully prevented the decline in Txnip after TNFα stimulation (Fig. 5B), confirming that the mechanism is dependent upon proteasomal degradation. Txnip transcription was also decreased after TNFα stimulation (Fig. 5C), suggesting that there are multiple mechanisms for the decline of cellular Txnip. However, whereas NOS2-derived NO is known to decrease Txnip expression (8), we found that NOS2 is not expressed in A549 cells after a 1-h TNFα stimulation (Fig. 5D), and the NOS2 inhibitor 1400W had no effect on TNFα-induced Txnip degradation (Fig. 5E). Collectively, these results suggest that the rapid decrease in cellular Txnip levels after TNFα cytokine activation is NOS2-independent and predominantly due to Txnip degradation by the ubiquitin-proteasome pathway.
FIGURE 5.
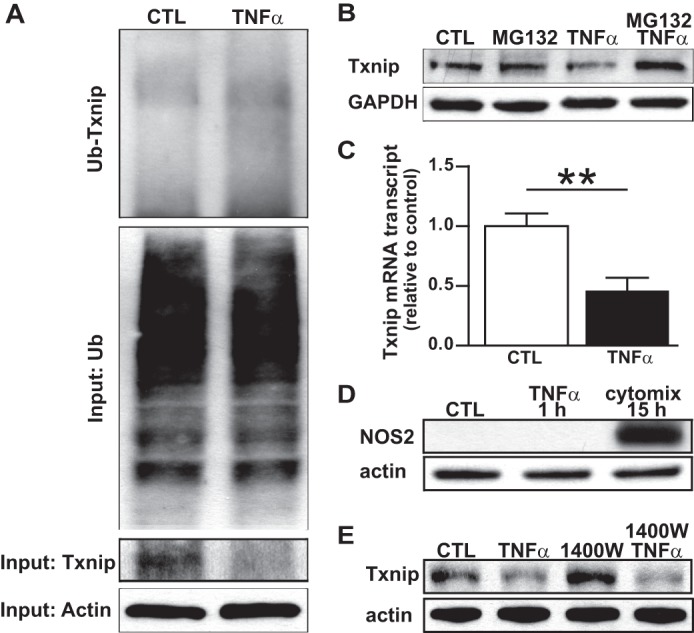
Mechanism(s) of cytokine-induced Txnip degradation. A, Txnip ubiquitination (Ub-Txnip) in A549 cells ± 1 h stimulation with TNFα (10 ng/ml) as demonstrated by Txnip immunoblots prepared from ubiquitin IP eluates. Total (input) Txnip is shown below the ubiquitin blots. B, Txnip immunoblots prepared from A549 cells treated with the proteasome inhibitor MG132 and stimulated with TNFα (10 ng/ml) for 1 h. C, Txnip mRNA as quantified by RT-PCR in A549 cells ± 1 h stimulation with TNFα (10 ng/ml). Mean ± S.E. (error bars), **, p < 0.01. D, NOS2 immunoblots prepared from A549 cells stimulated with TNFα (10 ng/ml) for 1 h, or with a NOS2-inducing cytokine mix of TNFα (10 ng/ml), IL-1β (10 ng/ml), IFN-γ (50 ng/ml), and LPS (500 ng/ml) for 15 h. E, Txnip immunoblots prepared from A549 cells treated with the NOS2 inhibitor 1400W (100 μm) and stimulated with TNFα (10 ng/ml) for 1 h.
Txnip Regulates Cytokine-induced NF-κB Denitrosylation and Activation
To further address whether Txnip functionally regulates cytokine-induced SNO-p65 denitrosylation, we overexpressed Txnip in A549 cells via lentiviral vector. In a manner analogous to the effects of the organogold TrxR inhibitors, Txnip overexpression fully prevented the TNFα-induced denitrosylation of SNO-p65 in A549 cells (Fig. 6, A and B). Similarly, Txnip overexpression significantly attenuated NF-κB DNA binding (Fig. 6C) whereas it had no effect on IκBα degradation or p65 nuclear translocation (Fig. 6D). These findings confirm that cytokine-induced decrease in Txnip is central to Trx1-mediated denitrosylation of SNO-p65 which is essential for the subsequent activation of NF-κB in the respiratory epithelium.
FIGURE 6.
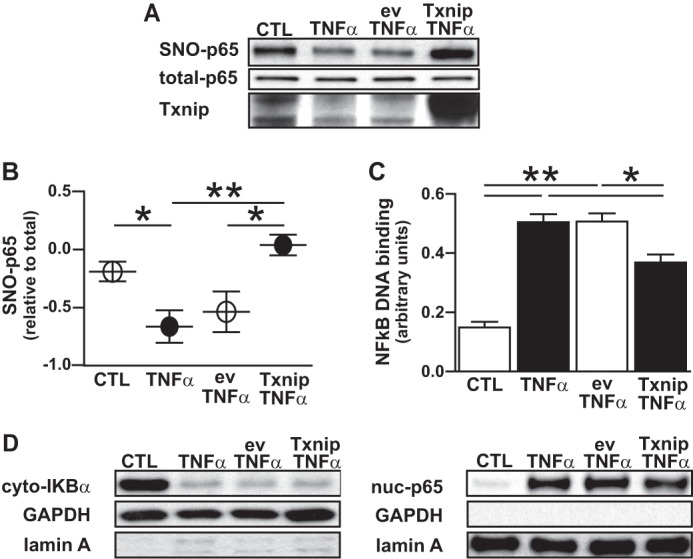
Txnip inhibits cytokine-induced denitrosylation and activation of NF-κB p65. A549 cells were infected with lentivirus that expressed Txnip (Txv) or an empty vector control (ev). Thirty-six hours after infection, cells were stimulated with TNFα (10 ng/ml) for 1 h followed by cell lysate preparation. A, SNO-p65 as determined in whole cell lysate by biotin switch assay. B, predicted marginal means of the ratio of SNO-p65/total-p65 Western blot densitometry values, n = 4, mean ± S.E. (error bars). *, p < 0.05; **, p < 0.01. C, NF-κB p65 oligonucleotide binding as quantified in nuclear lysates by TransAm assay, n = 4, mean ± S.E. *, p < 0.05; **, p < 0.01. D, IκBα and p65 immunoblots prepared from cytoplasmic and nuclear lysates, respectively.
DISCUSSION
It is increasingly recognized that S-nitrosylation is a critical mechanism for the post-translational regulation of immune response proteins. Whereas numerous mechanisms that target proteins for S-nitrosylation have been identified (e.g. via NOS protein-protein interaction) (13, 14), denitrosylating pathways, in particular those tied to specific stimuli, have only begun to be elucidated. In this study, we determined that the mechanism by which cytokine stimulation of the respiratory epithelium reverses constitutive and inhibitory S-nitrosylation of NF-κB p65 occurs through activation of the oxidoreductase Trx1. We further link Trx-mediated p65 denitrosylation to LPS-induced airway inflammation in vivo, thus emphasizing the relevance of S-nitrosylation as a regulatory pathway in the pulmonary immune response.
We and others have previously demonstrated that S-nitrosylation is a crucial mechanism by which NF-κB activity is modulated. In addition to the p50/p65-activating heterodimer, other proteins in the NF-κB pathway have been identified as targets of inhibitory S-nitrosylation including IκB kinase β (IKKβ) and MyD88 (in the TLR4 pathway) (15, 16). In the case of IKKβ and analogous to p65, S-nitrosylation is present under basal conditions with cytokine stimulation inducing rapid denitrosylation and IKK activation. S-nitrosylation thus appears to serve as a corollary inhibitory mechanism in the NF-κB pathway that functions in parallel with the well delineated cytoplasmic sequestering of the p50/p65-activating heterodimer by IκBα (17). This premise is supported by the present data where TrxR inhibitors and Txnip overexpression, while inhibiting NF-κB p65 denitrosylation and p50/p65 DNA binding, had no effect on TNFα-induced IκBα degradation or p65 nuclear translocation (Fig. 6D). In this regard, it is important to note that additional modification of the target cysteine in p65 (Cys38) can occur after denitrosylation, in particular sulfhydration which was recently shown to facilitate NF-κB p50/p65 DNA binding in cytokine-activated macrophages (18).
Trx is a well known mediator of NF-κB activation. Trx1 binds the p50/p65 heterodimer directly, reducing the reactive thiol in the Rel DNA binding domain, thereby facilitating binding at target gene κB promoter sites (6). In activated cells (including TNFα-stimulated respiratory epithelium), Trx undergoes nuclear translocation where it interacts with NF-κB p50/p65 and other transcription factors either directly or in association with APE/Ref-1 (5, 19). Interestingly, Hirota et al. demonstrated that whereas overexpression of Trx in the nucleus enhances NF-κB activity, cytoplasmic overexpression has the opposite effect, suggesting that other proteins lying upstream in the NF-κB pathway are actually inhibited by thiol reduction (10).
Whereas Trx is classically described as a disulfide reductase, an evolving body of literature has characterized the ability of Trx to reduce SNOs as well. Unlike GSNOR, which indirectly decreases cell protein S-nitrosylation through GSNO metabolism, Trx appears to directly target specific SNO-proteins for Cys reduction (20). In this regard, two previously identified targets of Trx-mediated denitrosylation, caspase-3 and N-ethylmaleimide-sensitive factor, have been shown to require protein interaction with Trx for denitrosylation to occur (7, 21). Although we show here that p65 similarly interacts directly with Trx1, our data suggest that the primary mechanism regulating Trx-dependent p65 denitrosylation in the respiratory epithelium is not modification in Trx1-p65 binding, but rather degradation of the Trx-inhibiting protein Txnip. Our present findings thus extend the recent observations that suppression of Txnip is a mechanism by which Trx prevents indiscriminate protein S-nitrosylation and nitrosative stress in cytokine-stimulated, NOS2-expressing macrophages (8).
Originally described as a vitamin D-up-regulated protein (VDUP1), one of the principal functions of Txnip is to regulate Trx activity (11, 22). Indeed, the pathophysiological importance of Txnip has been demonstrated in numerous disease models where both oxidative stress and S-nitrosylation are known to play key roles (e.g. diabetes, carcinogenesis, and inflammation) (23–25). In the context of inflammation, Txnip has previously been shown to suppress cytokine-induced NF-κB activation (26), findings that are corroborated in the present study. However, we found the mechanism by which Txnip levels acutely decrease to be primarily through heightened ubiquitination and proteasomal degradation, with attenuated transcription playing a lesser role. These results are in contrast with the NOS2-dependent decrease in Txnip transcription seen after more prolonged cytokine stimulation (8). In support of our observations, Txnip has recently been shown to be targeted by the ubiquitin E3 ligase Itch through interaction with a conserved PPXY motif in the C terminus of Txnip (12). However, it remains to be determined how specific stimuli, including TNFα, regulate this process in the respiratory epithelium.
We have previously shown that SNOs suppress lung inflammation and that aerosolized LPS acutely decreases SNO-p65 levels, leading to NF-κB activation in the respiratory epithelium and initiation of the pulmonary immune response (4). Our results now indicate that Trx activation is the mediator of this response. With these data, NF-κB p65 can now be classified as the first protein in which the regulatory mechanisms governing both stimulus-coupled S-nitrosylation (via NOS2) (1, 3) and denitrosylation (via Trx1) have been defined. However, given the importance of S-nitrosylation in regulating the cellular immune response, it seems likely that additional SNO-modified immune response proteins will be identified. Pharmacological inhibitors of Trx/TrxR thus hold promise as anti-inflammatory agents that can be employed either alone or in conjunction with SNO enhancing therapies.
Acknowledgments
We thank Dr. Derek Cyr (Duke Institute for Genome Sciences and Policy) and Dr. John Preisser, Jr. (University of North Carolina Department of Biostatistics) for their invaluable help with the statistical methods.
This work was supported, in whole or in part, by National Institutes of Health Grants HL106121 (to M. W. F. and H. E. M.), HL 081672 (to W. M. F.), and HL092994 (H. E. M.).
- NF-κB
- nuclear factor κB
- ALI
- acute lung injury
- BALF
- bronchoalveolar lavage fluid
- DTPA
- diethylene triamine pentaacetic acid
- GSNO
- S-nitrosoglutathione
- GSNOR
- S-nitrosoglutathione reductase
- IP
- immunoprecipitation
- SNO
- S-nitrosothiol
- Trx
- thioredoxin
- TrxR
- thioredoxin reductase
- Txnip
- thioredoxin-interacting protein.
REFERENCES
- 1. Kelleher Z. T., Matsumoto A., Stamler J. S., Marshall H. E. (2007) NOS2 regulation of NF-κB by S-nitrosylation of p65. J. Biol. Chem. 282, 30667–30672 [DOI] [PubMed] [Google Scholar]
- 2. Marshall H. E., Stamler J. S. (2001) Inhibition of NF-κB by S-nitrosylation. Biochemistry 40, 1688–1693 [DOI] [PubMed] [Google Scholar]
- 3. Kelleher Z. T., Potts E. N., Brahmajothi M. V., Foster M. W., Auten R. L., Foster W. M., Marshall H. E. (2011) NOS2 regulation of LPS-induced airway inflammation via S-nitrosylation of NF-κB p65. Am. J. Physiol. Lung Cell Mol. Physiol. 301, L327–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marshall H. E., Potts E. N., Kelleher Z. T., Stamler J. S., Foster W. M., Auten R. L. (2009) Protection from lipopolysaccharide-induced lung injury by augmentation of airway S-nitrosothiols. Am. J. Respir. Crit. Care Med. 180, 11–18 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 5. Harper R., Wu K., Chang M. M., Yoneda K., Pan R., Reddy S. P., Wu R. (2001) Activation of nuclear factor-κB transcriptional activity in airway epithelial cells by thioredoxin but not by N-acetylcysteine and glutathione. Am. J. Respir. Cell Mol. Biol. 25, 178–185 [DOI] [PubMed] [Google Scholar]
- 6. Matthews J. R., Wakasugi N., Virelizier J. L., Yodoi J., Hay R. T. (1992) Thioredoxin regulates the DNA binding activity of NF-κB by reduction of a disulphide bond involving cysteine 62. Nucleic Acids Res. 20, 3821–3830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benhar M., Forrester M. T., Hess D. T., Stamler J. S. (2008) Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 320, 1050–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Forrester M. T., Seth D., Hausladen A., Eyler C. E., Foster M. W., Matsumoto A., Benhar M., Marshall H. E., Stamler J. S. (2009) Thioredoxin-interacting protein (Txnip) is a feedback regulator of S-nitrosylation. J. Biol. Chem. 284, 36160–36166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hollingsworth J. W., Li Z., Brass D. M., Garantziotis S., Timberlake S. H., Kim A., Hossain I., Savani R. C., Schwartz D. A. (2007) CD44 regulates macrophage recruitment to the lung in lipopolysaccharide-induced airway disease. Am. J. Respir. Cell Mol. Biol. 37, 248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirota K., Murata M., Sachi Y., Nakamura H., Takeuchi J., Mori K., Yodoi J. (1999) Distinct roles of thioredoxin in the cytoplasm and in the nucleus. J. Biol. Chem. 274, 27891–27897 [DOI] [PubMed] [Google Scholar]
- 11. Nishiyama A., Matsui M., Iwata S., Hirota K., Masutani H., Nakamura H., Takagi Y., Sono H., Gon Y., Yodoi J. (1999) Identification of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 274, 21645–21650 [DOI] [PubMed] [Google Scholar]
- 12. Zhang P., Wang C., Gao K., Wang D., Mao J., An J., Xu C., Wu D., Yu H., Liu J. O., Yu L. (2010) The ubiquitin ligase itch regulates apoptosis by targeting thioredoxin-interacting protein for ubiquitin-dependent degradation. J. Biol. Chem. 285, 8869–8879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim S. F., Huri D. A., Snyder S. H. (2005) Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 310, 1966–1970 [DOI] [PubMed] [Google Scholar]
- 14. Nishida M., Ogushi M., Suda R., Toyotaka M., Saiki S., Kitajima N., Nakaya M., Kim K. M., Ide T., Sato Y., Inoue K., Kurose H. (2011) Heterologous down-regulation of angiotensin type 1 receptors by purinergic P2Y2 receptor stimulation through S-nitrosylation of NF-κB. Proc. Natl. Acad. Sci. U.S.A. 108, 6662–6667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Into T., Inomata M., Nakashima M., Shibata K., Häcker H., Matsushita K. (2008) Regulation of MyD88-dependent signaling events by S-nitrosylation retards Toll-like receptor signal transduction and initiation of acute-phase immune responses. Mol. Cell. Biol. 28, 1338–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reynaert N. L., Ckless K., Korn S. H., Vos N., Guala A. S., Wouters E. F., van der Vliet A., Janssen-Heininger Y. M. (2004) Nitric oxide represses inhibitory IκB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 101, 8945–8950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karin M., Yamamoto Y., Wang Q. M. (2004) The IKK NF-κB system: a treasure trove for drug development. Nat. Rev. Drug Discov. 3, 17–26 [DOI] [PubMed] [Google Scholar]
- 18. Sen N., Paul B. D., Gadalla M. M., Mustafa A. K., Sen T., Xu R., Kim S., Snyder S. H. (2012) Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 45, 13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ando K., Hirao S., Kabe Y., Ogura Y., Sato I., Yamaguchi Y., Wada T., Handa H. (2008) A new APE1/Ref-1-dependent pathway leading to reduction of NF-κB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 36, 4327–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benhar M., Thompson J. W., Moseley M. A., Stamler J. S. (2010) Identification of S-nitrosylated targets of thioredoxin using a quantitative proteomic approach. Biochemistry 49, 6963–6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ito T., Yamakuchi M., Lowenstein C. J. (2011) Thioredoxin increases exocytosis by denitrosylating N-ethylmaleimide-sensitive factor. J. Biol. Chem. 286, 11179–11184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Junn E., Han S. H., Im J. Y., Yang Y., Cho E. W., Um H. D., Kim D. K., Lee K. W., Han P. L., Rhee S. G., Choi I. (2000) Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J. Immunol. 164, 6287–6295 [DOI] [PubMed] [Google Scholar]
- 23. Nishizawa K., Nishiyama H., Matsui Y., Kobayashi T., Saito R., Kotani H., Masutani H., Oishi S., Toda Y., Fujii N., Yodoi J., Ogawa O. (2011) Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis 32, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 24. Yamawaki H., Pan S., Lee R. T., Berk B. C. (2005) Fluid shear stress inhibits vascular inflammation by decreasing thioredoxin-interacting protein in endothelial cells. J. Clin. Invest. 115, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yoshihara E., Fujimoto S., Inagaki N., Okawa K., Masaki S., Yodoi J., Masutani H. (2010) Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat. Commun. 1, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwon H. J., Won Y. S., Suh H. W., Jeon J. H., Shao Y., Yoon S. R., Chung J. W., Kim T. D., Kim H. M., Nam K. H., Yoon W. K., Kim D. G., Kim J. H., Kim Y. S., Kim D. Y., Kim H. C., Choi I. (2010) Vitamin D3 up-regulated protein 1 suppresses TNF-α-induced NF-κB activation in hepatocarcinogenesis. J. Immunol. 185, 3980–3989 [DOI] [PubMed] [Google Scholar]