

Background: A hallmark property of infectious prion protein (PrPSc) is its resistance to proteases.
Results: We show that the extent of PrPSc proteolytic resistance can be reversibly altered by changing salt concentration.
Conclusion: Thus, protease resistance is not an intrinsic property of the infectious agent, but it depends upon the environmental conditions.
Significance: These findings have practical and conceptual implications for understanding the mechanism of prion formation and clearance.
Keywords: Amyloid, Neurodegenerative Diseases, Prions, Protein Degradation, Protein Misfolding, Protease Resistance, Transmissible Spongiform Encephalopathies
Abstract
Transmissible spongiform encephalopathies are neurodegenerative diseases caused by prions in mammals. An aberrantly folded protein (PrPSc) is the main component of these proteinaceous infectious particles. Prions exhibit strong resistance to protease digestion, which is typically exploited for biochemical discrimination from its native cellular form (PrPC). This classical feature has been partially challenged by the isolation of sizeable amounts of protease-sensitive PrPSc isoforms that self-propagate in vivo. Here, we report that the degree of PrPSc protease resistance is highly dependent on the concentration of salt in the solution. Similar changes were observed in PrPSc obtained from different strains and species. Strikingly, the effect of salt is reversible and is associated with changes on the size of PrPSc particles, but surprisingly, the more protease-sensitive species consists of a larger size. These findings shed light on the mechanistic aspects of prion proteolysis and should be considered when assessing samples of biomedical relevance.
Introduction
Prions are abnormally folded proteinaceous infectious agents able to self-propagate in their hosts (1). In mammals, prions are associated with transmissible spongiform encephalopathies, a group of neurodegenerative diseases affecting a wide variety of species, including humans (2). The main or sole component of the infectious agent in transmissible spongiform encephalopathies is a misfolded conformation (PrPSc)2 of an endogenous protein termed PrPC that is expressed mainly in the brain. Although the exact role of PrPC in the normal physiology of the brain is still controversial (3), there is extensive evidence that its conversion into PrPSc causes all transmissible spongiform encephalopathies, not only the infectious forms (4). In vitro replication of mammalian infectious prions was first achieved by cyclic PrPSc-seeded conversion of PrPC from uninfected brain extracts (5, 6). Follow-up approaches showed later that highly purified PrPC and bacterial recombinantly expressed PrP can also be converted into an infectious form when RNA and lipids molecules are present in the mixture (7–9), lending strong support to the protein-only hypothesis about the nature of prions (10).
Histopathological analyses have shown the accumulation of PrPSc deposits spread within the infected brains, sometimes in the form of amyloid-like plaques, resembling those observed in other pathologies such as Alzheimer disease (4). When purified from brains, PrPSc is known to exist as amorphous insoluble aggregates that form rod-like particles upon protease treatment (11). Biochemical studies have shown that PrPSc consists of a broad size of aggregates, and smaller sized oligomers are believed to be the most efficient self-propagating species in vivo (12). The unusual biochemical features of PrPSc aggregates have precluded access to the structural details of these infectious particles (13). Alternative approaches have therefore relied on low-resolution techniques such as transmission electron microscopy and atomic force microscopy (4, 13). Size fractionation and protease-based assays have also complemented the biochemical analysis of PrPSc (14). These approaches have also been critical for the characterization of different prion strains, which are thought to be different PrP conformers that emerge from the same primary sequence (15).
Brain-derived and recombinant PrPSc are known to withstand high concentrations of protease treatment (14, 16). The protease-resistant subpopulation of PrPSc (PrPres) encompasses a C-terminal PrP fragment starting at around residue 90, with the exact position depending upon the prion strain. Although protease resistance is typically used as a biomarker for identifying and typifying prions, the mechanistic details responsible for acquisition of this property have not yet been elucidated. Protease-sensitive fractions coexisting with PrPres have been proposed as equally relevant in terms of infectivity, but the exact proportion of protease-sensitive and -resistant forms is yet unclear (17, 18). Some studies have shown that PrPSc protease resistance may be altered by environmental conditions (19). In particular, the concentrations of sodium chloride and several metals can greatly change PrPSc sensitivity to proteolysis (19). Kosmotropic salts and copper can also induce misfolding of recombinant PrP into PrPSc-like species in vitro (20).
Here, we study in detail the influence of the environment on PrPSc proteolysis. We found that protease resistance is an externally modifiable feature of PrPSc that is highly dependent on ionic strength and protease concentration. Moreover, we provided evidence that ionic strength modulates sensitivity to proteolysis without affecting PrPres intrinsic features.
EXPERIMENTAL PROCEDURES
PrPSc Purification from Infected Brains
Brains loaded with 263K prion strain from terminally ill Syrian golden hamsters were collected for purification. To assess the effect of protease resistance on PrPSc-enriched samples, we modified a previous protocol for prion purification from brains bypassing proteolytic digestion (21). Four diseased brains were homogenized in 11.1% sarkosyl dissolved in nanopure water at 11.1% g/ml concentration. The mixture was divided in two equal volume halves and supplemented with 0.1 volumes of 200 mm Tris-HCl, pH 7.4, in the presence or absence of 1.5 m NaCl. Large pieces of debris were centrifuged at 800 × g for 45 s at 4 °C, and supernatant was carefully layered on an ∼3.6-ml sucrose cushion containing 20% sucrose, 20 mm Tris-HCl, pH 7.4, Complete® protease inhibitor cocktail from Roche Applied Science with and without 150 mm NaCl. Both mixtures were centrifuged at 150,000 × g for 3 h at 4 °C. Each pellet was recovered and resuspended in 2.5 ml of 0.1% Z3-14 detergent containing 20 mm Tris-HCl, pH 7.4, protease inhibitors, and the presence or absence of 150 mm NaCl. The resuspended pellet was then layered over a 3-ml 20% sucrose cushion supplemented with 0.1% Z3-14 followed by ultracentrifugation at 150,000 × g for 3 h at 4 °C. This last step was repeated once, and both pellets were resuspended in deionized water. The mixture was layered over a 3-ml 20% sucrose solution in water with or without 150 mm NaCl and ultracentrifuged at 150,000 × g for 3 h at 4 °C. The pellets were finally resuspended in 200 μl of 20 mm Tris-HCl, pH 7.4, with or without 150 mm NaCl followed by 20 s of intermittent sonication until the pellets got clarified in the solution. When noted, purified PrPSc samples in NaCl-containing solutions were dialyzed two times overnight at 4 °C against a 250 dilution factor in 20 mm Tris-HCl, pH 7.4.
Purification of Recombinant PrP
Mouse and hamster recombinant PrP were purified using a previously reported protocol (21). PrP-overexpressed Escherichia coli cell pellets were thawed and resuspended in buffer A (50 mm Tris-HCl, pH 8.0, 1 mm EDTA, and 100 mm NaCl). Cells were lysed by adding 0.5 mg/ml lysozyme and subsequently sonicated. The released inclusion bodies were pelleted by centrifugation at 22,000 × g and then washed twice with buffer A supplemented with 0.05% (v/v) Triton X-100. The inclusion bodies containing recPrP were solubilized for 2 h with buffer B (10 mm Tris-HCl, 100 mm Na2HPO4, pH 8.0, 100 mm NaCl, 10 mm β-mercaptoethanol, 6 m guanidine hydrochloride) and then purified by using standard immobilized metal affinity chromatography procedure. Briefly, the sample was bound to nickel-Sepharose 6 Fast Flow resin (GE Healthcare) in batch mode for 1 h at room temperature and then washed with buffer B. recPrP was on-column refolded for 6 h and eluted with buffer B supplemented with 500 mm imidazole and without guanidine hydrochloride. The main peak was collected and quickly filtered to remove aggregates. The sample was buffer-exchanged using Zeba desalting columns (Pierce), further concentrated to ∼0.5 mg/ml, and flash-frozen at −80 °C. Protease inhibitors (Complete® protease inhibitor cocktail from Roche Applied Science) were used throughout the purification to minimize degradation. The protein was confirmed to be monomeric and properly folded by SDS-PAGE, Western blotting, and circular dichroism.
Protease Digestion Assay
PrPSc-containing samples were diluted at 1:20 ratio in 20 mm Tris-HCl, pH 7.4, in the absence or presence of 150 mm NaCl. The dilutions were treated with different protease concentrations using a 20 mg/ml proteinase K (PK) stock dissolved in water. Reactions were prepared at a 1:20 ratio of PK/diluted-PrPSc (v/v) in 20 mm Tris-HCl, pH 7.4, with or without 150 mm NaCl, and incubated at 37 °C for 4 h at 600 rpm agitation. For analysis, samples were boiled at 95 °C for 10 min in denaturing loading buffer (NuPAGE from Invitrogen), enriched with 100 mm DTT (final concentration) followed by SDS-PAGE and immunoblotting. Briefly, samples were electrophoresed in 12% SDS-acrylamide gels (Invitrogen) followed by transfer to nitrocellulose membrane (GE Healthcare) for 1 h at 4 °C in a buffer tank (Bio-Rad) containing 10% methanol, 250 mm Tris, and 2 m glycine. Membranes were blocked in 2% milk in washing buffer (saline buffer, 0.05% Tween 20) followed by incubation with monoclonal anti-PrP 3F4 antibody (Millipore) or polyclonal R20 (22) for 1 h at room temperature or 6D11 overnight. Membranes were washed three times for 10 min with washing buffer and then incubated in anti-mouse horseradish peroxidase-labeled IgG antibody for 1 h. After three 10-min rinses with washing buffer, membranes were treated with ECL-developing mix (GE Healthcare) and visualized on a Gel Doc station (Bio-Rad). For densitometry analysis, the density of the band intensity was measured by using ImageJ (open source image processor).
Equilibrium Sedimentation Analysis
Purified PrPSc (50 μl) was diluted with 1 volume of 20 mm Tris-HCl, pH 7.4, in the absence or presence of 150 mm NaCl. The 100-μl mix was layered over a sucrose gradient cushion prepared as follows: stock solutions of sucrose in 20 mm Tris-HCl, pH 7.4, with or without 150 mm NaCl, were prepared at concentrations ranging from 5 to 70% (g/ml) in 5% increments. Starting from the 70% stock, 300 μl of each stock solution were carefully layered over in an ultracentrifuge tube in decreasing order of sucrose content without mixing the solutions. Once PrPSc dilution was carefully added (without mixing), the tubes were ultracentrifuged at 150,000 × g for 24 h at 4 °C. After centrifugation, the sedimentation gradient mix was carefully collected in 315-μl samples each representing different sucrose concentrations. Samples were boiled at 95 °C for 10 min in DTT-enriched SDS-loading buffer and analyzed by SDS-PAGE followed by immunoblotting as described above.
Protein Misfolding Cyclic Amplification (PMCA)
PMCA of PrPSc was conducted as reported previously (21). Briefly, brains from uninfected 4–5-week-old Syrian gold hamsters were cold-homogenized in saline phosphate buffer with 1% Triton X-100 and 150 mm NaCl and centrifuged to remove debris. The supernatant was used as normal brain homogenate substrate for PrPSc amplification. 10 μl of PrPSc (with or without 150 mm NaCl) were added to 90 μl of normal brain homogenate, and then the mixture was serially diluted until 10−10. Each tube was supplemented with Teflon beads, and PMCA cycles were run for 42 h at 37 °C in a microplate horn attached to a Q700 sonicator (QSonica). Settings included cycles of 20 s sonication at 250 watts followed by 29.6 min of incubation. Samples were submitted to PK digestion using 50 μg/ml PK for 1 h at 37 °C and 450 rpm agitation and then immunoblotted as described above.
Brain Preparations of Different Prion Strains
Brain homogenates (10% g/ml) of diseased brains from animals inoculated intracerebrally with different prion strains (263K and Hyper from hamster and ME7, RML, and 301C from mouse) were prepared in cold saline phosphate solution and centrifuged for 1 min at 800 × g. Supernatants were ultracentrifuged at 150,000 × g for 3 h at 4 °C. Pellets were recovered and resuspended twice in 20 mm Tris-HCl, pH 7.4, in the presence or absence of 150 mm NaCl. Suspensions were protease-digested for 4 h at 37 °C at different PK concentrations followed by immunoblotting.
RESULTS
PrPSc Becomes Protease-sensitive in the Absence of Salt
To enrich our samples in undigested PrPSc, we modified a previously reported protocol to purify prions from diseased brains (21) by removing the proteolytic step and adding extra washes with the nondenaturing ionic detergent sarkosyl to remove nonspecifically bound contaminants. Without protease treatment, this procedure allowed us to purify PrPSc in the presence or absence of salts without affecting its yield. The “salt-free” PrPSc (sfPrPSc) sample was purified in solutions containing buffers without NaCl from the beginning to avoid carrying salts from the brain homogenate throughout the procedure.
Previous results showed that the ionic strength alters the proteolytic resistance of PrPSc molecules (19). We investigated the effect of salt on PrPSc protease resistance using different concentrations of PK (Fig. 1). As expected, PrPSc in the presence of physiological concentrations of NaCl showed great resistance to protease digestion even until 1 mg/ml PK (Fig. 1A) Conversely, salt-free PrPSc sample was readily digested under these conditions (Fig. 1A). To determine whether the absence of salts completely abolished the formation of PrPres, we incubated PrPSc and sfPrPSc at very low concentrations of protease (Fig. 1B). Interestingly, the typical signal associated with PrPres (exhibiting higher electrophoretical mobility as compared with full-length PrP) appeared in both samples at PK concentrations lower than 100 μg/ml. This result suggests that the absence of sodium chloride makes PrPSc less resistant to PK digestion, but does not alter the cleavage site of the protein. The use of 3F4 anti-PrP antibody that recognizes the middle sequence region of PrP will not detect proteolytic fragments of lower molecular weight, which may appear at higher concentrations of protease. To test this idea, we probed our blots using the C-terminal anti-PrP antibody R20 and found no signal, confirming the total absence of detectable sfPrPSc fragments upon digestion with high PK concentrations (data not shown).
FIGURE 1.
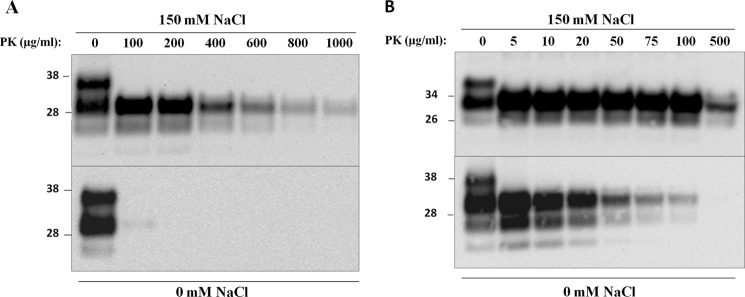
PrPSc resistance to proteolytic degradation depends on NaCl concentration. PrPSc was purified from diseased brains in the absence or presence of 150 mm NaCl (see “Experimental Procedures”). A and B, equal amounts of both PrPSc samples were submitted to proteolytic treatment in different reactions using high (A) and low (B) concentration ranges of PK for 4 h at 37 °C with agitation at 600 rpm. Immunoblotting was conducted using 3F4 antibody. Molecular sizes of protein standards are indicated on the left of each panel.
We then checked the salt-mediated interconvertibility between protease-resistant and -sensitive forms of PrPSc by extensively dialyzing PrPSc against salt-free buffer solution while adding concentrated NaCl to sfPrPSc. Desalting PrPSc resulted in a near identical profile of proteolytic stability to that exhibited by sfPrPSc that significantly deviated from the pattern obtained with salt-containing PrPSc, suggesting that the process is reversible (Fig. 2A). On the other hand, we added increasing concentrations of NaCl to sfPrPSc and studied the resistance to PK at the highest protease concentration that showed PrPres signal (1 mg/ml, Fig. 1A). The results showed that PrPSc progressively acquired resistance to PK digestion starting at around 75 mm NaCl, and the extent of protease resistance was directly proportional to the salt concentration (Fig. 2B). These results suggest that protease-sensitive and -resistant isoforms of PrPSc are interchangeable and that the proportion of them is highly dependent on the salt concentration. To rule out the interference of salts on PK activity, we performed protease digestion assays in the absence or presence of NaCl using natively folded recPrP (hamster PrP) purified from bacteria. We found that PK was able to digest recPrP with the same efficiency regardless of the salt content (Fig. 3).
FIGURE 2.

Reversibility of NaCl-dependent protease resistance of PrPSc. A, PrPSc purified in the presence of 150 mm NaCl was extensively dialyzed (two times) against 20 mm Tris-HCl, pH 7.4, and then submitted to protease digestion with the indicated concentrations of PK for 4 h at 37 °C with 600 rpm agitation. Protease-treated samples were immunoblotted using 3F4 antibody. B, densitometry analysis of the percentage of PrPres (% of PrPres) upon protease digestion. The density of the band intensity was measured and normalized to percentages. Densitometry as a function of PK concentration is shown for PrPSc purified in the presence (triangles) or absence (circles) of 150 mm NaCl (values calculated from Fig. 1B) as well as for the sample in 150 mm NaCl after dialysis (squares). C, PrPSc purified in the absence of salts was supplemented with different concentrations of NaCl (0, 1, 10, 30, 75, 150, 500, and 1000 mm), and samples were submitted to protease digestion at 1000 μg/ml for 4 h at 37 °C with 600 rpm agitation.
FIGURE 3.
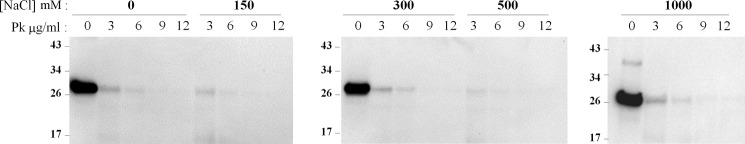
PK activity is not altered by the NaCl concentration. The effect of NaCl on the intrinsic activity of PK was assessed using purified hamster recPrP as substrate. Soluble, folded recPrP was dialyzed for 4 h at room temperature and supplemented with different concentrations of NaCl ranging from 0 to 1000 mm. Samples were treated with increasing PK concentrations at 37 °C for 30 min at 600 rpm agitation. PK-digested samples were immunoblotted as specified under “Experimental Procedures.”
Salt Changes Size Distribution of PrPSc Aggregates
PrPSc consists of an ensemble of particles with a broad range of sizes, ranging from small oligomers (∼400 kDa) to large aggregates with an estimated molecular size of >15 MDa (12, 23). Oligomers have been typically thought as less stable and more protease-sensitive than their higher molecular weight counterparts (12, 17). To study whether the changes in protease sensitivity induced by different salt concentrations were associated with salt-dependent size shifts, we analyzed the density-associated size distribution of PrPSc and sfPrPSc by equilibrium sedimentation fractionation using sucrose gradients. This technique has proven useful in isolating PrPSc oligomers of distinct size that were shown to be highly infectious in animal models (12, 18, 23). The results showed that both PrPSc and sfPrPSc were distributed mostly among highly dense portions of the gradient, spanning from fractions 9 to 15 (Fig. 4). To our surprise, however, sfPrPSc showed a shift toward higher density fractions, suggesting the presence of significantly bigger species. Indeed, although salted PrPSc peaked in fraction 12 (Fig. 4A), corresponding to an estimated size of 11 MDa (as estimated by using molecular size standards), sfPrPSc showed a peak at fraction 15 (Fig. 4B), indicating a size larger than 15 MDa. Interestingly, when 150 mm NaCl was added to sfPrPSc, the size distribution shifted to a lower molecular weight (peak in fraction 13), showing a pattern closer to that obtained when the protein was originally prepared in the presence of salt (Fig. 4C). Overall, these results suggest that the salt-dependent effects on PrPSc protease resistance are associated with changes on the size distribution of the particles, but surprisingly, the conditions promoting increase in protease resistance lead to a smaller size of aggregates. As with the studies with protease resistance, the salt-induced changes on PrPSc size distribution were reversible.
FIGURE 4.
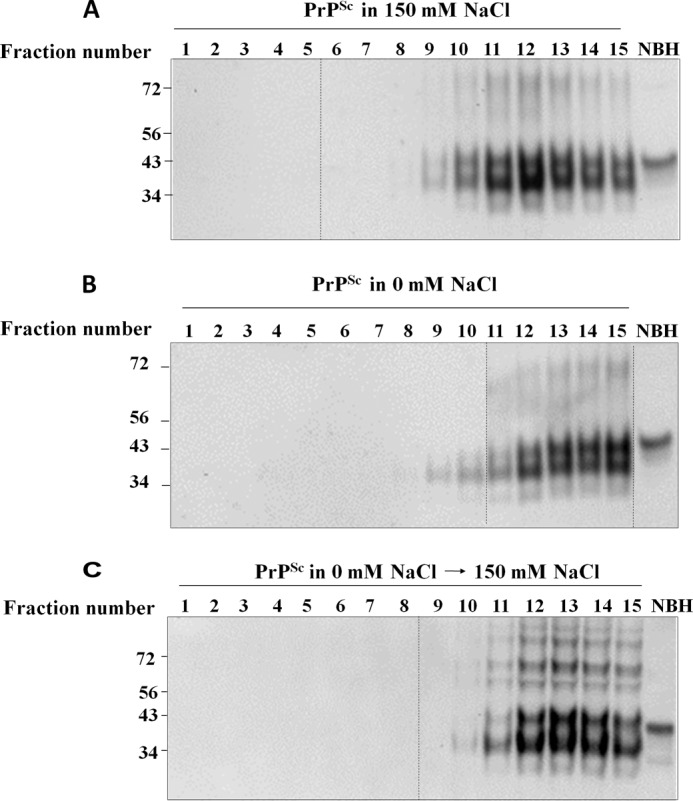
Size distribution of PrPSc aggregates purified in the presence or absence of NaCl. A and B, equal amounts of PrPSc purified in the presence (A) or in the absence (B) of 150 mm NaCl were submitted to equilibrium centrifugation on sucrose gradients as described under “Experimental Procedures.” C, also, PrPSc purified in the absence of salt was supplemented with 150 mm NaCl to analyze reversibility of the size distribution pattern. Samples corresponding to different sucrose densities (Fraction number) were analyzed by immunoblotting. Fractions from 1 to 15 span sucrose densities from 5 to 70% (g/ml). Untreated brain homogenate (NBH) was run as loading control. None of the samples were treated with PK. Numbers on the left correspond to molecular size markers, expressed in kDa. Dotted lines indicate blot splicing.
Salt-dependent Sensitivity of PrPSc to Proteolytic Digestion Is Not NaCl-specific
PrP primary sequence has many charged residues that can be affected by surrounding ionic species. To assess the specificity of Na+ and Cl− on the observed effects on protease resistance, we enriched sfPrPSc with different salts and performed protease digestion assays (Fig. 5). We initially maintained the sodium cation and replace chloride by bromide (NaBr) or fluoride (NaF). We observed that in the presence of these salts, protease resistance was reestablished to similar levels as those observed for NaCl. When keeping chloride and replacing the sodium cation by potassium (KCl) or calcium (CaCl2), we observed a smaller, but detectable effect only in the case of KCl. CaCl2 seemed not to be competent in promoting PrPSc protease resistance. At present the reason why this salt does not induce the protease-resistant state of PrPSc is unknown. However, it needs to be mentioned that calcium is a known activator of proteinase K (24).
FIGURE 5.
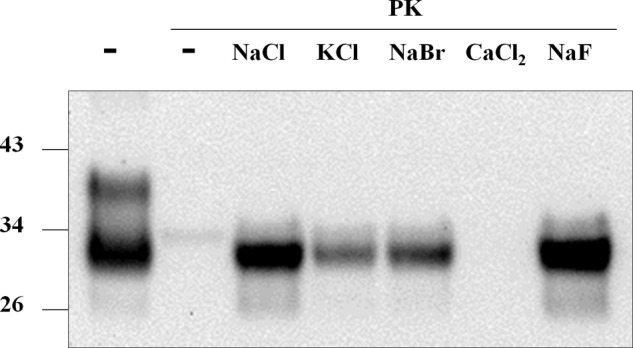
Effect of different salts on PrPSc protease resistance. Pure PrPSc purified in the absence of salt was supplemented with sodium bromide (NaBr), sodium chloride (NaCl), calcium chloride (CaCl2), or potassium chloride (KCl) at 150 mm and sodium fluoride (NaF) at 96 mm (saturation at the reaction temperature). Reactions were submitted to protease digestion (1000 μg/ml PK) followed by immunoblotting.
The Absence of Salts Does Not Modify the Self-propagation Ability of PrPSc in Vitro
To test whether the presence of salts irreversibly alters PrPSc ability to self-propagate, we performed experiments using the PMCA technique (5). PMCA has been shown to recapitulate key aspects of prion biology, such as indefinite propagation of inoculum infectivity in vitro (6, 25) as well as high fidelity of prion strain replication (26). We ran PMCA experiments using PrPSc and sfPrPSc as inoculums and normal hamster brain homogenate as substrate. We performed one round of amplification of serially diluted inoculums and observed that both samples were amplified equally well (Fig. 6), suggesting that salts do not alter the hallmark property of prion self-replication. However, because the PMCA reaction was done in the presence of NaCl, we cannot rule out that removal of salt altered the prion replication capacity of sfPrPSc, which was reverted after the addition of NaCl during PMCA. Nevertheless, the data indicate that any alteration of seeding activity in the absence of salt would be reversible.
FIGURE 6.

In vitro replication of PrPSc by PMCA. PrPSc purified with or without 150 mm NaCl was serially diluted into normal brain homogenate and used as inoculum for seeding PMCA as described under “Experimental Procedures.” Tubes were incubated for 42 h (84 PMCA cycles), and samples were treated with PK except for the normal brain homogenate (NBH) used as a migration control.
Salt Modulation of PrPSc Sensitivity to Proteolysis Is Not Prion Strain-dependent
Co-factor molecules have been proposed to have relevant roles on PrP aggregation and prion propagation (8, 9). Recently, several groups have shown that different prion strains react differentially to the presence or absence of several co-factor molecules in PMCA experiments (27, 28). To evaluate the strain specificity with respect to the effects of salts on PrPSc proteolysis, we analyzed two additional prion strains from hamster and three from mouse. Before protease digestion, brain homogenates of these strains were centrifuged and washed to decrease the salt content from the brains. As shown in Fig. 7, the absence of NaCl markedly increased the sensitivity to PK digestion of Hyper prion strain (from hamster) in a similar manner to that observed with the 263K strain. When the same treatment was applied to three different prion strains from mouse (ME7, RML, and 301C), we observed similar results (Fig. 7). These data strongly suggest that salt-dependent modulation of protease resistance of PrPSc is species- and prion strain-independent.
FIGURE 7.
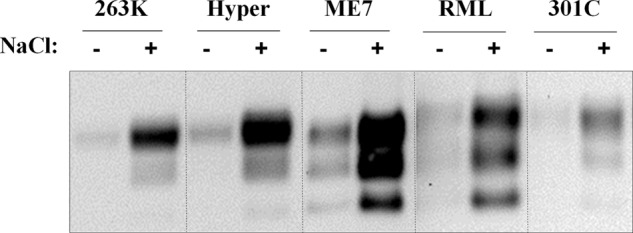
Effect of salts on protease resistance of PrPSc obtained from different prion strains and species. Two different prion strains from hamster (263K and Hyper) and three from mouse (ME7, RML, and 301C) were assayed for protease resistance in the presence (+) or absence (−) of 150 mm NaCl. Samples were PK-digested for 4 h at 37 °C at protease concentrations optimal for detectability of each prion strain: 600 μg/ml (263K, Hyper, and 301C) and 200 μg/ml (ME7 and RML). Dotted lines separate samples that are coming from different blots.
DISCUSSION
It is generally thought that once prions are formed, they adopt a very stable state that makes them highly invulnerable to typical decontamination procedures such as high temperature and alcohol treatments, among others (29–31). This stability is classically reflected by the strong resistance to proteolytic digestion exhibited by most known prion strains. The biochemical discrimination of prions from their normally folded counterpart is typically performed by protease treatment, taking into account that PrPC is highly sensitive to protease digestion as compared with PrPSc. Furthermore, proteolytic treatment of PrPSc yields a lower molecular weight fragment that is the actual protease-resistant segment within the PrP sequence and can therefore be easily identified through electrophoretic mobility. This hallmark feature has been widely exploited for detecting prions in clinically relevant samples and for prion strain typing. However, various studies have reported the presence of protease-sensitive prions (sPrPSc) (17, 18, 32, 33), which defies the classical view of prions as highly resistant to proteolysis. The existence of sPrPSc complicates the biochemical detection of the infectious agent, which could constitute a potential health-related problem.
In this study, we showed that the salt concentration can greatly modulate the sensitivity of PrPSc to proteolytic digestion. This effect did not alter other prion properties such as self-propagation and digestion profile and was observed in prions from various strains and species. In our hands, PK intrinsic activity is salt-insensitive toward recPrP at the concentrations utilized in the present experiments (Fig. 3), which suggests that the effects of NaCl are most likely exerted at the level of PrPSc rather than to the protease itself. This was also confirmed by Nishina et al. (19) through quantitative measurement of PK activity on chromogenic model peptides at different NaCl concentrations. We also ruled out that the effects of salts are due to differences in initial concentration between PrPSc and sfPrPSc because their levels were always equilibrated before starting each reaction. The fact that sfPrPSc still exhibited protease resistance at lower PK concentrations indicates that this feature is not abolished in the absence of salts, but rather modulated to different extents. The PK digestion profile and approximate cleavage site on PrPSc were also unaffected by NaCl, indicating that salts do not seem to structurally modify PrPSc architecture. This is not surprising because NaCl is a rather neutral salt with little to no denaturing capabilities. The only known salts to have deleterious effects on prions are those with strong denaturant properties, such as guanidine hydrochloride and guanidine thiocyanate. In these cases, total denaturation of prions is typically an irreversible process (34). Both sfPrPSc and PrPSc were shown to be interconvertible by means of adding/removing salts. In our hands, this effect was very reproducible, and it happened fast. The rapid reversibility between sfPrPSc and PrPSc states, observed during our experiments, further indicates that no large changes at the conformational level affect PrPSc when in the presence/absence of salts.
A possible mechanism by which salts can alter PrPSc protease resistance is through modification of its size distribution, thereby indirectly affecting their PK sensitivity by shifting PrPSc toward more oligomeric, protease-sensitive particles (18). Fractionation experiments using sedimentation analysis initially allowed the isolation of aggregates with distinct sizes that exhibited differential resistance to protease treatment (17, 18). Some of these fractions were particularly sensitive to proteases and were called protease-sensitive PrPSc (sPrPSc). These experiments indicate that sPrPSc is a subpopulation within the PrPSc aggregate continuum. To our surprise, we found that removing salts actually increased the size of the aggregates. A possible explanation for this result may lie in the highly charged nature of the PrP sequence. In the absence of salts, attractive electrostatic groups can easily interact with each other and form larger particles, whereas in the presence of salts, shielding effects of ions affecting exposed PrPSc charged surfaces may counteract potential particle attractions. However, it is unclear whether the changes observed in the size distribution of PrPSc particles explain satisfactorily the increased protease sensitivity of sfPrPSc.
Another possibility is that some salts may partially stabilize PrPSc particles at specific concentration ranges. While assessing the effect of different salts on protease digestion, we found that supplementing sfPrPSc with NaF gave a slightly higher PK-resistant signal than with NaCl, whereas treatment with NaBr yielded a lower signal. In the context of the Hofmeister series, the stabilizing effects of these anions would be in decreasing order F− > Cl− > Br−, which matches the intensity of the signals observed in our data (Fig. 5). We also observed that calcium did not reestablish PrPSc protease resistance. Interestingly, calcium is known to be a highly destabilizing cation within the series, whereas in addition being an activator of PK activity (24). We previously found that NaF efficiently promoted the conversion of recPrP to a PrPSc-like state, probably by salting-out effects (20). Although the concentrations used in that work were higher than those reported here, a similar salt-based stabilization mechanism may be operating with PrPSc, and additional experiments are ongoing to explore this scenario.
Our findings indicate that the protease resistance of PrPSc is not an intrinsic property of the infectious agent, but it actually depends upon the environmental conditions in the solution. This conclusion has many important practical implications. (i) The biological clearance of PrPSc, at least through proteolytic removal, may be highly dependent on local environmental conditions; (ii) detection of PrPSc by means of analyzing the amount of protein remaining after protease digestion might be inaccurate and dependent on the experimental conditions used to prepare the material; (iii) characterization and typing of prion strains through protease sensitivity studies has to be done carefully and controlling the solution conditions, particularly the ionic strength; and (iv) our data may open a new target for prion treatment by identifying molecules altering PrPSc protease resistance under physiological conditions, which could be used to decrease prion stability in vivo.
The fact that changes in the concentration of such a simple molecule as NaCl can dramatically alter PrPSc sensitivity to protease digestion suggests that this classical feature of PrPSc may be more labile than previously thought. It is possible to speculate that other, yet unknown, physiologic factors may have a similar effect in vivo. Identification of such factors may be important to understand the mechanisms controlling the biological stability and clearance of prions.
Acknowledgment
We are grateful to Dr. Rodrigo Morales (University of Texas Medical School at Houston) for help with the experiments of ultracentrifugation in sucrose gradients.
This work was supported, in whole or in part, by National Institutes of Health Grants R01NS049173 and P01AI077774 (to C. S.).
- PrP
- prion protein
- PrPSc
- infectious misfolded prion protein
- PrPC
- normal cellular prion protein
- PrPres
- protease-resistant fragment of PrPSc
- sPrPSc
- protease-sensitive prions
- sfPrPSc
- salt-free PrPSc
- recPrP
- recombinant PrP
- PMCA
- protein misfolding cyclic amplification
- PK
- proteinase K.
REFERENCES
- 1. Prusiner S. B., Gabizon R., McKinley M. P. (1987) On the biology of prions. Acta Neuropathol. 72, 299–314 [DOI] [PubMed] [Google Scholar]
- 2. Soto C., Saborío G. P. (2001) Prions: disease propagation and disease therapy by conformational transmission. Trends Mol. Med. 7, 109–114 [DOI] [PubMed] [Google Scholar]
- 3. Zomosa-Signoret V., Arnaud J. D., Fontes P., Alvarez-Martinez M. T., Liautard J. P. (2008) Physiological role of the cellular prion protein. Vet. Res. 39, 9. [DOI] [PubMed] [Google Scholar]
- 4. Caughey B., Baron G. S., Chesebro B., Jeffrey M. (2009) Getting a grip on prions: Oligomers, amyloids, and pathological membrane interactions. Annu. Rev. Biochem. 78, 177–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Saborio G. P., Permanne B., Soto C. (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810–813 [DOI] [PubMed] [Google Scholar]
- 6. Castilla J., Saá P., Hetz C., Soto C. (2005) In vitro generation of infectious scrapie prions. Cell 121, 195–206 [DOI] [PubMed] [Google Scholar]
- 7. Deleault N. R., Harris B. T., Rees J. R., Supattapone S. (2007) Formation of native prions from minimal components in vitro. Proc. Natl. Acad. Sci. U.S.A. 104, 9741–9746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang F., Wang X., Yuan C.-G., Ma J. (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327, 1132–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deleault N. R., Lucassen R. W., Supattapone S. (2003) RNA molecules stimulate prion protein conversion. Nature 425, 717–720 [DOI] [PubMed] [Google Scholar]
- 10. Diaz-Espinoza R., Soto C. (2010) Generation of prions in vitro and the protein-only hypothesis. Prion 4, 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prusiner S. B., McKinley M. P., Bowman K. A., Bolton D. C., Bendheim P. E., Groth D. F., Glenner G. G. (1983) Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 35, 349–358 [DOI] [PubMed] [Google Scholar]
- 12. Silveira J. R., Raymond G. J., Hughson A. G., Race R. E., Sim V. L., Hayes S. F., Caughey B. (2005) The most infectious prion protein particles. Nature 437, 257–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Diaz-Espinoza R., Soto C. (2012) High-resolution structure of infectious prion protein: the final frontier. Nat. Struct. Mol. Biol. 19, 370–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sajnani G., Requena J. R. (2012) Prions, proteinase K and infectivity. Prion 6, 430–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collinge J., Clarke A. R. (2007) A general model of prion strains and their pathogenicity. Science 318, 930–936 [DOI] [PubMed] [Google Scholar]
- 16. McKinley M. P., Bolton D. C., Prusiner S. B. (1983) A protease-resistant protein is a structural component of the scrapie prion. Cell 35, 57–62 [DOI] [PubMed] [Google Scholar]
- 17. Pastrana M. A., Sajnani G., Onisko B., Castilla J., Morales R., Soto C., Requena J. R. (2006) Isolation and characterization of a proteinase K-sensitive PrPSc fraction. Biochemistry 45, 15710–15717 [DOI] [PubMed] [Google Scholar]
- 18. Sajnani G., Silva C. J., Ramos A., Pastrana M. A., Onisko B. C., Erickson M. L., Antaki E. M., Dynin I., Vázquez-Fernández E., Sigurdson C. J., Carter J. M., Requena J. R. (2012) PK-sensitive PrP is infectious and shares basic structural features with PK-resistant PrP. PLoS Pathog. 8, e1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nishina K., Jenks S., Supattapone S. (2004) Ionic strength and transition metals control PrPSc protease resistance and conversion-inducing activity. J. Biol. Chem. 279, 40788–40794 [DOI] [PubMed] [Google Scholar]
- 20. Diaz-Espinoza R., Mukherjee A., Soto C. (2012) Kosmotropic anions promote conversion of recombinant prion protein into a PrPSc-like misfolded form. PLoS One 7, e31678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Morales R., Duran-Aniotz C., Diaz-Espinoza R., Camacho M. V., Soto C. (2012) Protein misfolding cyclic amplification of infectious prions. Nat. Protoc. 7, 1397–1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caughey B., Raymond G. J., Ernst D., Race R. E. (1991) N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 65, 6597–6603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tixador P., Herzog L., Reine F., Jaumain E., Chapuis J., Le Dur A., Laude H., Béringue V. (2010) The physical relationship between infectivity and prion protein aggregates is strain-dependent. PLoS Pathog. 6, e1000859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bajorath J., Hinrichs W., Saenger W. (1988) The enzymatic activity of proteinase K is controlled by calcium. Eur. J. Biochem. 176, 441–447 [DOI] [PubMed] [Google Scholar]
- 25. Saá P., Castilla J., Soto C. (2006) Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252 [DOI] [PubMed] [Google Scholar]
- 26. Castilla J., Morales R., Saá P., Barria M., Gambetti P., Soto C. (2008) Cell-free propagation of prion strains. EMBO J. 27, 2557–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saá P., Sferrazza G. F., Ottenberg G., Oelschlegel A. M., Dorsey K., Lasmézas C. I. (2012) Strain-specific role of RNAs in prion replication. J. Virol. 86, 10494–10504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gonzalez-Montalban N., Lee Y. J., Makarava N., Savtchenko R., Baskakov I. V. (2013) Changes in prion replication environment cause prion strain mutation. FASEB J. 27, 3702–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Safar J., Roller P. P., Gajdusek D. C., Gibbs C. J., Jr., (1993) Thermal stability and conformational transitions of scrapie amyloid (prion) protein correlate with infectivity. Protein Sci. 2, 2206–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prior F., Fernie K., Renfrew A., Heneaghan G. (2004) Alcoholic fixation of blood to surgical instruments-a possible factor in the surgical transmission of CJD? J. Hosp. Infect. 58, 78–80 [DOI] [PubMed] [Google Scholar]
- 31. Cohen F. E., Prusiner S. B. (1998) Pathologic conformations of prion proteins. Annu. Rev. Biochem. 67, 793–819 [DOI] [PubMed] [Google Scholar]
- 32. Colby D. W., Wain R., Baskakov I. V., Legname G., Palmer C. G., Nguyen H. O., Lemus A., Cohen F. E., DeArmond S. J., Prusiner S. B. (2010) Protease-sensitive synthetic prions. PLoS Pathog. 6, e1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim C., Haldiman T., Cohen Y., Chen W., Blevins J., Sy M. S., Cohen M., Safar J. G. (2011) Protease-sensitive conformers in broad spectrum of distinct PrPSc structures in sporadic Creutzfeldt-Jakob disease are indicator of progression rate. PLoS Pathog. 7, e1002242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kocisko D. A., Lansbury P. T., Jr., Caughey B. (1996) Partial unfolding and refolding of scrapie-associated prion protein: evidence for a critical 16-kDa C-terminal domain. Biochemistry 35, 13434–13442 [DOI] [PubMed] [Google Scholar]