

Background: The metalloprotease ADAM17 emerged as the main sheddase of several cytokines and cytokine receptors.
Results: The acidophilic kinase PLK2 interacts with and phosphorylates ADAM17 in mammalian cells.
Conclusion: PLK2 represents a novel cellular interaction partner of ADAM17 modulating its activity.
Significance: Regulation of ADAM17 activity is essential for inflammatory responses.
Keywords: ADAM ADAMTS, Metalloprotease, Phosphorylation, Shedding, Tumor Necrosis Factor (TNF), ADAM17, PLK2
Abstract
ADAM17 (a disintegrin and metalloprotease 17) controls pro- and anti-inflammatory signaling events by promoting ectodomain shedding of cytokine precursors and cytokine receptors. Despite the well documented substrate repertoire of ADAM17, little is known about regulatory mechanisms, leading to substrate recognition and catalytic activation. Here we report a direct interaction of the acidophilic kinase Polo-like kinase 2 (PLK2, also known as SNK) with the cytoplasmic portion of ADAM17 through the C-terminal noncatalytic region of PLK2 containing the Polo box domains. PLK2 activity leads to ADAM17 phosphorylation at serine 794, which represents a novel phosphorylation site. Activation of ADAM17 by PLK2 results in the release of pro-TNFα and TNF receptors from the cell surface, and pharmacological inhibition of PLK2 leads to down-regulation of LPS-induced ADAM17-mediated shedding on primary macrophages and dendritic cells. Importantly, PLK2 expression is up-regulated during inflammatory conditions increasing ADAM17-mediated proteolytic events. Our findings suggest a new role for PLK2 in the regulation of inflammatory diseases by modulating ADAM17 activity.
Introduction
Proteolysis of transmembrane proteins controls a broad range of physiological functions. 10% of the cellular secretome comprise ectodomains derived from proteolytically cleaved transmembrane proteins (1). Members of the ADAM (a disintegrin and metalloprotease) family catalyze hydrolysis of numerous transmembrane proteins and have functional relevance in homeostatic and pathophysiologic processes (2, 3). ADAM17 was discovered as the key protease involved in processing the membrane-bound precursor of TNFα to its mature soluble form (4, 5). To date, in addition to pro-TNFα, more than 70 ADAM17 substrates have been described including l-selectin, IL-6 receptor, and ligands of the EGF receptor (6, 7). The lethal phenotype of ADAM17 knock-out mice underlines the important physiological function of ADAM17 during development and is most likely caused by compromised EGF receptor-ligand shedding (8). The phenotype of conditional knock-out mice on immune cells highlights the central function of ADAM17 in modulating inflammatory reactions by regulating the bioavailability of membrane-bound cytokines, cytokine receptors, and chemokines. Injection of bacteria or purified LPS into mice lacking ADAM17 expression on leukocytes led to higher survival rates and decreased plasma TNFα levels compared with wild type mice (9, 10). Given the importance of ADAM17-mediated proteolysis during inflammatory reactions, it is of high interest to identify molecular modulators of this enzyme. Transgenic mice overexpressing ADAM17 are viable with no morphological defects and moreover do not show enhanced substrate cleavage, highlighting the importance of post-translational regulation of ADAM17 (11). ADAM17-mediated shedding can be induced by a number of stimuli that do not influence the ratio between the pro- and mature forms of the enzyme (12); e.g., pro-inflammatory cytokines (interferon-γ, TNFα, interleukin-1β), Toll-like receptor ligands (LPS, poly(I:C)), ligands of purinergic receptors, ligands of G-protein-coupled receptors, and activators of intracellular kinases like ERK (13), p38 MAPK (14), 1,3-phosphoinositide-dependent protein kinase-1 (15), and PKCs (16–18). Phosphorylation of the cytoplasmic domain is regarded as one hallmark of ADAM17 activation (15, 19, 20). However, many studies have failed to support this hypothesis because ADAM17 mutants lacking the cytoplasmic portion can rescue ADAM17 deficiency in knock-out fibroblasts in short term shedding assays (12, 21–23).
The family of Polo-like kinases (PLKs)2 comprises serine/threonine kinases that are evolutionary conserved from yeast to human (24, 25). Five mammalian PLKs have been discovered to date, which share the same domain topology comprising a conserved kinase domain at the N terminus and one or two highly conserved regions of 30 amino acids, termed Polo boxes, in the noncatalytic C-terminal domain (26–28). The Polo box domains (PBD) function as critical regulators for PLK kinase activity and mediate substrate recognition and subcellular localization (29, 30). Despite the well documented contribution of PLK1 in regulating cell cycle progression (31, 32), the functional role of PLK2 remains largely elusive. In recent studies, it was demonstrated that transcriptional up-regulation of plk2 is regulated by x-ray irradiation of thyroid cells (33), activation of the p53 pathway (34), and Toll-like receptor activation of dendritic cells (35). Therefore, it is highly likely that PLK2 is connected to cellular stress responses and inflammation, which concomitantly represent physiological activators of ADAM17 (7, 36).
In the present work, we characterize a novel pathway involved in ADAM17 activation, whereby PLK2 was identified as a specific intracellular binding partner for ADAM17. We mapped the C-terminal Polo box domains of PLK2 as a binding platform for ADAM17 and demonstrate that co-expression of PLK2 and ADAM17 resulted in serine phosphorylation of the cytoplasmic portion of the protease accompanied by increased shedding of ADAM17 substrates. In summary, our data strongly suggest that PLK2 activity is involved in modulating ADAM17-mediated proteolysis.
EXPERIMENTAL PROCEDURES
Reagents
The following reagents were purchased from Sigma: PMA, anisomycin, concanavalin A-Sepharose, LPS Escherichia coli O111:B4, and polyethyleneimine. The following additional reagents were used: protein G Dynabeads (Life Technologies), protease inhibitor mixture (Roche Applied Science), 1,10-phenanthroline (Calbiochem), and BI 2536 (Axon Medchem). The hydroxamate based ADAM inhibitors GW208264 and GI254023 were synthesized by Iris Biotech.
Antibodies
The following antibodies were used: ADAM17 (clone 10.1) and ADAM10 (clone 608) rabbit polyclonal antibodies were generated by peptide immunization of rabbits. Both antibodies were raised against the extracellular portion of either ADAM10 or ADAM17; the ADAM17 K133 rabbit polyclonal antibody was generated by ADAM17 cDNA immunization of rabbits and recognized the extracellular portion of ADAM17; anti-FLAG M2 (Sigma), anti-actin (Sigma), anti-phosphoserine (Sigma), anti-phosphothreonine (Calbiochem), and anti-phosphoERK, anti-ERK, anti-phospho-p38 MAPK, and anti-p38MAPK were obtained from Cell Signaling.
cDNA Constructs and Cloning
The cDNA for full-length PLK2 (NM_152804.2) was amplified from murine brain cDNA and cloned into the pcDNA4TO vector backbone containing an additional 5′ 3× FLAG epitope. The N-terminal truncated pcDNA4TO-PBD expression vector was generated comprising only the PBDs of PLK2 by using the following forward primer: 5′-GATCGCGGCCGCCGATGCAATCCGGATGATAGTCAG-3′, and reverse primer: 5′-GATCGTTTAAACTCAGTTACATCTCTGTAAGAGCATG-3′. The PLK2 cDNA was also subcloned into the pQCXIP retroviral vector by standard PCR techniques. To generate the kinase dead PLK2-KM mutant, lysine 108 was exchanged to methionine by using the site-directed mutagenesis kit (Fermentas) as instructed. The same strategy was used to generate the constitutive active PLK2-TE mutant where threonine 236 was mutated to glutamic acid. The cDNA for full-length PLK1 (NM_011121) was amplified from murine brain cDNA, and the cDNA for full-length PLK3 (NM_013807.2) was amplified from murine embryonic fibroblast cDNA. Both PLK1 and PLK3 cDNAs were cloned into the pcDNA4TO vector backbone containing an additional 5′-3× FLAG epitope. The cDNA for full-length cyclophilin A (Ppia, NM_008907) was amplified from a prey cDNA library clone isolated from the split ubiquitin yeast two-hybrid analysis with ADAM17 as a bait and cloned into the pcDNA4TO vector backbone containing an additional 5′-3× FLAG epitope.
Full-length murine ADAM17 cDNA lacking the signal peptide was cloned into the yeast expression vector pBT3-SUC with a 5′ and 3′ SfiI restriction. The plasmid pcDNA3.1-mADAM17 was used as template to subclone the ADAM17 sequence into the pQCXIH vector backbone by using the PmeI restriction sites. The C-terminally truncated ADAM17 deletion mutants were cloned with a 5′ NotI and 3′XhoI restriction site into the pcDNA3.1(+) using the following primers: forward, 5′-GATCGCGGCCGCACCATGAGGCGGCGTCTCC-3′, and reverse, 5′-GATCCTCGAGTTAGTCCAGTTTCTTATCCACAC-3′ (for ADAM17Δ700); 5′-GATCCTCGAGTTACTGGGGTGCAGGAAAG-3′ (for ADAM17Δ735); 5′-GATCCTCGAGTTAGTCCTTCTCAAAACCATCATC-3′ (for ADAM17Δ784); 5′-GATCCTCGAGTTAGTCTGTGAGATCCTCAAAG-3′ (for ADAM17Δ801); 5′-GATCCTCGGTTAGGCCGCCTTTTCACTCC-3′ (for ADAM17Δ811); and 5′-GATCCTCGAGTTAAACTCGGCTCTGACGCTG-3′ (for ADAM17Δ821). To generate the deletion mutants ADAM17-D4–ADAM17-D6, where 10–15 amino acids where stepwise deleted from the intracellular domain, the site-directed mutagenesis kit (Fermentas) was used according to the manufacturer's instructions: forward primer, 5′-TCACATGCAGATGACGGTTTTG-3′, and reverse primer, 5′-TGGCGGCATCATGGCAGCTG-3′ for ADAM17-D4 lacking the amino acids Val751-Asp772; forward primer, 5′-TCATTCAAGCTGCAGCGTCAGAGC-3′, and reverse primer, 5′-CTTGGCAGCTGTGCTGCTGTTG-3′ for ADAM17-D5 lacking the amino acids Ser773–Lys793; forward primer, 5′-TCATTCAAGCTGCAGCGTCAGAGC-3′, and reverse primer, 5′-CTTGGCAGCTGTGCTGCTGTTG-3′ for ADAM17-D6 lacking the amino acids Ser794–Ala810. ADAM17[S794A], ADAM17[S811A], and ADAM17[S822A] mutant constructs, in which serine residues were mutated to alanine, were generated by using the site-directed mutagenesis kit (Fermentas) as instructed. The Myc-ADAM17wt was generated by subcloning the cDNA of ADAM17 without the signal peptide in the pcDNA3.1(+) spIL-6R-Myc vector, leading to generation of a N-terminally Myc-tagged ADAM17. The Myc-ADAM17ΔPro (lacking the prodomain, amino acids Met1–Lys223) was generated as Myc-ADAM17wt.
ADAM17 and PLK2 luciferase fusion constructs were cloned into the Gateway© entry vector pENTR4 and subcloned into pSPICA-C1 and C2 (containing N- or C-terminal luciferase fragment; kindly provided by Yves Jacob, Institut Pasteur Paris) by homologue recombination using LR clonase II (Invitrogen). The pcDNA3.1(−)-mADAM10 expression vector was generously provided by Prof. Dr. Paul Saftig (Biochemical Institute, Christian-Albrechts-Universität, Kiel, Germany). Murine ERK (Addgene plasmid 14440) and murine p38α-MAPK (Addgene plasmid 20351) expression plasmids were obtained from Addgene. All coding sequences were verified by DNA sequencing.
Split Ubiquitin Yeast Two-hybrid System
A murine brain cDNA library (Mobitec) fused N-terminally to NubG was transformed into the yeast reporter strain NMY32 expressing the fusion protein ADAM17-Cub-LexA-VP17 as a bait. The split ubiquitin yeast two-hybrid screen was performed according to the manufacturer's instructions (Mobitec). Briefly, autoactivation of the bait was excluded prior screening on selective media lacking leucine, tryptophane, and histidine. Expression of the ADAM17-Cub-LexA-VP17 bait protein was analyzed by using the supplied positive (NubI) and negative (NubG) controls. cDNA library transformants were selected on SD Leu−Trp−His− plates containing 5 mm 3-aminotriazole to reduce background activity. Library transformants were isolated and transformed in E. coli XL1blue by electroporation. All isolated plasmids harboring an insert with a size over 250 bp were selected for sequencing. The sense isolated library plasmids were retransformed into the NMY32 yeast strain expressing either ADAM17 or ADAM10 as a bait. When the specific interaction with ADAM17 was confirmed, the cDNA clone was selected for further investigation.
Cell Lines and Transfection
All cells (HEK293T, NIH3T3, Neuro2A, mEF) were cultured in DMEM high glucose (PAA Laboratories) supplemented with 10% FCS and 1% penicillin/streptomycin at 37 °C, 5% CO2 atmosphere, and 95% relative humidity. Cells were transiently transfected with Turbofect (Fermentas, Thermo Scientific). Retrovirus was produced as described previously (37). NIH3T3 or mEFs were transduced with either pQCXIP empty vector or pQCXIP containing murine PKL2 and were selected with 3 μg/ml puromycin.
Generation of Bone Marrow-derived Macrophages (BMDMs) and Bone Marrow-derived Dendritic Cells (BMDCs)
BMDMs as well as BMDCs were differentiated as described previously (38, 39). Cells were maintained over 7 days in the differentiation medium, subsequently detached using Accutase (PAA Laboratories), and seeded on 6-well plates at a density of 0.5 × 106 cells/milliliter. The following day, cells were stimulated for the indicated time periods with LPS (1 μg/ml). TNFα, TNF-RI, and TNF-RII ELISAs were performed according the manufacturer's instructions (R&D Systems). BI 2536 or the indicated metalloprotease inhibitors were added 10 min prior stimulation with LPS or PMA.
RT-PCR and Real Time PCR Analysis
RNA was extracted using the GeneJET RNA purification kit (Fermentas, Thermo Scientific). cDNA was synthesized from 2 μg of RNA using the RevertAidTM Moloney MuLV reverse transcriptase (Fermentas, Thermo Scientific). The mRNA transcripts were amplified by PCR using the following primer pairs: PLK2_fwd, CGGTGCTGAAATACTTTTCTCAT; PLK2_rev, TCGGGAGATCACCACCAT, ADAM17_fwd TCCCTGTCTCTGTTTCATCACA; and ADAM17_rev, AGGTACTGGCGGCATCAT, and quantitative PCR was performed using the LightCycler® 480 Probes Master on a LightCycler480® real time PCR system (Roche Applied Science) according to the manufacturer's instructions. For assay design, the Universal Probe Library System was used to amplify intron-spanning regions for PLK2 and ADAM17. Relative amounts of target gene mRNA were normalized to the housekeeping gene GAPDH. For quantification, the LightCycler® 480 software 1.5.0 (Roche Diagnostics) was used. The relative expression levels were calculated by the 2−ΔΔCt method. Comparative real time PCR results were performed in triplicate.
Co-immunoprecipitations
HEK293T cells were transiently co-transfected with plasmids encoding for ADAM17 (4 μg) and PLK kinases (1 μg). 24 h post-transfection, the cells were washed with ice-cold PBS and incubated for 1 h on ice in IP buffer (0,1% Triton X-100, 150 mm NaCl, 50 mm Tris-HCl, pH 7.4) containing 10 mm 1,10-phenanthroline plus complete protease inhibitor mixture. The lysates were centrifuged (15,000 × g, 15 min, 4 °C) and subsequently precleared with protein G-Sepharose (1 h, 4 °C). Immunoprecipitations were performed with anti-FLAG (M2) or anti-ADAM17 (K133) antibodies (overnight, 4 °C) (23). Immunocomplexes were precipitated with 30 μl of protein G Dynabeads for 30 min at room temperature. Beads were washed three times with IP buffer and two times with PBS, eluted in 2× SDS sample buffer containing 50 mm β-mercaptoethanol, and incubated at 95 °C for 5 min prior to Western blot analysis.
Dephosphorylation and Deglycosylation Analysis
2.5 × 106 HEK293T cells were lysed for 1 h at 4 °C in IP buffer (0,1% Triton X-100, 150 mm NaCl, 50 mm Tris-HCl, pH 7.4, 10 mm 1,10-phenanthroline plus complete protease inhibitor mixture). Glycoproteins (200 μg of total protein) were captured on concanavalin A-Sepharose (30 μl; Sigma) overnight at 4 °C. Sepharose beads were denatured at 100 °C for 10 min in 1× deglycosylation lysis buffer (New England Biolabs) and treated with N-glycosidase F (PNGase F) or endoglycosidase H (EndoH) according to the manufacturer's instructions (New England Biolabs). For dephosphorylation analysis, calf intestine phosphatase (Fermentas) was used. HEK293T cell lysates were prepared as described above. Concanavalin A-enriched lysates were resuspended in FastAP buffer containing 30 μl of concanavalin A-enriched lysate and 5 μl of FastAP. Control reaction was performed in the absence of FastAP. After 1 h of incubation at 37 °C, the reaction was stopped by heating at 100 °C for 5 min.
Gaussia princeps Luciferase Complementation Assay
HEK293T cells were seeded at a density of 200,000 cells/12-well plates. 24 h later, cells were transiently transfected with ADAM17-luciferase fusion plasmids in presence or absence of the PLK2-luciferase fusion plasmid as described (40). The luciferase assay was performed 2 days after transfection as described previously (23).
Immunofluorescence and Confocal Microscopy
HEK293T cells transiently transfected with ADAM17 as well as PLK2 and were seeded at a density of 50,000 cells/well in a 12-well plate on glass coverslips. 24 h later, cells were washed three times with PBS and fixed with 4% paraformaldehyde/PBS for 20 min at room temperature. Fixed cells were permeabilized for 5 min with PBS containing 0.2% saponin and blocked for 1 h at 37 °C in staining buffer (PBS containing 3% BSA and 0.2% saponin). The primary antibodies were diluted 1:100 (anti-A17 K133) or 1:1,000 (anti-FLAG M2) and incubated for 1 h at room temperature in staining buffer. The coverslips were washed three times with PBS, stained for 1 h at room temperature with secondary antibody (1:250) in staining buffer, washed twice, and finally stained with DAPI solution. Stained coverslips were mounted in mounting medium (Dako), viewed, and photographed with an FV1000 confocal laser scanning microscope (Olympus) equipped with a U Plan S Apo 100 × oil immersion objective (N.A. 1.40). Digital images were processed and merged using FV10-ASW 2.0 Viewer (Olympus).
LPS Injection
For the experimental endotoxemia studies (approved by the German Animal Welfare Committee, Number V312-72241.121-3 (60-5/11)), C57BL/6 mice (Charles River) were injected intravenously with 2.5 mg/kg LPS (0111:B4; Sigma-Aldrich). 1 h after LPS injection, mice were sacrificed, and tissues were snap frozen in 300 μl of TRIzol® (Invitrogen) for RNA preparation. All mice were maintained under conventional conditions with water and food ad libitum.
Mass Spectrometry-based Identification of Phosphorylation Sites
For MS-based identification of phosphosites, ADAM17 fused C-terminally to the PC tag was used. HEK293T cells were transfected with or without PLK2 and lysed 48 h later in MS buffer (0,5% Nonidet P-40, 150 mm NaCl, 50 mm Tris-HCl, pH 7.4, 4 mm CaCl2,10 mm 1,10-phenanthroline plus complete protease inhibitor mixture). ADAM17 was immunoprecipitated with an anti-PC antibody (41, 42) and separated by standard SDS-PAGE (12.5%). The corresponding bands were in-gel digested with trypsin. Briefly, bands were excised into 2-mm3 cubes and destained with 30% acetonitrile (ACN) in 50 mm ammonium bicarbonate. Samples were reduced with 10 mm dithiothreitol at 56 °C for 45 min and alkylated using iodoacetamide (55 mm) at room temperature for 30 min in the dark. Prior to digest, gel pieces were washed with ammonium bicarbonate and shrunk with ACN. The gels were rehydrated with 50 ng of trypsin in 25 mm ammonium bicarbonate with the addition of 10% ACN and left to digest overnight at 37 °C. Samples were extracted using 1% formic acid, 50% ACN in 1% formic acid, or 100% ACN. Samples were dried in a vacuum centrifuge and resuspended in 3% ACN in 0.1% trifluoroacetic acid.
Peptides were separated and analyzed with an UltiMate 3000 RS Nano/Cap system (Dionex, Idstein, Germany) coupled online to an Orbitrap LTQ Velos (Thermo, Bremen, Germany). Samples were desalted for 7 min (Acclaim PepMap100 C-18, 300-μm inner diameter × 5 mm, 5 μm, 100 Å; Dionex) at a flow rate of 30 μl/min using 3% ACN, 0.1% TFA. An Acclaim PepMap100 C-18 column (75-μm inner diameter × 150 mm, 3 μm, 100 Å) was used for analytical separation using buffers A (0.05% formic acid) and B (80% ACN, 0.04% formic acid) at a flow rate of 300 μl/min. For elution, a linear gradient from 5–10% B (6–7 min), 12–50% B (7–127 min), and 55–90% B (127–135 min) followed by an isocratic wash (90% B, 135–145 min) and column re-equilibration (5% B, 145–160 min) was applied.
MS scans were acquired in the mass range of 300–2,000 m/z at a resolution of 60,000. The five most intense signals were subjected to CID and HCD fragmentation using a dynamic exclusion of 90 s and a repeat count of 3. For MS/MS measurements, an isolation width of 2.0 Da was used for CID, whereas for HCD spectra 3.0 Da was used. Normalized collision energy was set to 35 and 40 for CID and HCD, respectively. For CID fragmentation, multistage activation was enabled. HCD spectra were acquired at a resolution of 7,500.
HCD and CID MS2 spectra were searched separately using Sequest and Mascot (2.2.7) via Proteome Discoverer 1.3.0.339 (DBVersion: 78) against the reviewed and canonical human database (08.03.2103) with common contaminants, mouse ADAM17 (Q9Z0F8), and recombinant ADAM17 appended to the database (26,226 sequences). The data were searched with 5-ppm MS tolerance and 0.5- and 0.02-Da tolerance for CID and HCD spectra, respectively. Spectra were searched with both tryptic and semitryptic (two missed cleavages allowed) enzyme specificity. Carbamidomethylation of cysteine residues (+57.021 Da) was set a fixed modification, oxidation (+15.995 Da) on methionine residues, and phosphorylation (+79.966 Da) of serine, threonine, and tyrosine residues were set as dynamic modifications. Combined spectra were validated using percolator. Event detector, precursor ion area detector, and Phospho RS 3.0 were implemented using default settings.
RESULTS
PLK2 Interacts with ADAM17 in Yeast
Making use of the split ubiquitin membrane yeast two-hybrid technology, we aimed to identify novel proteins interacting with ADAM17. To this end, we constructed an ADAM17 bait by fusing the full-length murine cDNA N-terminally to Cub-LexA, thus generating ADAM17-Cub-LexA (Fig. 1A). We confirmed the specificity of the employed method by co-expression of ADAM17-Cub-LexA with a noninteracting membrane protein (negative control, NubG) or with the ADAM17 substrate pro-TNFα (NubG-mbTNFα), which led to robust split ubiquitin formation and activation of the yeast reporter system (Fig. 1B). To identify novel ADAM17 interaction partners, we screened a murine brain cDNA library with the cDNAs inserted downstream of the NubG sequence. Two isolated cDNAs encoded PLK2, a member of the Polo-like kinase family. Re-expression of NubG-PLK2 and ADAM17-Cub-lexA in yeast verified the specific interaction between the two proteins. However, co-transformation of ADAM10-Cub-LexA with NubG-PLK2 did not lead to colony formation upon selection, indicating that PLK2 is a specific interaction partner for ADAM17 in yeast (Fig. 1C).
FIGURE 1.

PLK2 was identified in the Split ubiquitin yeast two-hybrid screen as ADAM17-binding protein. A, schematic overview of the ADAM17-Cub-LexA bait protein used in this study. ADAM17 cDNA lacking the native signal peptide was fused to Cub, followed by an artificial transcription factor (LexA). The numbers of amino acids of ADAM17 and Cub-LexA portions are indicated. ySP, SUC2 (yeast) cleavable signal peptide; ECD, extracellular domain; TM, transmembrane region; ICD, cytoplasmic portion. B, co-expression of ADAM17-Cub-LexA with a noninteracting yeast membrane protein fused to the NubI (+) or NubG (−) domains. Only co-expression of ADAM17-Cub-LexA with NubI resulted in split ubiquitin formation and activation of the yeast reporter system (panel I). This occurs because NubI associates with Cub independent of additional protein-protein interaction. In contrast NubG only activates the yeast reporter system when additional protein-protein interaction takes place. Co-transformation of ADAM17-Cub-LexA with NubG did not lead to colony formation on minimal agar plates supplemented with 5 mm 3-aminotriazole and lacking tryptophan (−trp), leucine (−leu), and histidine (−his) (panel II). Co-transformation of ADAM17-Cub-LexA but not ADAM10-Cub-LexA with the prey NubG-mbTNFα (membrane-bound TNFα) led to split ubiquitin formation and activation of the yeast reporter system (panel III). C, ADAM17-Cub-LexA or ADAM10-Cub-LexA were co-transformed with the identified Nub-PLK2 prey in NMY51 yeast. Transformation of bait and prey was verified on agar plates (SC) lacking tryptophan (−trp) and leucine (−leu) (upper panel). Interaction of ADAM17-Cub-LexA with Nub-PLK2 was shown under selective pressure on minimal agar plates lacking tryptophan (−trp), leucine (−leu), histidine (−his), and adenine (−ade) in comparison to the negative control where ADAM10-Cub-LexA was used as a bait (lower panel).
PLK2 and ADAM17 Interact and Co-localize in Human Cells
Because ADAM17 interacted with the cytoplasmic kinase PLK2 in the split ubiquitin membrane yeast two-hybrid system, we sought to validate the interaction in mammalian cells. To this end, ADAM17 and the ADAM17Δcyto mutant, which lacks the intracellular domain, were transiently overexpressed in the presence or absence of FLAG-PLK2 in HEK293T cells, and co-immunoprecipitations were performed (Fig. 2, A and B). ADAM17 was co-precipitated with PLK2 (Fig. 2A), and vice versa PLK2 was found in the precipitate after ADAM17 pull-down (Fig. 2B). Importantly, the ADAM17Δcyto mutant did not precipitate PLK2, clearly demonstrating that the cytoplasmic tail of ADAM17 is necessary for the interaction. Next, we used the split luciferase complementation assay to further study ADAM17-PLK2 complex formation in intact HEK293T cells (40). We therefore fused fragments of luciferase N-terminally to PLK2 (N2-PLK2) and C-terminally to either ADAM17 (ADAM17-C1) or ADAM17Δcyto (ADAM17 Δcyto-C1) and co-transfected both fusion proteins in HEK293T cells (Fig. 2, C and D). Only co-expression of ADAM17-C1 and N2-PLK2 resulted in activation of luciferase activity (Fig. 2E). Upon co-expression of ADAM17Δcyto-C1 with N2-PLK2, no luminescence was measured, again indicating that the cytoplasmic tail of ADAM17 is important for the interaction of ADAM17 and PKL2 (Fig. 2E).
FIGURE 2.

PLK2, a new ADAM17 interaction partner. A, co-IP of ADAM17 and ADAM17Δcyto with FLAG-PLK2 in HEK293T cells. FLAG-PLK2 was precipitated using the anti-FLAG (M2) antibody. Co-precipitation of ADAM17 but not ADAM17Δcyto was shown by Western blot using an anti-ADAM17 antibody raised against the extracellular portion (clone 10.1). Single transfections of ADAM17 and ADAM17Δcyto served as specificity controls. B, HEK293T cells were transfected as described in A, and ADAM17 was precipitated using the anti-ADAM17 antibody K133. Co-precipitation of FLAG-PLK2 with ADAM17 was monitored by Western blot analysis using the anti-FLAG (M2) antibody. FLAG-PLK2 was not detected in the precipitate of ADAM17Δcyto expressing cells. C, scheme of the constructs used in the luciferase-based protein complementation assay. ADAM17 or ADAM17Δcyto were fused to the inactive luciferase fragment C1. PLK2 was fused to the inactive luciferase fragment N2. Numbers of amino acids of ADAM17, PLK2, and luciferase portions are indicated. D, ADAM17-C1 or ADAM17Δcyto-C1 was co-expressed with N2-PLK2 in HEK293T cells. ADAM17-C1, ADAM17Δcyto-C1, and N2-PLK2 single transfections served as luminescence controls. Expression of all fusion proteins was verified by Western blot (WB) using anti-ADAM17 (clone 10.1) and anti-FLAG (M2) antibodies. E, HEK293T cells were transfected as described in D. 48 h post-transfection, cells were lysed to determine reconstituted luciferase reporter activity. The gray line represents the threshold of 3.5. The luciferase reporter assay was measured in three in-plate replicates of at least three independent experiments; one representative experiment is shown. F, co-localization of ADAM17 with PLK2 in transiently transfected HEK293T cells. Cells were fixed, permeabilized, and immunostained with antibodies against ADAM17 (K133-Alexa488) and PLK2 via anti-FLAG (M2-Alexa594) antibody.
Next, we addressed whether ADAM17 co-localized with PLK2. For this purpose, co-transfected HEK293T cells were used to visualize ADAM17 and PLK2 by confocal microscopy. ADAM17 overexpression led to intracellular perinuclear staining as reported previously (13). The simultaneous expression of FLAGPLK2 revealed co-localization of both proteins mainly in intracellular vesicular compartments. No redistribution of ADAM17 to the cell surface after PLK2 co-transfection was observed, indicating no obvious role in cell trafficking (Fig. 2F). Based on these data obtained with independent experimental systems, we conclude that ADAM17 and PLK2 form a complex in mammalian cells.
The Polo Box Domains of PLK2 Are Crucial for the Interaction with ADAM17
To determine which part of PLK2 mediated the interaction, we co-expressed ADAM17 together with full-length or an N-terminal truncated versions of PLK2, comprising only the conserved PBDs, which are considered to be essential for Polo-like kinase substrate recognition. As shown in Fig. 3A, both PLK2 and PBD co-precipitated with ADAM17. In contrast, FLAG-tagged cyclophilin A, which was used as negative control, showed no co-precipitation. More importantly, co-expression of PBD with ADAM10 did not lead to complex formation, indicating that PLK2 specifically interacted with ADAM17 via the PBD domains (Fig. 3B). Two other members of the Polo box domain family, namely PLK1 and PLK3, failed to co-precipitate with ADAM17 (Fig. 3C), additionally highlighting the specificity of the PLK2/ADAM17 interaction.
FIGURE 3.

The PLK2-Polo box domains are crucial for ADAM17 binding. A, ADAM17 was co-expressed in HEK293T cells in presence of FLAG-cyclophilin A (Cyl.A), a truncated PLK2 mutant comprising only the PBD and wt PLK2. Co-immunoprecipitation of ADAM17 by using the anti-FLAG (M2) antibody was analyzed by Western blot analysis and revealed that the PBD region of PLK2 is sufficient to bind ADAM17. Cyclophilin A served as negative control. B, co-IP of ADAM17 and ADAM10 with FLAG-PBD (PLK2) in HEK293T cells. FLAG-PBD was precipitated using the anti-FLAG (M2) antibody. Co-precipitation of ADAM17 but not ADAM10 was shown by Western blot using an anti-ADAM17 antibody raised against the extracellular portion (clone 10.1) or an anti-ADAM10 antibody raised against the extracellular portion (clone 608). Single transfections of ADAM17 and ADAM10 served as specificity controls (n = 2). C, ADAM17 was co-expressed in HEK293T cells in presence of the PLK2 paralogues FLAG-PLK1 and FLAG-PLK3. Co-immunoprecipitations were performed as described in A and verified a strong interaction of ADAM17 with PLK2 but not PLK1 or PLK3 (n = 3).
ADAM17 Binding Is Independent of the S(pT/pS)(P/X) Motif
PBDs act as phosphopeptide-binding modules engaging the consensus motif (S(pT/pS)(P/X)). Thus, PLK2 binds target proteins, which have already been phosphorylated by an upstream “priming kinase,” adding further phosphate groups (43). To determine the binding motif in the ADAM17 cytoplasmic tail responsible for PLK2 interaction, we constructed several ADAM17 truncation mutants (Fig. 4A) and tested their ability to engage PLK2. Expression of PLK2 in the presence of wt ADAM17, ADAM17Δ821, ADAM17Δ811, and ADAM17Δ801 led to complex formation, whereas the truncation mutants ADAM17Δ784 and ADAM17Δ735 did not associate with PLK2 (Fig. 4B). We therefore concluded that the binding interface for PLK2 is located downstream of Asp783 in the ADAM17 cytoplasmic portion. Within this region, we identified the 788SSTA792 sequence resembling the PBD consensus binding motif (44). To test whether 788SSTA792 mediated binding to PLK2, we generated an ADAM17 deletion mutant in which the 788SSTA792 sequence stretch was deleted. As shown in Fig. 4C, the ADAM17ΔSSTA mutant co-immunoprecipitated with PLK2 as well as wt ADAM17. Thus, the ability of PLK2 to bind ADAM17 was independent of the canonical S(pT/pS)(P/X) motif, implying the existence of a noncanonical interaction site. To further map the PLK2 interaction region within the ADAM17 cytoplasmic tail, we generated three additional deletion mutants termed ADAM17-D4 (ΔVal751–Asp772), ADAM17-D5 (ΔSer773–Lys793), and ADAM17-D6 (ΔSer794–Ala810) (Fig. 4A). Upon co-transfection with PLK2, only the ADAM17-D6 deletion mutant failed to co-precipitate with PLK2 (Fig. 4D), indicating that the PLK2 binding epitope within the cytoplasmic portion of ADAM17 resides between Ser794 and Ala810.
FIGURE 4.

The amino acid stretch Ser794–Ala810 within the cytoplasmic tail of ADAM17 binds to PLK2. A, schematic representation of truncated and deletion mutants within the cytoplasmic domain of ADAM17 used to map the interaction interface required for PLK2 binding. A gray box highlights the identified binding epitope for PLK2. B, Western blots of co-immunoprecipitates and lysates of HEK293T cells transiently co-transfected with the ADAM17 mutants represented in A in presence or absence of FLAG-PLK2. Precipitation of PLK2 with the anti-FLAG antibody (M2) led to co-precipitation of ADAM17Δ801, ADAM17Δ811, and ADAM17Δ821. C, Western blots of co-immunoprecipitates and lysates of HEK293T cells transiently co-transfected with the ADAM17 deletion mutant ADAM17ΔSSTA in presence or absence of FLAG-PLK2. D, Western blots of co-immunoprecipitates and lysates of HEK293T cells transiently co-transfected with the ADAM17 deletion mutants represented in A in presence or absence of FLAG-PLK2. Precipitation of PLK2 with the anti-FLAG antibody (M2) led to co-precipitation of ADAM17-D4 and ADAM17-D5. Single transfections of ADAM17 mutants served as specificity controls.
PLK2 Interacts with and Phosphorylates ADAM17
We observed that co-expression of ADAM17 with PLK2 resulted in a pronounced shift to lower SDS gel mobility without changes in total expression, suggestive of phosphorylation (Fig. 5A). Notably, we did not observe a similar SDS gel mobility shift after co-expression of ADAM17 with ERK or p38-MAPK, even after stimulation with PMA or anisomycin (Fig. 5B). The PLK2 kinase-dead mutant K108M (KM) had no effect on the electrophoretic mobility of ADAM17, whereas the constitutively active PLK2 mutant T236E (TE) caused a SDS gel shift similar to that of wt PLK2. Both PLK2-KM and PLK2-TE co-precipitated with ADAM17 (Fig. 5C). ADAM17 was detected by Western blot analysis with an α-ADAM17 (clone 10.1) antibody in two distinct forms: a 130-kDa proform and a 110-kDa form, which might represent (i) the mature protein devoid of the prodomain or (ii) a differently post-translationally modified isoform of ADAM17. Of note, the 110-kDa ADAM17 form was detected in most but not in all experiments. Notably, when ADAM17 was co-transfected with wt PLK2 or PLK2-TE, the ratio between the 130- and 110-kDa ADAM17 forms shifted significantly toward the proform, which might indicate that PLK2 co-expression either inhibited ADAM17 maturation or affected post-translational modification of the protease. To address this question, we constructed two Myc-tagged ADAM17 variants termed MycADAM17 (control) or MycΔPro-ADAM17 (mutant lacking the prodomain), which allowed the distinction between the pro- and mature forms of ADAM17 by Western blot analysis (Fig. 5D). Previous studies suggest that full-length ADAM17 is degraded when expressed without the prodomain in insect cells (45). However, in our experimental setting, we did not observe enhanced degradation of the MycΔPro-ADAM17 mutant compared with wt ADAM17. Co-transfection of MycADAM17 or MycΔPro-ADAM17 with PLK2 resulted in SDS gel mobility shifts as compared with the single transfectants. The 110-kDa ADAM17 form, which was strongly diminished after PLK2 co-transfection, did not correspond to the mature form of ADAM17 as judged by SDS-PAGE mobility of the MycΔPro-ADAM17 mutant (Fig. 5D). To exclude that maturation defects of MycΔPro-ADAM17 led to altered electrophoretic migration, we compared the electrophoretic mobility of MycΔProADAM17 with MycADAM17 cleaved in vitro by furin (Fig. 5E). Furin-cleaved ADAM17 showed the same migration behavior as MycΔPro-ADAM17. Therefore, we hypothesized that PLK2 was not involved in maturation of ADAM17. To investigate, if PLK2 influenced the glycosylation pattern of ADAM17, we tracked ADAM17 maturation by EndoH or PNGase F digestion. Co-expression of ADAM17 with PLK2, kinase-dead PLK2-KM as well as constitutively active PLK2-TE revealed the presence of almost equivalent populations of EndoH-sensitive ADAM17, although some EndoH-resistant material was visible. These results reflect the predominant localization of ADAM17 in the ER (Fig. 5F).
FIGURE 5.

Phospho-dependent binding of PLK2 to ADAM17. A, gel mobility shift of ADAM17 induced by co-transfected wt PLK2. Lysates containing 50 μg of protein were resolved directly by SDS-PAGE on an 8% polyacrylamide gel (n = 3). WB, Western blot. B, ADAM17 was co-expressed with plasmids encoding for murine ERK1 (T7-tagged), murine p38α (FLAG-tagged), murine PLK2 (FLAG-tagged), and murine constitutive active murine PLK2-TE (FLAG-tagged). 24 h after transfection cell culture medium was replaced by growth media containing DMSO (untreated), 100 nm PMA or 1 μm anisomycin and stimulated for 1 h prior to lysis. Lysates containing 10 μg of protein were resolved by SDS-PAGE on a polyacrylamide gel for detection of ADAM17, PLK2, ERK, phospho-ERK, p38α, phospo-p38α, and actin. C, gel mobility shift of ADAM17 induced by co-transfection of wt PLK2 or constitutively active (TE) PLK2 in HEK293T cells, but not by kinase-dead (KM) PLK2. Co-immunoprecipitations of ADAM17 verified the interaction with the PLK2 mutants. ADAM17 was visualized by using the anti-ADAM17 (clone 10.1) antibody, which detects under reducing conditions two specific bands: the first migrates as a 130-kDa protein (corresponding to the proform of ADAM17), and the second was detectable at 110 kDa. D, scheme of the Myc-tagged ADAM17 variants. Myc tag was inserted between the signal peptide and the prodomain of wt ADAM17 (part i) or between the signal peptide and a mutant lacking the complete prodomain (part ii). RVKR, furin cleavage site at the boundary of the pro- and catalytic domain of ADAM17. Both ADAM17 variants were co-transfected with PLK2 and resolved directly by SDS-PAGE on an 8% polyacrylamide gel followed by Western blot by using an anti-Myc antibody. Myc-ADAM17 as well as MycΔpro-ADAM17 displayed a gel mobility shift. Myc-ADAM17 was detected as two bands (n = 3). E, HEK293T cells were transiently transfected with MycADAM17 and MycΔPro-ADAM17. 48 h after transfection cells were lysed, and ADAM17 was immunoprecipitated with anti-Myc-Sepharose. In addition, precipitated MycADAM17 was cleaved in vitro with furin as indicated. Western blot analysis of ADAM17 was performed using either the anti-Myc or anti-ADAM17–10.1 antibody (n = 2). F, HEK293T cells were co-transfected with mock pcDNA4TO, FLAG-PLK2, FLAG-PLK2-KM, and FLAG-PLK2-TE. ADAM17 was enriched by concanavalin A-Sepharose from 200 μg of total protein. Immunoprecipitates were incubated with EndoH or PNGase F and subsequent analyzed by Western blot using the anti-ADAM17 (clone 10.1) antibody; c, control; H, EndoH; P, PNGase F (n = 3).
Identification of a Novel ADAM17 Phosphorylation Site Used by PLK2
Treatment of Concanavalin A-enriched lysates with calf intestinal alkaline phosphatase abolished the PLK2-induced gel shift of ADAM17, pointing to phosphorylation of ADAM17 by this kinase (Fig. 6A). To directly monitor phosphorylation of the ADAM17 intracellular domain (ADAM17-ICD), we used LC-MS-based phosphoproteomics. We identified three serine residues, namely Ser794, Ser811, and Ser822, which were phosphorylated in the sample when both ADAM17 fused to the PC tag and PLK2 were present. We detected also one phosphorylated serine within the PC epitope tag of ADAM17 (Ser829), possibly occurring because of the highly flexible nature of the C-terminal tag (Table 1 and Fig. 6B). To verify the results obtained from LC-MS-based phosphoproteomics, we mutated each of the three potentially phosphorylated serine residues in the ADAM17-ICD to alanine and demonstrated that Ser794 but not Ser811 and Ser822 was phosphorylated after PLK2 co-expression as judged by SDS gel mobility shift (Fig. 6C) Interestingly, binding of ADAM17[S794A] to PLK2 was also strongly diminished, indicating that phosphorylation of Ser794 stabilized the ADAM17-PLK2 protein complex (Fig. 6C).
FIGURE 6.

Phospho-dependent binding of PLK2 to ADAM17. A, HEK293T cells were co-transfected with pcDNA4TO, FLAG-PLK2, FLAG-PLK2-KM, and FLAG-PLK2-TE. 48 h after transfection, cells were lysed and ADAM17 was enriched by concanavalin A (ConA)-Sepharose from 200 μg of total protein. Immunoprecipitates were incubated with calf intestinal alkaline phosphatase (CIAP) for 1 h and subsequently analyzed by Western blot using the anti-ADAM17 (clone 10.1) antibody. B, MS/MS spectra (HCD) of phosphopeptides identified in ADAM17 + PLK2 samples via database searching. For clarity not all matched b- and y-ions are shown. C, ADAM17[S794A], ADAM17[S811A], and ADAM17[S822A] single phosphomutants of ADAM17 were co-transfected in presence or absence of PLK2. Binding to PLK2 was analyzed by anti-FLAG (M2) co-immunoprecipitation and subsequent Western blot analysis of ADAM17. Gel mobility shift of ADAM17 induced by co-transfected wt PLK2 was completely abolished in the [S794A] mutant (input, αA17) but not in the other two phosphomutant-ADAM17 variants. Binding of ADAM17[S794A] to PLK2 was strongly reduced (co-IP, αA17).
TABLE 1.
Phosphopeptides and corresponding nonphosphorylated peptides identified by LC-MS/MS in ADAM17 or ADAM17 and PLK2 samples
Phosphorylated peptides are indicated in bold type. Extracted ion chromatographs of the phosphorylated and nonphosphorylated peptides and corresponding MS2 spectra are shown in Fig. 6B. (p) indicates PO43−43- group.
| Residues | Sequence | Modifications | ADAM17 + PLK2 | ADAM17 |
|---|---|---|---|---|
| 794–805 | SFEDLTDHPVTR | Yes | Yes | |
| 794–805 | SFEDLTDHPVTR | S1 (p) | Yes | No |
| 806–813 | SEKAASFKLQR | Yes | Yes | |
| 806–813 | SEKAASFKLQR | S6 (p) | Yes | No |
| 809–813 | AASFKLQR | Yes | Yes | |
| 809–813 | AASFKLQR | S3 (p) | Yes | No |
| 817–836 | QSRVDSKETECGSEDQVDPR | Yes | Yes | |
| 817–836 | QSRVDSKETECGSEDQVDPR | S13 (p) | Yes | No |
| 817–836 | QSRVDSKETECGSEDQVDPR | S6 (p)/S13 (p) | Yes | No |
| 820–836 | VDSKETECGSEDQVDPR | Yes | Yes | |
| 820–836 | VDSKETECGSEDQVDPR | S10 (p) | Yes | No |
| 820–841 | VDSKETECGSEDQVDPRLIDGK | Yes | Yes | |
| 820–841 | VDSKETECGSEDQVDPRLIDGK | S10 (p) | Yes | No |
PLK2 Facilitates the Sheddase Activity of ADAM17
We next investigated whether the proteolytic activity of ADAM17 was regulated by PLK2. NIH3T3 and immortalized murine embryonic fibroblasts (mEF) represent a useful tool to study regulation of ADAM17 activity because both cell lines endogenously express the protease in addition to the endogenous ADAM17 substrate TNF-RI. Furthermore, we extended our analysis to murine Neuro2A cells because PLK2 has crucial functions in modulating synaptic plasticity in neuronal cells (43, 46). As shown in Fig. 7A, constitutive release of TNF-RI was significantly inhibited in presence of the pan-PLK inhibitor BI 2536, which has shown good tolerability in mice and humans and is currently being tested as an anti-tumor agent in phase II clinical trials (47). Interestingly, PMA-induced TNF-RI release was not significantly affected by BI 2536 treatment (Fig. 7A). Next, we stably transfected wt mEF cells with PLK2 leading to 12-fold up-regulation of PLK2 mRNA as compared with mock transfected mEFs (Fig. 7B). When we evaluated the constitutive release of TNF-RI into the cell culture supernatant, we observed an ∼2–3-fold increase of soluble TNF-RI compared with mock transfected cells (Fig. 7C). We confirmed that ADAM17 activity was also enhanced in stably transfected NIH3T3 (Fig. 7D), as well as transiently transfected Neuro2A cells (Fig. 7E). In contrast to wt PLK2, shedding activity was not increased when the inactive PLK2-KM variant was expressed in Neuro2A cells (Fig. 7E).
FIGURE 7.
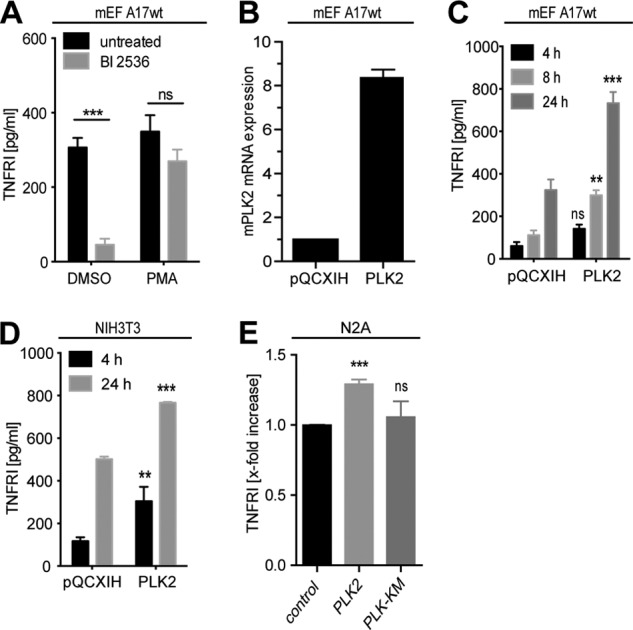
Activity analysis of ADAM17. A, activity of ADAM17 was studied through the analysis of ADAM17-specific shedding events. wt mEFs expressing the endogenous ADAM17 substrate TNFRI were seeded at a density of 500,000 cells. After 24 h, cell culture medium was replaced by medium containing either 1 μm of the pan-PLK inhibitor BI 2536 (gray bars) or DMSO (vehicle control, black bars). Media were conditioned for 6 h, and afterward 100 nm PMA was added for 1 h. B, wt mEFs were stably transfected with wt PLK2 or mock plasmid (pQXCIP). Quantitative RT-PCR analysis was performed to quantify PLK2 expression levels in the stable transfected mEF cells. One representative experiment is shown. C, stably transfected wt PLK2-mEF cells as well as mock transfected cells (pQXCIP-mEFs) were seeded at a density of 250,000 cells/12 wells. 24 h later the cell culture medium was replaced and conditioned for the indicated time periods. D, stably transfected wt PLK2-NIH3T3 cells as well as mock transfected cells (pQXCIP-NIH3T3) were seeded at a density of 500,000 cells. 24 h later, the cell culture medium was replaced and conditioned for the indicated time periods. E, Neuro2A cells were transiently transfected with PLK2 or PLK2KM and seeded at a density of 1 × 106 cells. 24 h later, the cell culture medium was replaced and conditioned for 24 h (n = 3). All error bars denote S.E. **, p < 0.01; ***, p < 0.001; ns, not significant. For the ELISA experiment, we show the mean of three independent experiments performed in duplicate.
PLK2 Inhibition Prevents LPS-induced Pro-TNFα Release in Primary Cells
We measured relative expression of plk2 and adam17 mRNA in a panel of mouse tissues and could demonstrate high expression of both transcripts in colon, heart, and brain where strong adverse effects have been reported in ADAM17-deficient mice (8, 48, 49) (Fig. 8A). Inflammatory challenge of C57/Bl6N mice with a single bolus injection of LPS led to massive up-regulation of plk2 mRNA after only 1 h in lymphoid organs but not in the liver (Fig. 8B). Notably, adam17 mRNA was not significantly altered by LPS injection in our experimental setting (Fig. 8C). Finally, we extended our analysis to primary BMDC as well as BMDM. Activation of Toll-like receptor 4 signaling by LPS led to an up-regulation of plk2-mRNA, which peaked after 4 h in BMDMs and after 2 h in BMDCs and decreased at later time points (Fig. 8D). To determine whether PLK2 played a role in the LPS-induced activation of ADAM17, we applied the pan-PLK inhibitor BI 2356 to BMDCs and BMDMs. LPS treatment led to enhanced shedding of pro-TNFα and TNF-RI from the cell surface of BMDMs and BMDCs, which was efficiently blocked by the combined ADAM17 and ADAM10 inhibitor GW208264 but not by the ADAM10 inhibitor GI254023, corroborating its dependence on ADAM17. As shown in Fig. 8 (E and F), blocking PLK activity by BI 2356 led to significant inhibition of TNFα as well as TNF-RI release from the cell surface of BMDMs and BMDCs.
FIGURE 8.

PLK inhibition affects pro-inflammatory responses. A, quantitative RT-PCR analysis of RNA isolated from several tissues from C57/Bl6N mice. plk2 and adam17 expression levels were normalized against gapdh mRNA. The corresponding spleen signal was fixed at 100%, and all other signals were expressed as a proportion of this (n = 3). One representative result from three replicate experiments is shown. B and C, C57/Bl6N mice were challenged for 1 h with LPS. Expression levels of plk2 and adam17 mRNA was determined by quantitative RT-PCR (n = 2 untreated mice; n = 3 LPS-treated mice). D, BMDMs as well as BMDCs were seeded at a density of 1 × 106/6-well and incubated for indicated time periods with 1 μg/ml LPS. plk2 mRNA was quantified by quantitative RT-PCR. One representative experiment is shown (n = 3). E and F, BMDMs and BMDCs were seeded as described in D. After 24 h, cell culture medium was replaced, and the cells were stimulated with 1 μg/ml LPS (12 h) in presence or absence of the metalloprotease or PLK inhibitors BI 2536 (BI, 1 μm), GI254023X (GI, 3 μm), and GW208264 (GW, 3 μm). Inhibitors were added 10 min prior to stimulation. Soluble TNFα (D and E) or sTNFRI (D) concentrations were determined by ELISAs in the supernatants (n = 3). All error bars denote S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant. For the ELISA experiments, we show the means of three independent experiments performed in duplicate.
DISCUSSION
In this study, we identified PLK2 as a binding partner of the major TNFα sheddase ADAM17, and in subsequent studies we established a role for PLK2 in TNFα signaling, most likely by controlling phosphorylation and activation of ADAM17. Despite numerous studies addressing the function of PLK1 in mitotic entry, spindle pole assembly, and cytokinesis, the biological functions of its paralogs, PLK2, PLK3, and PLK4, are poorly understood, and their cellular targets remain largely unknown (47, 50). Although they are believed to be less important than PLK1 in mitosis regulation, their function in nondividing cells such as dendritic cells and neurons are only beginning to emerge (30, 35, 46). We have demonstrated that PLK inhibition suppresses pro-TNFα and TNF-RI shedding, which all represent in vivo validated ADAM17 substrates. These findings are of high clinical relevance because proteolytically cleaved (soluble) TNFα is critically involved in development and maintenance of inflammation. Consequently, blockade of TNFα has proven to be effective in ameliorating numerous inflammatory disorders that are characterized by dysbalanced TNF signaling (51, 52). Our results indicate that chemical PLK inhibition skews the balance between pro- and anti-inflammatory TNFα signaling strongly toward the anti-inflammatory side by suppressing TNFα shedding through ADAM17. Of note, PLK2 and PLK4 kinase activities have been defined as essential components of antiviral pathways by modulating type I interferon-induced gene transcription. Specifically, Toll-like receptor stimulation has been shown to activate PLK2 and 4, which was accompanied by subsequent phosphorylation of PLK target proteins and finally controlling expression of antiviral cytokines (35). Importantly, mRNA expression levels of inflammatory cytokines including IL-1β, IL-1α, or TNFα were not affected by PLK inhibition via BI 2356 (35). However, IL-1β as well TNFα bioavailability is largely controlled by caspase-1 or ADAM17-mediated proteolysis rather than by transcriptional up-regulation (9, 53). Therefore, the molecular link between PLK2 and ADAM17 we established in this study might represent a novel mechanism by which PLK2 controls inflammatory responses.
PLK2-deficient mice display mild developmental disorders because 30% of them were born with open eye lids and are smaller compared with littermate controls (54). Remarkably, both the open eye as well as the growth retardation phenotype was also reported in ADAM17-deficient animals reflecting the importance of PLK2 biology in controlling ADAM17 activity.
Using mass spectrometry we identified three serine residues in the ADAM17 cytoplasmic tail, Ser794, Ser811, and Ser822, as being phosphorylated upon PLK2 co-transfection. In addition, we observed a pronounced gel mobility shift of ADAM17 after co-transfection with wt PLK2 but not the kinase-dead PLK2-KM mutant consistent with extensive conformational changes. A point mutation of Ser794 to Ala794 (S794A) abolished the mobility shift of ADAM17, indicating that Ser794 phosphorylation is a prerequisite for PLK2-induced conformational changes. Previous studies have defined several phosphorylation sites in the ADAM17 cytoplasmic tail, the most prominent being Thr735 (13, 20, 55). ADAM17 cytoplasmic tail phosphorylation is evoked by a wide range of stimuli including the bacterial outer membrane component LPS (55). The MAPKs ERK and p38 have been defined as main regulators of LPS-induced ADAM17 activity in primary monocytes (55, 56). Here, we demonstrated that PLK2 was involved in serine phosphorylation of ADAM17, which represents to our knowledge the first report linking PLK biology to ADAM proteases. For the kinases PLK2 and PLK3, overlapping substrate specificity has been suggested because it was previously shown for α-synuclein, which represents a common PLK2/PLK3 substrate (57, 58). However, in our experiments ADAM17 interacted only with PLK2, and consequently PLK3 co-transfection did not induce a pronounced gel-mobility shift of ADAM17. Therefore, we suggest that ADAM17 is a specific substrate for PLK2. However, we cannot exclude that PLK3 might also functionally compensate in the absence of PLK2. As it was shown for other substrates of PLKs, ADAM17 binding is mediated by the C-terminal Polo box domains, which are classified as Ser(P)/Thr(P) binding modules (46, 59). In contrast to other phospho-dependent binding domains (WW domains, FHA, WD40), PBDs are unique to the Polo-like kinase family and play a crucial role in subcellular localization and substrate binding (60). We determined that PLK2 binding critically depends on the small amino acid stretch within the ADAM17 cytotail comprising Ser794–Ala810. Remarkably, this sequence did not contain the canonical PBD binding core sequence S(pT/pS)(P/X) described for protein-protein interaction, implying at least for PLK2, the existence of a noncanonical substrate recognition motif (44). Point mutation of Ser794 to Ala (S794A) dramatically reduced PLK2-dependent ADAM17 binding, suggesting that (i) PLK2 binds to previously primed, phosphorylates ADAM17 cytoplasmic tail, and further phosphorylates ADAM17 at multiple sites, which leads to stabilization of the complex, or (ii) ADAM17 serves as a PLK2 substrate even in the absence of phosphorylation.
Further investigations will reveal whether PLK2 binding to ADAM17 depends on an upstream “priming kinase” as was shown for the PLK2 substrate SPAR (spine-associated Rap GTPase-activating protein) (43). The impact of phosphorylation in regulating ADAM17 activity is still a matter of debate. Direct phosphorylation is supposed to be a molecular hallmark of ADAM17 activation. However, this hypothesis has been increasingly challenged by previous studies showing that short term activation of ADAM17 by several stimuli was completely independent of cytoplasmic phosphorylation (23, 61). In our experimental system, PLK inhibition interfered with constitutive (long term), as well as LPS-driven proteolysis of various ADAM17 substrates. However, PMA-induced ADAM17 activation was only mildly affected by BI 2356, demonstrating that distinct intracellular pathways control constitutive and stimulated shedding. We are currently investigating whether a direct interaction between ADAM17 and PLK2 is a prerequisite for the enhanced sheddase activity. In addition to direct post-translational modification via phosphorylation, it is possible that (unknown) ADAM17 regulators are direct targets of kinases, like PLK2, and mediate ADAM17 activation (62).
In conclusion, our results demonstrate that PLK2 is a novel intracellular binding partner for the major sheddase ADAM17. PLK2-dependent phosphorylation of ADAM17 at Ser794 led to a pronounced SDS gel mobility shift. Additionally, the global blockade of PLKs dampened TNFRI, TNFRII, and pro-TNFα release from the cell surface of primary macrophages and dendritic cells implicating a new pro-inflammatory function of PLKs. Our findings have clinical implications because inhibitors for PLKs are in clinical trials for cancer therapy, and in particular BI 2536 therapy resulted in clinical benefit in patient suffering from non-small cell lung, pancreatic, and prostate cancer (63, 64). Our study of PLK2-mediated ADAM17 phosphorylation expands the potential role of PLK2 beyond mitotic entry and cytokinesis to inflammatory diseases of the body.
Acknowledgments
We thank J. Blenis for providing the expression plasmid pcDNA3.1-ERK-T7, R. Davis for providing the expression plasmid pcDNA3.1-p38α-MAPK, J. Prox for providing the expression yeast plasmid pBTR3-ADAM10-Cub-LexA, I. Lorenzen for providing the expression plasmid ADAM17-PC, M. Müller for providing the expression plasmid ADAM17-C1, C. Desel for excellent advice in confocal microscopy, and A. Jeschke, M. Mannbar, D. Dohr, and E. Schulz for technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft (Grant SFB 877 for Projects A1, B8, and Z2; Grant SFB 841 for Project C1; Seneszenz und Autoimmunität Grant RA2404/1-1; and the Cluster of Excellence—“Inflammation at Interfaces”).
- PLK
- Polo-like kinase
- TNFR
- TNFα receptor
- PBD
- Polo box domain
- Cub
- C-terminal portion of ubiquitin
- Nub
- N-terminal portion of ubiquitin
- PMA
- phorbol 12-myristate 13-acetate
- BMDM
- bone marrow-derived macrophage
- BMDC
- bone marrow-derived dendritic cell
- PNGase F
- N-glycosidase F
- EndoH
- endoglycosidase H
- ACN
- acetonitrile
- mEF
- murine embryonic fibroblast
- IP
- immunoprecipitation.
REFERENCES
- 1. Fong K. P., Barry C., Tran A. N., Traxler E. A., Wannemacher K. M., Tang H. Y., Speicher K. D., Blair I. A., Speicher D. W., Grosser T., Brass L. F. (2011) Deciphering the human platelet sheddome. Blood 117, e15–e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Blobel C. P. (2005) ADAMs. Key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43 [DOI] [PubMed] [Google Scholar]
- 3. Saftig P., Reiss K. (2011) The “A Disintegrin And Metalloproteases” ADAM10 and ADAM17. Novel drug targets with therapeutic potential? Eur. J. Cell Biol. 90, 527–535 [DOI] [PubMed] [Google Scholar]
- 4. Black R. A., Rauch C. T., Kozlosky C. J., Peschon J. J., Slack J. L., Wolfson M. F., Castner B. J., Stocking K. L., Reddy P., Srinivasan S., Nelson N., Boiani N., Schooley K. A., Gerhart M., Davis R., Fitzner J. N., Johnson R. S., Paxton R. J., March C. J., Cerretti D. P. (1997) A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 385, 729–733 [DOI] [PubMed] [Google Scholar]
- 5. Moss M. L., Jin S. L., Milla M. E., Bickett D. M., Burkhart W., Carter H. L., Chen W. J., Clay W. C., Didsbury J. R., Hassler D., Hoffman C. R., Kost T. A., Lambert M. H., Leesnitzer M. A., McCauley P., McGeehan G., Mitchell J., Moyer M., Pahel G., Rocque W., Overton L. K., Schoenen F., Seaton T., Su J. L., Becherer J. D. (1997) Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 385, 733–736 [DOI] [PubMed] [Google Scholar]
- 6. Gooz M. (2010) ADAM-17. The enzyme that does it all. Crit. Rev. Biochem. Mol. Biol. 45, 146–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheller J., Chalaris A., Garbers C., Rose-John S. (2011) ADAM17. A molecular switch to control inflammation and tissue regeneration. Trends Immunol. 32, 380–387 [DOI] [PubMed] [Google Scholar]
- 8. Peschon J. J., Slack J. L., Reddy P., Stocking K. L., Sunnarborg S. W., Lee D. C., Russell W. E., Castner B. J., Johnson R. S., Fitzner J. N., Boyce R. W., Nelson N., Kozlosky C. J., Wolfson M. F., Rauch C. T., Cerretti D. P., Paxton R. J., March C. J., Black R. A. (1998) An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284 [DOI] [PubMed] [Google Scholar]
- 9. Horiuchi K., Kimura T., Miyamoto T., Takaishi H., Okada Y., Toyama Y., Blobel C. P. (2007) Cutting edge. TNF-α-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J. Immunol. 179, 2686–2689 [DOI] [PubMed] [Google Scholar]
- 10. Long C., Wang Y., Herrera A. H., Horiuchi K., Walcheck B. (2010) In vivo role of leukocyte ADAM17 in the inflammatory and host responses during E. coli-mediated peritonitis. J. Leukocyte Biol. 87, 1097–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yoda M., Kimura T., Tohmonda T., Morioka H., Matsumoto M., Okada Y., Toyama Y., Horiuchi K. (2013) Systemic overexpression of TNFα-converting enzyme does not lead to enhanced shedding activity in vivo. PLoS One 8, e54412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le Gall S. M., Maretzky T., Issuree P. D., Niu X. D., Reiss K., Saftig P., Khokha R., Lundell D., Blobel C. P. (2010) ADAM17 is regulated by a rapid and reversible mechanism that controls access to its catalytic site. J. Cell Sci. 123, 3913–3922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Soond S. M., Everson B., Riches D. W., Murphy G. (2005) ERK-mediated phosphorylation of Thr735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 118, 2371–2380 [DOI] [PubMed] [Google Scholar]
- 14. Xu P., Liu J., Sakaki-Yumoto M., Derynck R. (2012) TACE activation by MAPK-mediated regulation of cell surface dimerization and TIMP3 association. Sci. Signal. 5, ra34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Q., Thomas S. M., Lui V. W., Xi S., Siegfried J. M., Fan H., Smithgall T. E., Mills G. B., Grandis J. R. (2006) Phosphorylation of TNF-α converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc. Natl. Acad. Sci. U.S.A. 103, 6901–6906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chalaris A., Rabe B., Paliga K., Lange H., Laskay T., Fielding C. A., Jones S. A., Rose-John S., Scheller J. (2007) Apoptosis is a natural stimulus of IL6R shedding and contributes to the proinflammatory trans-signaling function of neutrophils. Blood 110, 1748–1755 [DOI] [PubMed] [Google Scholar]
- 17. Chanthaphavong R. S., Loughran P. A., Lee T. Y., Scott M. J., Billiar T. R. (2012) A role for cGMP in inducible nitric-oxide synthase (iNOS)-induced tumor necrosis factor (TNF) α-converting enzyme (TACE/ADAM17) activation, translocation, and TNF receptor 1 (TNFR1) shedding in hepatocytes. J. Biol. Chem. 287, 35887–35898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dang M., Armbruster N., Miller M. A., Cermeno E., Hartmann M., Bell G. W., Root D. E., Lauffenburger D. A., Lodish H. F., Herrlich A. (2013) Regulated ADAM17-dependent EGF family ligand release by substrate-selecting signaling pathways. Proc. Natl. Acad. Sci. U.S.A. 110, 9776–9781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Díaz-Rodríguez E., Montero J. C., Esparís-Ogando A., Yuste L., Pandiella A. (2002) Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735. A potential role in regulated shedding. Mol. Biol. Cell 13, 2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu P., Derynck R. (2010) Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 37, 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maretzky T., Zhou W., Huang X. Y., Blobel C. P. (2011) A transforming Src mutant increases the bioavailability of EGFR ligands via stimulation of the cell-surface metalloproteinase ADAM17. Oncogene 30, 611–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reddy P., Slack J. L., Davis R., Cerretti D. P., Kozlosky C. J., Blanton R. A., Shows D., Peschon J. J., Black R. A. (2000) Functional analysis of the domain structure of tumor necrosis factor-α converting enzyme. J. Biol. Chem. 275, 14608–14614 [DOI] [PubMed] [Google Scholar]
- 23. Schwarz J., Broder C., Helmstetter A., Schmidt S., Yan I., Müller M., Schmidt-Arras D., Becker-Pauly C., Koch-Nolte F., Mittrücker H. W., Rabe B., Rose-John S., Chalaris A. (2013) Short-term TNFα shedding is independent of cytoplasmic phosphorylation or furin cleavage of ADAM17. Biochim. Biophys. Acta 1833, 3355–3367 [DOI] [PubMed] [Google Scholar]
- 24. Clay F. J., McEwen S. J., Bertoncello I., Wilks A. F., Dunn A. R. (1993) Identification and cloning of a protein kinase-encoding mouse gene, Plk, related to the polo gene of Drosophila. Proc. Natl. Acad. Sci. U.S.A. 90, 4882–4886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sunkel C. E., Glover D. M. (1988) polo, a mitotic mutant of Drosophila displaying abnormal spindle poles. J. Cell Sci. 89, 25–38 [DOI] [PubMed] [Google Scholar]
- 26. Cheng K. Y., Lowe E. D., Sinclair J., Nigg E. A., Johnson L. N. (2003) The crystal structure of the human Polo-like kinase-1 Polo box domain and its phospho-peptide complex. EMBO J. 22, 5757–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. García-Alvarez B., de Cárcer G., Ibañez S., Bragado-Nilsson E., Montoya G. (2007) Molecular and structural basis of Polo-like kinase 1 substrate recognition. Implications in centrosomal localization. Proc. Natl. Acad. Sci. U.S.A. 104, 3107–3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kothe M., Kohls D., Low S., Coli R., Cheng A. C., Jacques S. L., Johnson T. L., Lewis C., Loh C., Nonomiya J., Sheils A. L., Verdries K. A., Wynn T. A., Kuhn C., Ding Y. H. (2007) Structure of the catalytic domain of human Polo-like kinase 1. Biochemistry 46, 5960–5971 [DOI] [PubMed] [Google Scholar]
- 29. Elia A. E., Rellos P., Haire L. F., Chao J. W., Ivins F. J., Hoepker K., Mohammad D., Cantley L. C., Smerdon S. J., Yaffe M. B. (2003) The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 115, 83–95 [DOI] [PubMed] [Google Scholar]
- 30. Evers D. M., Matta J. A., Hoe H. S., Zarkowsky D., Lee S. H., Isaac J. T., Pak D. T. (2010) Plk2 attachment to NSF induces homeostatic removal of GluA2 during chronic overexcitation. Nat. Neurosci. 13, 1199–1207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Barr F. A., Silljé H. H., Nigg E. A. (2004) Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5, 429–440 [DOI] [PubMed] [Google Scholar]
- 32. Petronczki M., Lénárt P., Peters J. M. (2008) Polo on the rise. From mitotic entry to cytokinesis with Plk1. Dev. Cell 14, 646–659 [DOI] [PubMed] [Google Scholar]
- 33. Shimizu-Yoshida Y., Sugiyama K., Rogounovitch T., Ohtsuru A., Namba H., Saenko V., Yamashita S. (2001) Radiation-inducible hSNK gene is transcriptionally regulated by p53 binding homology element in human thyroid cells. Biochem. Biophys. Res. Commun. 289, 491–498 [DOI] [PubMed] [Google Scholar]
- 34. Burns T. F., Fei P., Scata K. A., Dicker D. T., El-Deiry W. S. (2003) Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol. Cell Biol. 23, 5556–5571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chevrier N., Mertins P., Artyomov M. N., Shalek A. K., Iannacone M., Ciaccio M. F., Gat-Viks I., Tonti E., DeGrace M. M., Clauser K. R., Garber M., Eisenhaure T. M., Yosef N., Robinson J., Sutton A., Andersen M. S., Root D. E., von Andrian U., Jones R. B., Park H., Carr S. A., Regev A., Amit I., Hacohen N. (2011) Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell 147, 853–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rose-John S. (2013) ADAM17, shedding, TACE as therapeutic targets. Pharmacol. Res. 71C, 19–22 [DOI] [PubMed] [Google Scholar]
- 37. Suthaus J., Tillmann A., Lorenzen I., Bulanova E., Rose-John S., Scheller J. (2010) Forced homo- and heterodimerization of all gp130-type receptor complexes leads to constitutive ligand-independent signaling and cytokine-independent growth. Mol. Biol. Cell 21, 2797–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Inaba K., Inaba M., Romani N., Aya H., Deguchi M., Ikehara S., Muramatsu S., Steinman R. M. (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J. Exp. Med. 176, 1693–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klug K., Ehlers S., Uhlig S., Reiling N. (2011) Mitogen-activated protein kinases p38 and ERK1/2 regulated control of Mycobacterium avium replication in primary murine macrophages is independent of tumor necrosis factor-α and interleukin-10. Innate Immun. 17, 470–485 [DOI] [PubMed] [Google Scholar]
- 40. Cassonnet P., Rolloy C., Neveu G., Vidalain P. O., Chantier T., Pellet J., Jones L., Muller M., Demeret C., Gaud G., Vuillier F., Lotteau V., Tangy F., Favre M., Jacob Y. (2011) Benchmarking a luciferase complementation assay for detecting protein complexes. Nat. Meth. 8, 990–992 [DOI] [PubMed] [Google Scholar]
- 41. Lorenzen I., Lokau J., Düsterhöft S., Trad A., Garbers C., Scheller J., Rose-John S., Grötzinger J. (2012) The membrane-proximal domain of A Disintegrin and Metalloprotease 17 (ADAM17) is responsible for recognition of the interleukin-6 receptor and interleukin-1 receptor II. FEBS Lett. 586, 1093–1100 [DOI] [PubMed] [Google Scholar]
- 42. Lorenzen I., Trad A., Grötzinger J. (2011) Multimerisation of A disintegrin and metalloprotease protein-17 (ADAM17) is mediated by its EGF-like domain. Biochem. Biophys. Res. Commun. 415, 330–336 [DOI] [PubMed] [Google Scholar]
- 43. Seeburg D. P., Feliu-Mojer M., Gaiottino J., Pak D. T., Sheng M. (2008) Critical role of CDK5 and Polo-like kinase 2 in homeostatic synaptic plasticity during elevated activity. Neuron 58, 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Elia A. E., Cantley L. C., Yaffe M. B. (2003) Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 299, 1228–1231 [DOI] [PubMed] [Google Scholar]
- 45. Leonard J. D., Lin F., Milla M. E. (2005) Chaperone-like properties of the prodomain of TNFα-converting enzyme (TACE) and the functional role of its cysteine switch. Biochem. J. 387, 797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee K. J., Lee Y., Rozeboom A., Lee J. Y., Udagawa N., Hoe H. S., Pak D. T. (2011) Requirement for Plk2 in orchestrated ras and rap signaling, homeostatic structural plasticity, and memory. Neuron 69, 957–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Strebhardt K. (2010) Multifaceted Polo-like kinases. Drug targets and antitargets for cancer therapy. Nat. Rev. Drug Discov. 9, 643–660 [DOI] [PubMed] [Google Scholar]
- 48. Chalaris A., Adam N., Sina C., Rosenstiel P., Lehmann-Koch J., Schirmacher P., Hartmann D., Cichy J., Gavrilova O., Schreiber S., Jostock T., Matthews V., Häsler R., Becker C., Neurath M. F., Reiss K., Saftig P., Scheller J., Rose-John S. (2010) Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 207, 1617–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. La Marca R., Cerri F., Horiuchi K., Bachi A., Feltri M. L., Wrabetz L., Blobel C. P., Quattrini A., Salzer J. L., Taveggia C. (2011) TACE (ADAM17) inhibits Schwann cell myelination. Nat. Neurosci. 14, 857–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Archambault V., Glover D. M. (2009) Polo-like kinases. Conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 10, 265–275 [DOI] [PubMed] [Google Scholar]
- 51. Feldmann M., Maini S. R. (2008) Role of cytokines in rheumatoid arthritis. An education in pathophysiology and therapeutics. Immunol. Rev. 223, 7–19 [DOI] [PubMed] [Google Scholar]
- 52. Mohler K. M., Sleath P. R., Fitzner J. N., Cerretti D. P., Alderson M., Kerwar S. S., Torrance D. S., Otten-Evans C., Greenstreet T., Weerawarna K. (1994) Protection against a lethal dose of endotoxin by an inhibitor of tumour necrosis factor processing. Nature 370, 218–220 [DOI] [PubMed] [Google Scholar]
- 53. Schroder K., Tschopp J. (2010) The inflammasomes. Cell 140, 821–832 [DOI] [PubMed] [Google Scholar]
- 54. Ma S., Charron J., Erikson R. L. (2003) Role of Plk2 (Snk) in mouse development and cell proliferation. Mol. Cell Biol. 23, 6936–6943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rousseau S., Papoutsopoulou M., Symons A., Cook D., Lucocq J. M., Prescott A. R., O'Garra A., Ley S. C., Cohen P. (2008) TPL2-mediated activation of ERK1 and ERK2 regulates the processing of pre-TNF α in LPS-stimulated macrophages. J. Cell Sci. 121, 149–154 [DOI] [PubMed] [Google Scholar]
- 56. Scott A. J., O'Dea K. P., O'Callaghan D., Williams L., Dokpesi J. O., Tatton L., Handy J. M., Hogg P. J., Takata M. (2011) Reactive oxygen species and p38 mitogen-activated protein kinase mediate tumor necrosis factor α-converting enzyme (TACE/ADAM-17) activation in primary human monocytes. J. Biol. Chem. 286, 35466–35476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mbefo M. K., Paleologou K. E., Boucharaba A., Oueslati A., Schell H., Fournier M., Olschewski D., Yin G., Zweckstetter M., Masliah E., Kahle P. J., Hirling H., Lashuel H. A. (2010) Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 285, 2807–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Salvi M., Trashi E., Cozza G., Franchin C., Arrigoni G., Pinna L. A. (2012) Investigation on PLK2 and PLK3 substrate recognition. Biochim. Biophys. Acta 1824, 1366–1373 [DOI] [PubMed] [Google Scholar]
- 59. van de Weerdt B. C., Littler D. R., Klompmaker R., Huseinovic A., Fish A., Perrakis A., Medema R. H. (2008) Polo-box domains confer target specificity to the Polo-like kinase family. Biochim. Biophys. Acta 1783, 1015–1022 [DOI] [PubMed] [Google Scholar]
- 60. Lee K. S., Grenfell T. Z., Yarm F. R., Erikson R. L. (1998) Mutation of the Polo-box disrupts localization and mitotic functions of the mammalian Polo kinase Plk. Proc. Natl. Acad. Sci. U.S.A. 95, 9301–9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hall K. C., Blobel C. P. (2012) Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735. PLoS One 7, e31600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Maretzky T., McIlwain D. R., Issuree P. D., Li X., Malapeira J., Amin S., Lang P. A., Mak T. W., Blobel C. P. (2013) iRhom2 controls the substrate selectivity of stimulated ADAM17-dependent ectodomain shedding. Proc. Natl. Acad. Sci. U.S.A. 110, 11433–11438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mross K., Frost A., Steinbild S., Hedbom S., Rentschler J., Kaiser R., Rouyrre N., Trommeshauser D., Hoesl C. E., Munzert G. (2008) Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 26, 5511–5517 [DOI] [PubMed] [Google Scholar]
- 64. Steegmaier M., Hoffmann M., Baum A., Lénárt P., Petronczki M., Krssák M., Gürtler U., Garin-Chesa P., Lieb S., Quant J., Grauert M., Adolf G. R., Kraut N., Peters J. M., Rettig W. J. (2007) BI 2536, a potent and selective inhibitor of Polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 17, 316–322 [DOI] [PubMed] [Google Scholar]