

Background: The role of homodimerization in the family of C2a-capped HAD phosphatases is unknown.
Results: Chronophin homodimerization is required for proper positioning of the substrate specificity loop and for substrate dephosphorylation.
Conclusion: The specificity of chronophin is allosterically controlled by a homophilic intermolecular interaction.
Significance: Our results reveal a general principle of how HAD hydrolase dimerization can contribute to substrate specificity.
Keywords: Enzyme Mechanisms, Enzyme Structure, Pyridoxal Phosphate, Serine/Threonine Protein Phosphatase, X-ray Crystallography, Haloacid Dehalogenase Phosphatase, Phosphatase Dimerization, Pyridoxal Phosphatase, Substrate Specificity
Abstract
Mammalian phosphatases of the haloacid dehalogenase (HAD) superfamily have emerged as important regulators of physiology and disease. Many of these enzymes are stable homodimers; however, the role of their dimerization is largely unknown. Here, we explore the function of the obligatory homodimerization of chronophin, a mammalian HAD phosphatase known to dephosphorylate pyridoxal 5′-phosphate (PLP) and serine/threonine-phosphorylated proteins. The exchange of two residues in the murine chronophin homodimerization interface (chronophinA194K,A195K) yields a constitutive monomer both in vitro and in cells. The catalytic activity of monomeric chronophin toward PLP is strongly impaired. X-ray crystallographic studies of chronophinA194K,A195K revealed that dimer formation is essential for an intermolecular arginine-arginine-tryptophan stacking interaction that positions a critical histidine residue in the substrate specificity loop of chronophin for PLP coordination. Analysis of all available crystal structures of HAD hydrolases that are grouped together with chronophin in the C2a-type structural subfamily uncovered a highly conserved mode of dimerization that results in intermolecular contacts involving the substrate specificity loop. Our results explain how the dimerization of HAD hydrolases contributes to their catalytic efficiency and substrate specificity.
Introduction
Enzymes of the haloacid dehalogenase (HAD)4-type constitute a large and ancient superfamily whose members are present in all three kingdoms of life. The majority of HAD enzymes are phosphatases known to cover an exceptionally broad substrate space, ranging from metabolites to macromolecules such as DNA and serine/threonine (Ser/Thr)- or tyrosine (Tyr)-phosphorylated proteins (1–3). A number of HAD phosphatases have been causally linked to human diseases, including cancer and cardiovascular, metabolic, and neurological disorders (4); however, very little is currently known about the regulation of these enzymes.
Contrasting their structurally highly diverse substrates, HAD phosphatases are remarkably similar in terms of topology and active site architecture even though their overall amino acid sequence identities are very low (3). A canonical, modified Rossmann fold positions the catalytic core residues that are distributed over four HAD motifs. The first aspartate in the strictly conserved DXDX(V/T) HAD phosphatase signature motif serves as the nucleophile and phosphoryl group acceptor that forms a phosphoaspartate intermediate during catalysis. This aspartate also coordinates the catalytically essential Mg2+ ion (1).
HAD phosphatases are additionally equipped with so-called cap domains. Unlike the structurally stereotypical buildup of the catalytic domain, caps are highly diversified modules that can be grouped into four classes, C0, C1, C2a, or C2b, according to their size, structure, and insertion site in the core domain (3). A primary cap function is to mediate solvent occlusion/inclusion during the catalytic cycle. In general, C1/C2-capped HAD phosphatases process small metabolites that can be sequestered within the active site by cap closure, thus ensuring efficient dephosphorylation. In contrast, macromolecules themselves can provide the necessary active site shielding and are preferentially processed by C0 (“capless”) phosphatases. Besides contributing to catalytic efficiency, caps supply substrate specificity determinants with residues that engage in substrate recognition and thereby establish phosphatase specificity (5–8). Interestingly, caps can also mediate the commonly found HAD phosphatase oligomerization: among the 20 structurally characterized human HAD phosphatases alone, 10 are oligomeric, and in four of these phosphatases, oligomerization is mediated by cap-cap interactions (4).
Chronophin is a homodimeric, C2a-capped HAD phosphatase (9–11) known to target Ser3-phosphocofilin (12–16) and the Ser/Thr-phosphorylated steroid receptor co-activators 1 and 3 (17). In addition to its emerging protein phosphatase activities, chronophin (gene name, pyridoxal phosphatase (PDXP)) acts as a small molecule phosphatase dedicated to the metabolism of pyridoxal 5′-phosphate (PLP) (10, 11). PLP is the biologically active form of vitamin B6 that functions as a cofactor in the catalysis of >160 different enzymatic reactions, in particular in the biosynthesis of neurotransmitters (18, 19). Chronophin has been suggested to link pathological alterations in vitamin B6 metabolism with cofilin-dependent actin dynamics in the rat hippocampus following status epilepticus (20, 21).
In the present study, we show that chronophin homodimerization is a prerequisite for its proper enzymatic function as a PLP phosphatase. These findings can be extended to dimeric C2a-capped HAD hydrolases in general and indicate a paradigmatic role for their dimerization in substrate recognition and thus in the control of catalytic efficiency and substrate specificity.
EXPERIMENTAL PROCEDURES
Database Searches
The Protein Data Bank was searched for structures of haloacid dehalogenase-like hydrolases using the Pfam entries PF13419, PF00702, PF13344, PF13242, PF08282, and PF12710. The search was conducted with a 90% sequence identity cutoff to reduce the number of multiple entries and mutant proteins and yielded 177 unique entries. Cap domain subtypes were determined manually and cross-validated using a recently published data set (22). The PISA online tool (23) was used to determine the oligomeric state and for dimer interface calculations (see supplemental Table S1).
DNA Constructs
Murine chronophin cDNA was reverse transcribed from adult mouse brain tissue. Total RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's instructions, and cDNA was obtained with the High Fidelity RNA PCR kit (Takara Bio Inc.) and oligo(dT) primers. The PCR product was subcloned into the BamHI and EcoRI restriction sites of pcDNA3 (Invitrogen) to construct untagged chronophin, into the KpnI and XhoI sites of pENTR3C (Invitrogen) followed by insertion via homologous recombination into pDEST27 (Invitrogen) to produce GST-tagged chronophin for expression in mammalian cells, or into the bacterial expression vector pETM11 (European Molecular Biology Laboratory) to create N-terminally His6-tagged chronophin for in vitro studies. The chronophinA194K,A195K (chronophinKK) construct was generated by site-directed mutagenesis.
Protein Expression and Purification
His6-tagged chronophin wild type or chronophinKK in pETM11 was transformed into BL21(DE3) cells (Stratagene) and expressed for 18 h at 20 °C after induction with 0.5 mm isopropyl β-d-1-thiogalactopyranoside. To increase solubility, chronophin was co-expressed with chaperones from the pG-Tf2 plasmid (Takara Bio Inc.) according to the instructions of the manufacturer. Cells were harvested at 8,000 × g for 10 min and lysed in 100 mm triethanolamine (TEA), 500 mm NaCl, 20 mm imidazole, 5 mm MgCl2, pH 7.4 in the presence of protease inhibitors (EDTA-free protease inhibitor tablets, Roche Applied Science) and 150 units/ml DNase I (Applichem) using a cell disruptor (Microfluidizer Processor M-110 P, Microfluidics). Cell debris was removed by centrifugation for 30 min at 30,000 × g. For purification, cleared supernatants were loaded on a HisTrap HP column operated on an ÄKTA liquid chromatography system (GE Healthcare) in binding buffer (50 mm TEA, 500 mm NaCl, 20 mm imidazole, 5 mm MgCl2, pH 7.4), and His6-tagged proteins were eluted using a linear gradient up to 50% elution buffer (50 mm TEA, 250 mm NaCl, 500 mm imidazole, 5 mm MgCl2, pH 7.4). Fractions containing His6-tagged chronophin were pooled, and the His6 tag was cleaved with tobacco etch virus protease for 2 days at 4 °C. Subsequently, cleaved protein was separated from uncleaved protein and from the His6-tagged tobacco etch virus protease on a HisTrap HP column. Untagged chronophin was further purified on a HiLoad 16/60 Superdex 200 prep grade size exclusion chromatography column (GE Healthcare) in buffer A (50 mm TEA, 250 mm NaCl, 5 mm MgCl2, 5% (v/v) glycerol, pH 7.4).
Analytical Size Exclusion Chromatography
Globular proteins of known molecular weight (Gel Filtration LMW Calibration kit, GE Healthcare) were used to calibrate a Superdex 200 10/300 GL column (GE Healthcare), and blue dextran was used to determine the column void volume. Protein elution volumes were measured by monitoring the absorption at 280 nm. The elution volumes were used to calculate the partition coefficient (Kav) with the formula Kav = (Ve − V0)/(Vt −V0) where Ve is the elution volume, V0 is the void volume, and Vt is the total volume of the column. The apparent molecular weight was then derived from the inverse logarithm of the partition coefficient.
Analytical Ultracentrifugation
Sedimentation velocity analytical ultracentrifugation was carried out using a Beckman Optima XL-I analytical ultracentrifuge (Beckman Coulter) with an eight-hole An-50 Ti rotor at 40,000 rpm at 20 °C. Four hundred microliters of dialyzed, purified recombinant protein dialyzed against buffer A without glycerol and reference buffer solution were loaded in standard double-sector charcoal-filled Epon centerpieces equipped with sapphire windows. Protein concentration corresponded to an A280 of 0.5–0.8. Data were collected in continuous mode at a step size of 0.003 cm using absorption optical detection at a wavelength of 280 nm. Data were analyzed using the NIH software SEDFIT to determine continuous distributions for solutions to the Lamm equation c(s) as described previously (24). Analysis was performed with regularization at confidence levels of 0.68 and floating frictional ratio (f/f0 ∼ 1.32 ± 0.02 for both chronophin wild type and mutant, suggesting a globular conformation), time-independent noise, baseline, and meniscus position to root mean square deviation (r.m.s.d.) values between 0.007 and 0.012. Consistent results were obtained in three independent experiments.
Atomic Force Microscopy (AFM)
Proteins were diluted between 30- and 100-fold from 30 μm stock solutions in AFM deposition buffer (25 mm HEPES, 50 mm KCl, 10 mm MgCl2, pH 7.5), immediately deposited onto freshly cleaved mica, rinsed with deionized water, and dried in a gentle stream of nitrogen. All images were collected on an MFP-3D-BIO atomic force microscope (Asylum Research) in oscillating mode using Olympus OMCL-AC240 silicon probes with spring constants of ∼2 newtons/m and resonance frequencies of ∼70 kHz. Images were captured using Asylum Research software on Igor Pro at a scan size of 2 × 2 μm2, a scan rate of 0.5 Hz, and a resolution of 1024 × 1024 pixels. For analysis, AFM images were flattened to third order. AFM volumes were measured using the NIH Image-based software ImageSXM and used to calculate protein molecular weights as described (25) with the formula V = 1.2 × (MW) − 5.9 where V is the AFM volume and MW is the molecular weight. Molecular weights (and error ranges) were derived from the center positions (and two standard deviations) of Gaussian fits with R2 > 0.79 or 0.96 for chronophinWT or chronophinKK, respectively) to the distributions of measured volumes using the software Origin (OriginLab, version 8.6). Consistent results were obtained from triplicate experiments for both chronophinWT and chronophinKK.
Isothermal Titration Calorimetry (ITC)
ITC experiments were performed on a MicroCal ITC200 microcalorimeter (GE Healthcare) at 25 °C and were analyzed using MicroCal Origin software. Prior to all ITC experiments, protein samples were extensively dialyzed overnight at 4 °C against filtered and degassed buffer A. Each titration experiment consisted of 2.5-μl injections of 300–600 μm BeF3− diluted in buffer A into the 280-μl sample cell containing 25–75 μm chronophin. Heats of dilution measurements were carried out as mentioned above by injecting BeF3− into buffer A. For each experiment, the binding enthalpy was directly measured, whereas the stoichiometry (N) and the dissociation constant (Kd) were obtained using the analysis software, assuming a single site binding model.
Crystallization and Data Collection
Proteins were concentrated to 8–10 mg/ml (as determined by absorption at 280 nm using a calculated molar extinction coefficient of 18,450 m−1·cm−1) in buffer A (chronophinKK) or in 10 mm TEA, 100 mm NaCl, 1 mm MgCl2 (chronophinWT) using 10,000 molecular weight cutoff centrifugal filter devices (Amicon Ultra-15, Millipore). All crystals were grown at 20 °C using the hanging drop vapor diffusion method by mixing equal volumes of protein solution with reservoir solution. ChronophinKK crystals were grown in 0.1 m MES at pH 6.5 with 25% (w/v) polyethylene glycol monomethyl ether 550 and appeared as thin plates after 3–4 days. ChronophinWT was crystallized in 0.1 m imidazole, 0.2 m NaCl, 1 m sodium tartrate supplemented with 1 mm BeF3− to obtain the chronophinWT-BeF3− structure, and cubic crystals appeared within 24 h. All crystals were cryoprotected for flash cooling in liquid nitrogen by soaking in mother liquor containing 30% (v/v) glycerol. All data sets were collected at beamline 14.1 (Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BESSY), Berlin, Germany). Data were processed using iMOSFLM (26) and Scala from the CCP4 program suite (27). All three structures were solved by molecular replacement with the program Phaser (28) using human pyridoxal 5′-phosphate phosphatase/chronophin (Protein Data Bank code 2OYC) as a search model. The chronophinWT/chronophinWT-BeF3− structures were refined at 2.2-Å resolution, and the chronophinKK structure was refined at 1.75-Å resolution with Phenix (29), incorporating torsion angle non-crystallographic symmetry restraints. Structural representations were generated with PyMOL (The PyMOL Molecular Graphics System, version 1.5.0.4, Schrödinger, LLC). PyMOL was also used to determine the r.m.s.d. of structural alignments.
In Vitro Phosphatase Assays
PLP dephosphorylation assays were conducted in 96-well microtiter plates. ChronophinWT or chronophinKK (100 nm/well) was preincubated for 10 min at 22 °C in buffer A supplemented with 0.001% (v/v) Triton X-100. The reactions were started by the addition of PLP (final concentrations ranging from 0 to 1,000 μm in a total volume of 50 μl) and stopped after 2 min by the addition of 100 μl of Biomol Green (Enzo Life Sciences). Color was allowed to develop for 10 min before the absorbance of the resulting phosphomolybdate complex was read at 620 nm on an Envision 2104 multilabel microplate reader (PerkinElmer Life Sciences). Free phosphate release was quantified using phosphate standard curves, and Vmax and Km values were calculated using GraphPad Prism version 6 (GraphPad Software Inc.). The lines were fitted by nonlinear regression using the least square fitting method.
Cell Transfection and Pulldown Assays
HEK293-AD cells (Stratagene) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) fetal calf serum (FCS), 2 mm l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were plated at a density of 4.5 × 105 cells in 6-well plates and transfected the next day using TransIT-LT1 (Mirus) with a total amount of 1 μg of DNA. The amount of transfected DNA was kept constant with empty vector. Cells were lysed in 50 mm TEA, 250 mm NaCl, 5 mm MgCl2, 1% (v/v) Triton X-100, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 50 μg/ml aprotinin, 1 mm 4-(2-aminoethyl)benzenesulfonyl fluoride (Pefabloc) for 15 min at 4 °C under constant rotation. After centrifugation at 15,000 × g for 15 min at 4 °C, the lysates were incubated for 1.5 h under constant rotation with glutathione-Sepharose 4B beads (GE Healthcare) pre-equilibrated with washing buffer (50 mm TEA, 250 mm NaCl, 5 mm MgCl2, 1% (v/v) Triton X-100). The beads were washed four times with washing buffer using a vacuum manifold, bead-associated proteins were eluted with Laemmli sample buffer, and eluates were separated by SDS-PAGE and blotted onto nitrocellulose membranes (Hybond C, Amersham Biosciences). Chronophin dimerization was assessed by Western blot analysis with chronophin-specific antibodies (clone C85E3, Cell Signaling Technology).
Accession Codes
The x-ray crystal structures of murine chronophinWT and murine chronophinKK have been deposited in the Protein Data Bank under accession codes 4BX3 (murine chronophin), 4BX2 (murine chronophin in complex with BeF3−), and 4BX0 (murine chronophinA194K,A195K).
Statistical Analysis
Pulldown and ITC experiments were analyzed with the two-tailed unpaired t test using GraphPad Prism version 6.
RESULTS
Homooligomer Formation Is a Common Feature of C2a-type HAD Hydrolases
We analyzed a data set comprising all available HAD hydrolase structures from various species (see “Experimental Procedures” for details). Of the 177 unique entries, 104 HAD hydrolases (∼59%) are likely to form homooligomers as compared with 72 structures that are assigned as monomers and with one heterooligomer. As shown in Table 1, C2a-type enzymes are particularly noteworthy because all available structures represent dimers (12 entries) or tetramers (five entries). However, the function of HAD hydrolase dimerization is unclear as all catalytic core residues are encoded in a single polypeptide chain, and the available structures of oligomeric HAD hydrolases show that the adjacent protomer does not contribute active site residues. To understand the role of HAD dimerization, we investigated mammalian chronophin as a representative C2a-type family member.
TABLE 1.
Oligomeric states of structurally characterized HAD-type hydrolases
The numbers of structures per structural subfamily and per oligomeric state are indicated. The corresponding Protein Data Bank entries and protein information can be found in supplemental Table S1.
| Structural HAD subfamily |
Structures/oligomeric state | |||||
|---|---|---|---|---|---|---|
| C0 | C1 | C1 + C2 | C2a | C2b | ||
| Monomers | 11 | 40 | 6 | − | 15 | 72 |
| Dimers | 3 | 43 | 6 | 12 | 11 | 75 |
| Trimers | − | 2 | − | − | − | 2 |
| Tetramers | 10 | 4 | − | 5 | 1 | 20 |
| Hexamers | − | 1 | − | − | − | 1 |
| Octamers | 4 | 1 | − | − | − | 5 |
| Dodecamers | − | 1 | − | − | − | 1 |
| Heterodimers | − | 1 | − | − | − | 1 |
| Structures/subfamily | 28 | 93 | 12 | 17 | 27 | 177 |
Crystallization and Structure Determination of Murine Chronophin
We determined the three-dimensional structure of murine chronophin by x-ray crystallography and refined it by molecular replacement with human chronophin (Protein Data Bank code 2OYC and Ref. 9) to 2.2-Å resolution with an Rwork of 17.1% and an Rfree of 20.7% (Protein Data Bank code 4BX3). Data collection and refinement statistics are given in Table 2. The alignment of human and murine chronophin structures (Protein Data Bank codes 2OYC and 4BX3) shows an almost perfect superposition with a r.m.s.d. of 0.61 Å determined for the Cα atoms of residues 1–290. Both orthologs crystallize as homodimers via the cap domain as shown for murine chronophin in Fig. 1A (see also Ref. 9). The cap consists of five central parallel β-strands that are connected by helices, and dimerization is mainly mediated by helix α6 following β-strand 3, by a β-hairpin (referred to as substrate specificity loop) inserted after β-strand 4, and by the ensuing helix α7. This substrate specificity loop also acts as a roof for the entrance to the active site.
TABLE 2.
Data collection and refinement statistics
r.m.s., root mean square.
| ChronophinWT | ChronophinWT-BeF3− | ChronophinKK | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.91841 | 0.91841 | 0.91841 |
| Space group | I23 | I23 | P2 |
| Unit cell parameters | |||
| a, b, c (Å) | 167.10, 167.10, 167.10 | 166.83, 166.83, 166.83 | 36.13, 91.82, 39.19 |
| α, β, γ (°) | 90.00, 90.00, 90.00 | 90.00, 90.00, 90.00 | 90.00, 98.28, 90.00 |
| Resolution range (Å)a | 44.66–2.19 (2.31–2.19) | 44.59–2.19 (2.31–2.19) | 33.32–1.75 (1.84–1.75) |
| Rsymb | 0.082 (0.858) | 0.116 (1.12) | 0.093 (0.658) |
| Rp.i.mc | 0.034 (0.358) | 0.048 (0.472) | 0.049 (0.347) |
| I/σId | 12.9 (2.2) | 10.2 (1.8) | 10.2 (2.3) |
| Completeness (%) | 99.9 (99.7) | 100 (100) | 99.8 (99.9) |
| Multiplicity | 6.8 (6.7) | 6.7 (6.6) | 3.5 (3.5) |
| Total reflections | 269,875 | 266,023 | 89,076 |
| Unique reflections | 39,743 (5,736) | 39,582 (5,743) | 25,494 (3,709) |
| Refinement | |||
| Wilson B-factor (Å2) | 47.0 | 41.1 | 19.1 |
| Average B-factor (Å2) | 48.4 | 45.5 | 28.6 |
| Macromolecules | 48.7 | 45.8 | 28.0 |
| Solvent | 40.6 | 39.2 | 34.2 |
| Rcryste | 0.1707 | 0.1678 | 0.1913 |
| Rfreee | 0.2067 | 0.2123 | 0.2363 |
| Number of non-hydrogen atoms | 4,645 | 4,676 | 2,496 |
| Macromolecules | 4,453 | 4,460 | 2,247 |
| Ligands | 14 | 28 | 7 |
| Water | 178 | 188 | 242 |
| r.m.s. deviations in | |||
| Bond lengths (Å) | 0.015 | 0.014 | 0.005 |
| Bond angles (°) | 1.640 | 1.567 | 1.006 |
| Planar groups (Å) | 0.010 | 0.010 | 0.004 |
| Dihedral angles (°) | 15.91 | 15.31 | 13.14 |
| Coordinate error (Å)f | 0.25 | 0.27 | 0.28 |
| Ramachandran statisticsg | |||
| Favored (%) | 96.92 | 98.29 | 99.32 |
| Allowed (%) | 2.22 | 1.54 | 0.34 |
| Outliers (%) | 0.85 | 0.17 | 0.34 |
| MolProbity clashscoreh | 9.83 | 11.83 | 11.63 |
a Numbers in parentheses refer to the respective highest resolution data shell in the data set.
b Rsym = ΣhklΣi|Ii − I|/ΣhklΣiIi where Ii is the ith measurement, and I is the weighted mean of all measurements of I.
c Rp.i.m. = Σhkl(1/(n − 1))1/2Σi|Ii − I|/ΣhklΣiIi where n is the multiplicity of the observed reflection.
d Indicates the average of the intensity divided by its S.D. value.
e Rcryst = Σ|Fo − Fc|/Σ|Fo| where Fo and Fc are the observed and calculated structure factor amplitudes. Rfree, same as Rcryst for 5% of the data randomly omitted from the refinement.
f Estimated coordinate error based on Rfree.
g Ramachandran statistics indicate the fraction of residues in the favored, allowed, and disallowed regions of the Ramachandran diagram as defined by MolProbity (54).
h Number of serious clashes per 1,000 atoms (54).
FIGURE 1.
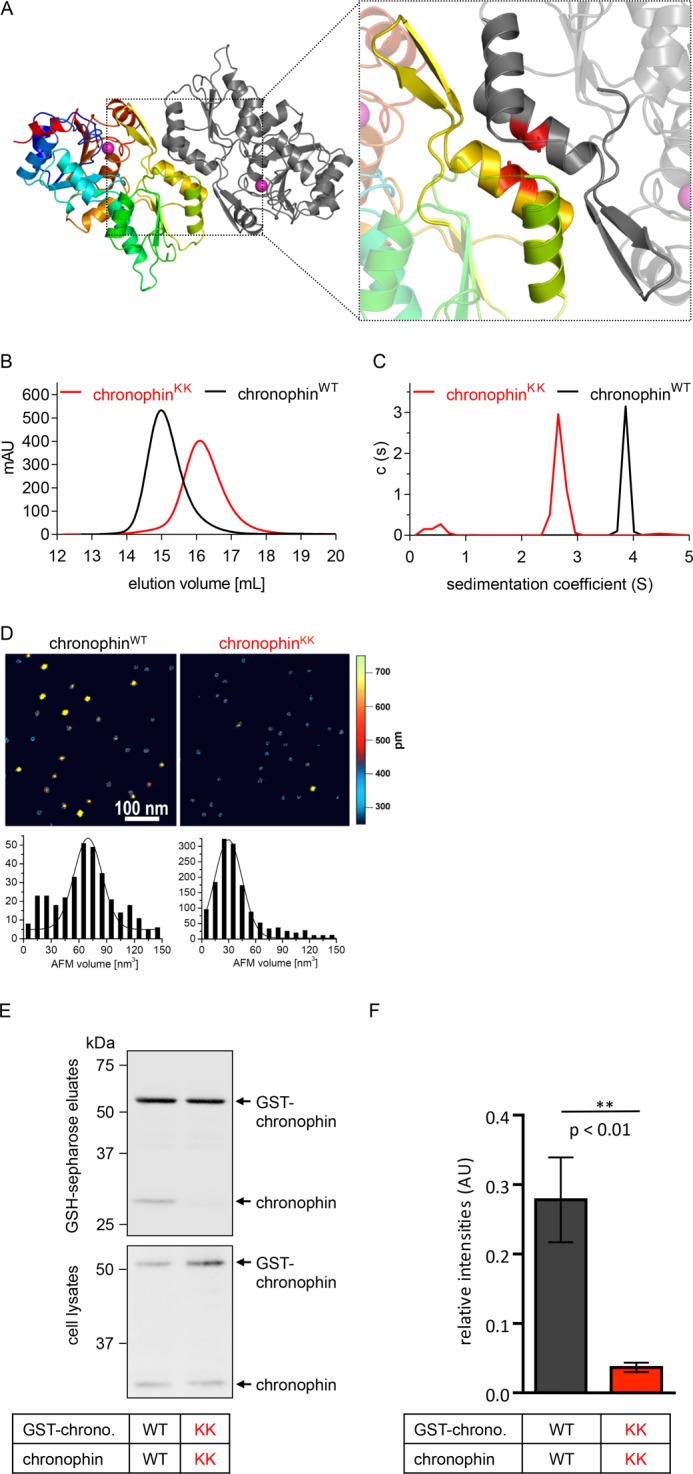
Characterization of chronophinKK. A, ribbon diagram of the dimeric murine chronophin structure (Protein Data Bank code 4BX3). One protomer is shown in rainbow colors from the N terminus (blue) to the C terminus (red); the other protomer is represented in gray. The cofactor Mg2+ (magenta-colored sphere) indicates the active site; the catalytic domain is shown in blue/cyan and orange/red. Dimerization is mediated by the capping domain (green/yellow). The enlarged area highlights helices α6 (green)/α7 (yellow) and the substrate specificity loop (yellow) that form the dimerization interface of chronophin. Ala194 and Ala195 in helix α7 that are mutated to lysines in chronophinKK are depicted as red sticks in both monomers. B, analytical size exclusion chromatography shows a peak elution volume of chronophinWT (theoretical molecular mass, 31.8 kDa) at 14.9 ml, corresponding to a calculated molecular mass of 60.7 kDa. ChronophinKK is detected at a peak elution volume of 16.1 ml, corresponding to a calculated molecular mass of 33.7 kDa. C, analytical ultracentrifugation data demonstrate different sedimentation coefficients for chronophinWT and chronophinKK that correspond to molecular masses of ∼56 and ∼32 kDa, respectively. D, atomic force microscopy images of chronophinWT and chronophinKK. Top, images are 500 × 500 nm with a height scale of 0.75 nm. Bottom, Gaussian fits to statistical volume distributions for chronophinWT and chronophinKK give maxima of 70 nm3 (337 particles) and 30 nm3 (1,409 particles), respectively, which translate into a molecular masses of 63 and 30 kDa, respectively. A fraction of chronophinWT also shows protein volumes consistent with a monomeric state. E, GST pulldown experiments. GST-tagged chronophinWT/chronophinKK (GST-chrono. WT/GST-chrono. KK) and untagged chronophinWT/chronophinKK (chronophin WT/chronophin KK) were co-expressed in HEK293-AD cells as indicated. GST-chronophin was precipitated with glutathione-Sepharose beads, bead-associated proteins were separated by SDS-PAGE, and chronophin was detected with a chronophin-specific antibody by immunoblotting. Western blots of the corresponding whole cell lysates are shown to assess protein expression levels in cells. F, densitometric quantification of Western blots. The band corresponding to untagged chronophin was normalized to the GST-chronophin signal in each lane and compared with the optical density of the signal corresponding to chronophinWT precipitated with GST-chronophinWT. The mean values ±S.E. (error bars) of the relative intensities are shown (n = 4). mAU, milliabsorbance units; AU, absorbance units.
Creation of a Monomeric Murine Chronophin Variant
To study the functional relevance of chronophin homodimerization, we exchanged the short, uncharged side chains of Ala194 and Ala195 in the murine chronophin dimerization interface (see Fig. 1A, inset) for the longer and charged side chains of Lys (chronophinKK) and compared the oligomeric states of recombinantly expressed, purified chronophinWT and chronophinKK by size exclusion chromatography. Fig. 1B shows that chronophin (molecular mass, 31.8 kDa) has a peak elution volume that corresponds to a calculated molecular mass of 60.7 kDa, indicating that the protein indeed forms a stable dimer in solution. In contrast, chronophinKK has a peak elution volume corresponding to a calculated molecular mass of 33.7 kDa, equivalent to chronophin in a monomeric state. Analytical ultracentrifugation sedimentation velocity experiments confirmed a monomeric molecular mass for chronophinKK (∼32 kDa), whereas wild type chronophin showed a sedimentation behavior consistent with a dimeric state (∼56 kDa) (Fig. 1C). Furthermore, in AFM imaging experiments, chronophinKK showed protein volumes than are consistent with a monomeric state of this variant (30 ± 13 kDa for chronophinKK compared with 63 ± 18 kDa for chronophinWT) (Fig. 1D).
To determine the oligomeric states of chronophinWT and chronophinKK in mammalian cells, we simultaneously expressed GST-tagged or untagged versions of chronophinWT and chronophinKK in HEK293 cells, performed pulldown binding experiments with glutathione-Sepharose beads, and subsequently probed bead-associated chronophin by Western blotting. The expectation is that GST-chronophinWT will pull down untagged chronophinWT, whereas the monomerized chronophinKK will prevent co-precipitation. Fig. 1E demonstrates that GST-chronophinWT (detectable at ∼60 kDa after separation of the bead eluates by SDS-PAGE and immunoblotting with α-chronophin antibodies) indeed associated with chronophinWT (detectable at ∼32 kDa) in pulldown binding assays, whereas an interaction of GST-chronophinKK with chronophinKK was not detectable. The results of four independent experiments are quantified in Fig. 1F. Together, these data clearly show that chronophinKK is a monomer both in vitro and in mammalian cells.
Enzymatic Properties of Monomeric Chronophin
We next investigated the effect of chronophin monomerization on its enzymatic activity toward PLP. Fig. 2 demonstrates that whereas homodimeric chronophin efficiently dephosphorylates PLP the activity of the monomeric chronophinKK variant is strongly impaired. This is mainly due to a ∼65-fold increase in Km, whereas the calculated Vmax remains largely unaffected. As a result, the catalytic efficiency (kcat/Km) of chronophinKK is reduced to ∼3.5% compared with chronophinWT. The catalytic constants of chronophinWT and chronophinKK toward PLP are summarized in Table 3.
FIGURE 2.
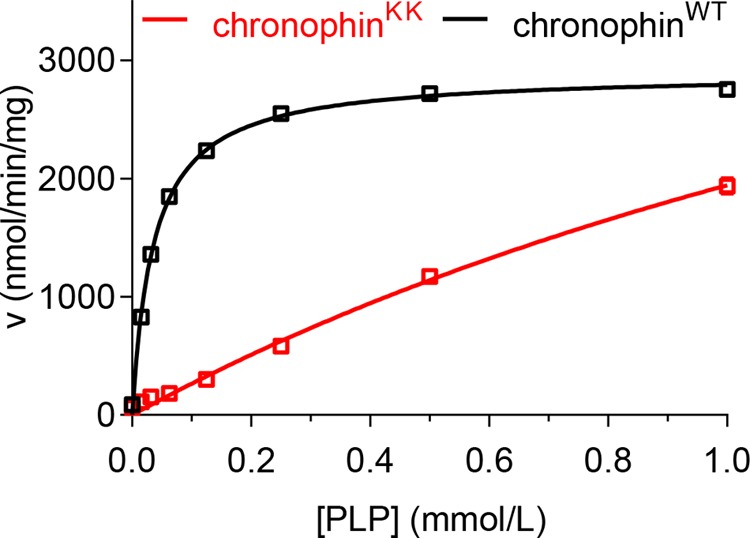
Analysis of chronophinKK phosphatase activity. In vitro PLP phosphatase assays were performed in 96-well microtiter plates in a total assay volume of 50 μl using recombinantly expressed, purified chronophinWT or chronophinKK (0.16 μg of protein/well) and 0–1 mm PLP. The reaction was stopped with malachite green, and released inorganic phosphate was determined by measuring A620. The enzyme velocities (v) toward increasing PLP concentrations are shown. Results represent mean values ± S.E. of three independent experiments performed with three independently purified batches of proteins. Note that most error bars are masked by the symbols.
TABLE 3.
Kinetic constants of chronophinWT and chronophinKK toward PLP
PLP dephosphorylation was measured in 96-well microtiter plates in a total assay volume of 50 μl using recombinantly expressed, purified chronophinWT or chronophinKK (0.16 μg of protein/well) and 0–1 mm PLP. The reaction was stopped with malachite green, and released inorganic phosphate was determined by measuring A620. The data are mean values ± S.E. of three independent experiments performed with three independently purified batches of proteins. Km, Michaelis-Menten constant; kcat, turnover number; kcat/Km, specificity constant. The kcat values were calculated from the maximum enzyme velocities using a molecular mass of 31.828 kDa for chronophinWT and 31.942 kDa for chronophinKK.
| ChronophinWT | ChronophinKK | |
|---|---|---|
| Km (μm) | 36 ± 1.2 | 2,387 ± 492 |
| kcat (s−1) | 1.5 ± 0.01 | 3.5 ± 0.6 |
| kcat/Km (s−1·m−1) | 4.3 × 104 | 0.2 × 104 |
Isothermal Titration Calorimetry of BeF3− Binding to ChronophinWT and ChronophinKK
To test whether the impaired catalytic efficiency of chronophinKK is due to rearrangements in catalytic core residues, we measured the binding of chronophin to BeF3−. BeF3− structurally mimics the phosphoaspartate transition state of HAD phosphatases by coordinating the catalytic core residues and the catalytically essential Mg2+ as a phosphate analog (30–36). Fig. 3 shows that the interaction of BeF3− with chronophinWT and chronophinKK can be optimally fitted to a one-site binding model. The stoichiometry of BeF3− binding is ∼0.85 for chronophinWT and chronophinKK, indicating an equimolar interaction in both cases (Table 4). The apparent deviation from the 1:1 binding ratio is likely due to protein precipitation issues during the experiment. Thus, chronophinWT and chronophinKK bind BeF3− with a comparable stoichiometry. Surprisingly, however, the BeF3− binding constant of chronophinKK is about 3-fold higher than that of chronophinWT. Furthermore, the quantity of released heat (ΔH), the entropic contribution (ΔS) upon BeF3− binding, and the resulting free Gibbs free energy (ΔG) values also differ significantly between chronophinWT and chronophinKK (Table 4). Together, the in vitro phosphatase and ITC experiments suggest that the chronophin homodimer interface in the cap domain exerts allosteric effects on the catalytic cleft of the enzyme.
FIGURE 3.
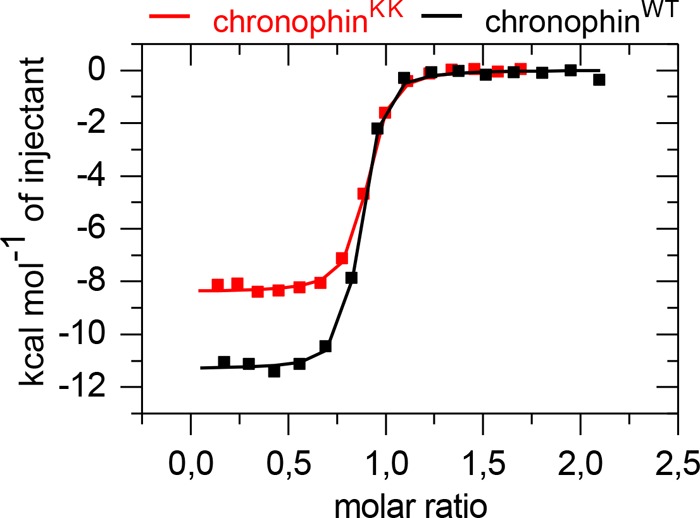
Isothermal titration calorimetry-derived binding isotherms of BeF3− titrated into chronophinWT (■) or chronophinKK (■). Measured binding enthalpies are plotted as a function of the molar ratio of BeF3− to the respective chronophin variant. Results are representative of three independent experiments performed with three independently purified batches of proteins.
TABLE 4.
Thermodynamic profiles of BeF3− binding to chronophinWT or chronophinKK
The data are mean values ± S.E. of three independent isothermal titration calorimetry experiments performed with three independently purified batches of proteins. ΔH, enthalpy; T·ΔS, entropy; N, stoichiometry; ΔG, Gibbs free energy; Kd, dissociation constant; ND, not determined.
| ChronophinWT | ChronophinKK | p value | |
|---|---|---|---|
| ΔH (kcal/mol) | −11.92 ± 0.33 | −8.72 ± 0.20 | 0.0011 |
| T·ΔS (kcal/mol) | −2.37 ± 0.23 | 0.23 ± 0.18 | 0.0009 |
| N | 0.87 ± 0.03 | 0.84 ± 0.01 | ND |
| ΔG (kcal/mol) | −9.55 ± 0.20 | −8.95 ± 0.02 | 0.0034 |
| Kd (nm) | 78 ± 13 | 215 ± 7 | 0.0008 |
Crystallization and Structure Determination of ChronophinKK
Therefore, we solved the structure of the monomerized chronophin variant. ChronophinKK crystallized in the space group P2 and could be refined to 1.75-Å resolution with an Rwork of 19.1% and an Rfree of 23.6% (Protein Data Bank code 4BX0 and Table 2). The r.m.s.d. of 0.47 Å for the Cα atoms of residues 1–290 between chronophinWT and chronophinKK clearly shows that the replacement of Ala194 and Ala195 with Lys residues in the cap domain had no impact on the overall fold of a chronophin protomer (Fig. 4A). However, the substrate specificity loop in chronophinKK is tilted by ∼25° compared with chronophinWT (measured between the Cα atoms of Asp182 and Pro187 of the respective molecules). The enlarged areas in Fig. 4A show that in chronophinWT residues Trp177 and Arg185 in the substrate specificity loop (Pro176–Pro187) of protomer A stack together with Arg163 of protomer B, which itself forms a hydrogen bond with the carbonyl oxygen atom in the backbone of Gly183 in protomer A. Importantly, the imidazole ring of His178 in the substrate specificity loop coordinates the pyridine ring of PLP by π-electron stacking (see Protein Data Bank code 2P69). In chronophinKK, the interprotomer contacts are lost, resulting in a tilting of the substrate specificity loop and consequently an altered orientation of the substrate-binding His178 residue (Fig. 4, A and B). These results demonstrate that contacts between the substrate specificity loop and the other protomer in the homodimer are crucial for the correct positioning of the substrate specificity loop and hence for the orientation of residues involved in substrate coordination.
FIGURE 4.
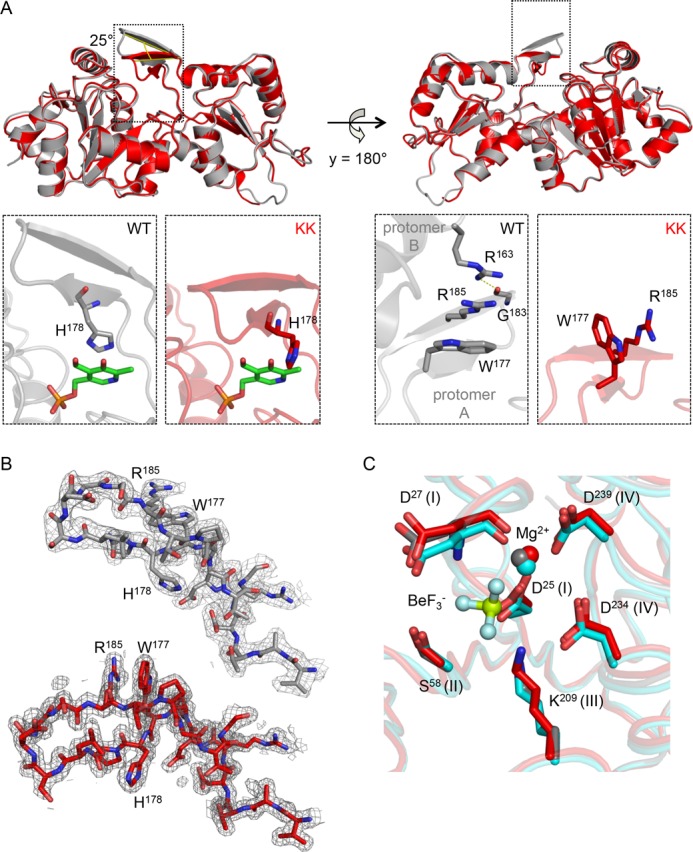
Structural comparison of murine chronophinWT and chronophinKK. A, upper left, both structures are shown as ribbon diagrams and are superimposed. ChronophinWT (Protein Data Bank code 4BX3) is in gray, and chronophinKK (Protein Data Bank code 4BX0) is in red. The substrate specificity loop in chronophinKK is tilted by ∼25° compared with chronophinWT as measured between the Cα atoms of Asp182 and Pro187 of the respective molecules. Lower left panels, detailed views show that in chronophinWT His178 of the substrate specificity loop coordinates the pyridine ring of PLP by π-electron stacking. PLP has been introduced from Protein Data Bank code 2P69 for visualization purposes. The altered substrate specificity loop positioning in chronophinKK causes a reorientation of His178 with its imidazole side chain now being almost perpendicular to the PLP pyridine ring. Right panels, back views of the structures. The detailed views in the lower panels reveal that in chronophinWT Trp177 and Arg185 in the substrate specificity loop of protomer A stack with Arg163 of protomer B (which is not shown in the upper panels). Arg163 additionally forms a hydrogen bond to the backbone carbonyl oxygen of Gly183 in the substrate specificity loop of protomer A. These interprotomer interactions are absent in monomeric chronophinKK. B, electron density maps of residues 170–190. Shown are the refined 2Fo − Fc electron density maps contoured at an r.m.s.d. of 1 overlaid on the models of chronophinWT (gray) and chronophinKK (red). C, superposition of the active sites of chronophinWT (gray; Protein Data Bank code 4BX3) and chronophinKK (red; Protein Data Bank code 4BX0). ChronophinWT is additionally shown in the BeF3−-liganded state (cyan; Protein Data Bank code 4BX2). Catalytically essential residues of the HAD motifs I–IV (roman numerals) are indicated.
To elucidate whether all catalytic core residues in monomeric chronophin are oriented correctly, we compared the HAD motif I–IV catalytic core residues of chronophinKK and chronophinWT with chronophinWT in complex with BeF3−. The chronophinWT-BeF3− structure was refined to 2.2-Å resolution with an Rwork of 16.8% and an Rfree of 21.2% (Protein Data Bank code 4BX2; see Table 2 for data collection and refinement statistics). (Note that we were unable to crystallize chronophinKK in complex with BeF3−. Co-crystallization attempts failed due to protein precipitation issues, and soaking experiments resulted in broken crystals). Fig. 4C shows that the positioning of these amino acids does not differ substantially between chronophinWT and chronophinKK and further confirms that the cofactor Mg2+ is properly coordinated. We conclude from these results that the impaired binding of BeF3− to monomeric chronophin (see Fig. 3 and Table 3) is not due to conformational changes in the active site residues of chronophinKK.
Together, the results of the steady-state enzyme kinetics, the ITC experiments, and the structural analyses support the conclusion that the core catalytic machinery of the monomeric chronophinKK variant is unaltered compared with homodimeric chronophin. However, the tilting of the substrate specificity loop and the subsequent reorientation of the PLP-binding His178 residue in the cap domain of the monomer lead to a marked increase in the Km value toward the substrate PLP. The reduced binding affinity (albeit identical binding stoichiometry) of chronophinKK to BeF3− compared with chronophinWT may thus indicate a diminished accessibility of the active site for BeF3− due to the tilted substrate specificity loop.
Potential General Role of Dimerization for the Function of C2-capped HAD Phosphatases
Are the mode and function of chronophin dimerization for the positioning of the substrate specificity loop/β-hairpin a unique feature of this phosphatase or a characteristic trait of C2a-capped HAD hydrolases in general? We addressed this question by performing a DALI structure similarity search with human chronophin (Protein Data Bank code 2OYC) as a search model. The results were manually curated to select members of the C2a subfamily of HAD hydrolases. In accordance with the results shown in Table 1, we found 16 structures in addition to human chronophin and the newly solved structure of murine chronophin. The r.m.s.d. values for structural alignments of all Cα atoms of these family members with murine chronophin indicate high structural homology, although the amino acid sequence identities between these proteins and murine chronophin are very low (ranging from 16.1 to 28.3% with the exception of human chronophin that is 91.2% identical with murine chronophin). PDBePISA analysis indicates that all 18 molecules are highly likely to exist as homodimers as judged by the buried surface areas of ∼1,000 Å2 at the respective dimer interfaces. Five of them possibly form tetramers (dimers of dimers). The C2a-capped HAD hydrolases, r.m.s.d. values, and results of the PDBePISA analysis are listed in Table 5.
TABLE 5.
Oligomeric states of structurally characterized C2a-type HAD phosphatases
r.m.s.d. values for structural alignments of all Cα atoms of family members with murine chronophin (Protein Data Bank code 4BX3) were calculated using PyMOL.
| UniProtKB (Protein Data Bank code) | Organism | r.m.s.d. compared with 4BX3 | Dimer interface (ΔiG)a,b |
|---|---|---|---|
| Å | Å2 (kcal/mol) | ||
| Q96GD0 (2OYC) | Homo sapiens | 0.67 | 1,036 (−16.4) |
| P60487(4BX3) | Mus musculus | − | 992 (−14.7) |
| Q9H0R4 (3HLT) | H. sapiens | 2.47 | 1,315 (−13.9) |
| Q3UGR5 (2HO4) | M. musculus | 3.08 | 1,300 (−13.6) |
| Q9H008 (2X4D) | H. sapiens | 2.17 | 1,307 (−17.9) |
| P36151 (3RF6) | S. cerevisiae | 10.61 | 1,866 (−25.7) |
| O32125 (3PDW) | Bacillus subtilis | 2.69 | 1,203 (−15.3) |
| Q11S56 (2HX1) | Cytophaga hutchinsonii | 2.44 | 980 (−13.7) |
| ATCC 33406 | 1,273 (−5.2) | ||
| Q836C7 (1YV9) | Enterococcus faecalis V583 | 1.66 | 1,151 (−12.2) |
| P0AF24 (2C4N) | E. coli K-12 | 2.02 | 1,097 (−23.8) |
| 1,072 (−6.6) | |||
| L7N4Y2 (4I9F) | Mycobacterium tuberculosis | 2.31 | 1,146 (−19.6) |
| O59622 (1ZJJ) | Pyrococcus horikoshii OT3 | 1.78 | 1,162 (−18.9) |
| Q8E044 (3EPR) | Streptococcus agalactiae | 1.69 | 1,143 (−12.9) |
| 1,048 (−5.8) | |||
| Q8DTD6 (1WVI) | Streptococcus mutans UA159 | 1.94 | 1,157 (−15.3) |
| 955 (−4.3) | |||
| Q97Q24 (1YDF) | Streptococcus pneumoniae | 2.13 | 1,080 (−12.7) |
| Q99ZW4 (1YS9) | Streptococcus pyogenes | 1.67 | 1,185 (−14.1) |
| 868 (−4.8) | |||
| Q9X264 (1VJR) | Thermotoga maritima | 1.91 | 1,033 (−14.7) |
| O29873 (3QGM) | Archaeoglobus fulgidus | 1.71 | 1,315 (−23.8) |
a Buried surface areas and ΔiG were calculated using the PDBePISA online tool. ΔiG, solvation-free energy gain upon formation of the interface (not taking into account the effect of satisfied hydrogen bonds and salt bridges across the interface). Negative ΔiG values correspond to hydrophobic interfaces or to positive protein affinity (23).
b Values in italics correspond to potential tetramer interfaces.
Fig. 5 shows the dimer interfaces of the structurally characterized homodimeric C2a-type HAD hydrolases identified in this analysis. All dimer interfaces are composed of two homologous α-helices in the cap domains and contain a β-hairpin structure that resembles the substrate specificity loop in chronophin. We note that although hydrogen bonds are present in the corresponding loop structure of the uncharacterized Saccharomyces cerevisiae protein YKR070W (Protein Data Bank code 3RF6) and the Escherichia coli protein NagD (Protein Data Bank code 2C4N (7)), they are not shown as β-sheets by the automatic secondary structure assignment because the participating residues are not adjacent to each other in the sequence. Nevertheless, the corresponding loop structure also participates in interprotomer contacts and may likewise also contribute to substrate specificity. Therefore, dimerization appears to be a general prerequisite for the proper positioning of the β-hairpin substrate specificity loop (or of corresponding elements) in all C2a-capped HAD hydrolases structurally characterized to date.
FIGURE 5.
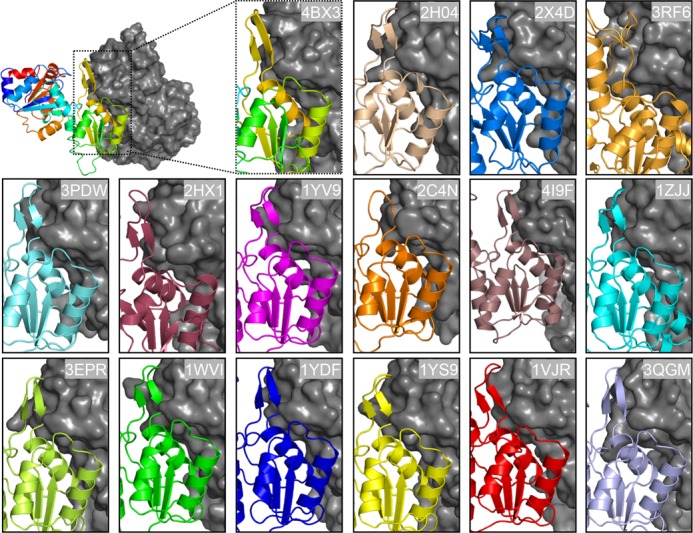
Homodimerization interfaces in C2a-type HAD hydrolases. Protomer A is shown in a colored ribbon representation, whereas protomer B is shown in gray surface representation. Protein Data Bank codes are indicated. The structures of human pyridoxal 5′-phosphate phosphatase/chronophin (Protein Data Bank code 2OYC) and human HDHD2 (Protein Data Bank code 3HLT) are very similar to the murine orthologs (Protein Data Bank codes 3BX4 and 2HO4) and are therefore not shown. All dimer interfaces are composed of two homologous helices and contain a β-hairpin structure that resembles the substrate specificity loop in chronophin. Although hydrogen bonds are present in the loops of Protein Data Bank codes 2C4N and 3RF6, they are not shown as β-sheets by the automatic secondary structure assignment because the participating residues are not adjacent to each other in the sequence.
DISCUSSION
Substrate specificity in capped HAD phosphatases is accomplished by cap movements that facilitate active site solvent exclusion and by specificity determinants encoded in the cap domain. We identify homodimerization via C2a-type caps as a previously unrecognized factor involved in substrate specificity control. We demonstrate that the constitutive homodimerization of chronophin is a prerequisite for the proper positioning of the substrate specificity loop and consequently for efficient PLP dephosphorylation. Thus, the specificity of chronophin toward PLP as a substrate depends on an allosteric effect induced by a homophilic intermolecular interaction. Crystal structure analyses of oligomeric C2a-type HAD hydrolases from different species reveal that the positioning of the β-hairpin/presumable substrate specificity loop via homodimerization is a conserved and common feature in this structural subfamily. Our results therefore suggest a general principle of how the dimerization of C2a-capped HAD hydrolases can contribute to substrate specificity.
Although the role of homodimerization in the C2a family has not been investigated so far, the relevance of oligomerization for C1- and C0-type HAD hydrolase functions was addressed in previous studies. For example, homodimerization of the C1-type Haemophilus influenzae P4 acid phosphatase is important for catalysis because side chains of one protomer stabilize the conformation of catalytic loop IV in the active site of the other protomer. Such intersubunit contacts that reach into the catalytic site may also be important for substrate recognition (37). The C1-capped mammalian cytosolic 5′-nucleotidase (cN-II) exists as a tetramer in its native form. Although a dimeric mutant of this protein is still active, monomeric cN-II is inactive, suggesting that enzymatic activity may be controlled by switching the oligomeric state, although the underlying mechanism is unknown (38, 39). Structural analysis of the tetrameric, C0-type (capless) HAD phosphatase KdsC from E. coli has revealed that protomers can act as cap surrogates to shield the active site of an adjacent protomer and to supply residues involved in substrate recognition (40). A similar tetrameric organization was reported for the Bacteroides thetaiotaomicron KDN-9-P phosphatase (41). Homooligomeric interfaces may also contribute to proper folding or stability of the active site as has been proposed for the bifunctional C0-type T4 polynucleotide kinase/phosphatase PNKP (42). Taken together, the constitutive dimer formation of oligomeric HAD phosphatases appears to be generally required for appropriate catalytic activity and can also contribute to substrate coordination. Our finding that homodimerization is necessary for the proper orientation of the β-hairpin/substrate specificity loop in the C2a-capped HAD phosphatase chronophin further advances our mechanistic understanding of HAD phosphatase specificity control.
Self-association of proteins to form dimers or higher order oligomers is frequently observed (43). Oligomerization provides a simple way to increase protein complexity and can result in structural (e.g. improved stability) and functional advantages (e.g. control over active site accessibility and specificity) (44). In some oligomeric proteins, protomers must be stably assembled to build a functional protein; this may be the case for the constitutively oligomeric HAD phosphatases characterized to date. Some “classical,” non-HAD-type phosphatases such as E. coli alkaline phosphatase are also known to form obligate homodimers and to lose structural stability and catalytic activity upon monomerization (45). In other homooligomeric proteins, the association and dissociation of protomers is reversible and can serve as a (concentration-sensing) mechanism to regulate enzyme activity (43, 46–48). Some receptor-like protein-tyrosine phosphatases utilize this mechanism of dynamic regulation. For example, protein-tyrosine phosphatase α, CD45, and SAP-1 are reversibly inhibited by dimer formation, which can be controlled by ligand binding (49), phosphorylation (50), or oxidative stress (51). Here, dimerization leads to a reciprocal occlusion of the catalytic site of each phosphatase protomer (52, 53). It is currently unknown whether those HAD phosphatases that are assigned as monomeric can associate to form homo- or heterooligomers in a stimulus-dependent manner in cells. Conversely, it is unclear whether oligomeric C2a-type HAD hydrolases such as chronophin can also be present in a monomeric form in vivo, for example below a threshold concentration, in particular subcellular compartments, or upon posttranslational modification.
Acknowledgments
We thank Anna-Karina Lamprecht for technical assistance and the BESSY staff at beamline BL14.1.
This work was supported in part by Deutsche Forschungsgemeinschaft Grants SFB688 (to A. G.) and FZ82 (to A. G., H. S., and I. T.).

This article contains supplemental Table S1.
The atomic coordinates and structure factors (codes 4BX3, 4BX2, and 4BX0) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HAD
- haloacid dehalogenase
- AFM
- atomic force microscopy
- ITC
- isothermal titration calorimetry
- PLP
- pyridoxal 5′-phosphate
- r.m.s.d.
- root mean square deviation
- chronophinKK
- chronophinA194K,A195K
- TEA
- triethanolamine
- cN-II
- cytosolic 5′-nucleotidase.
REFERENCES
- 1. Allen K. N., Dunaway-Mariano D. (2009) Markers of fitness in a successful enzyme superfamily. Curr. Opin. Struct. Biol. 19, 658–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aravind L., Koonin E. V. (1998) The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 23, 469–472 [DOI] [PubMed] [Google Scholar]
- 3. Burroughs A. M., Allen K. N., Dunaway-Mariano D., Aravind L. (2006) Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 361, 1003–1034 [DOI] [PubMed] [Google Scholar]
- 4. Seifried A., Schultz J., Gohla A. (2013) Human HAD phosphatases: structure, mechanism, and roles in health and disease. FEBS J. 280, 549–571 [DOI] [PubMed] [Google Scholar]
- 5. Lahiri S. D., Zhang G., Dunaway-Mariano D., Allen K. N. (2006) Diversification of function in the haloacid dehalogenase enzyme superfamily: the role of the cap domain in hydrolytic phosphoruscarbon bond cleavage. Bioorg. Chem. 34, 394–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lu Z., Dunaway-Mariano D., Allen K. N. (2005) HAD superfamily phosphotransferase substrate diversification: structure and function analysis of HAD subclass IIB sugar phosphatase BT4131. Biochemistry 44, 8684–8696 [DOI] [PubMed] [Google Scholar]
- 7. Tremblay L. W., Dunaway-Mariano D., Allen K. N. (2006) Structure and activity analyses of Escherichia coli K-12 NagD provide insight into the evolution of biochemical function in the haloalkanoic acid dehalogenase superfamily. Biochemistry 45, 1183–1193 [DOI] [PubMed] [Google Scholar]
- 8. Walldén K., Rinaldo-Matthis A., Ruzzenente B., Rampazzo C., Bianchi V., Nordlund P. (2007) Crystal structures of human and murine deoxyribonucleotidases: insights into recognition of substrates and nucleotide analogues. Biochemistry 46, 13809–13818 [DOI] [PubMed] [Google Scholar]
- 9. Almo S. C., Bonanno J. B., Sauder J. M., Emtage S., Dilorenzo T. P., Malashkevich V., Wasserman S. R., Swaminathan S., Eswaramoorthy S., Agarwal R., Kumaran D., Madegowda M., Ragumani S., Patskovsky Y., Alvarado J., Ramagopal U. A., Faber-Barata J., Chance M. R., Sali A., Fiser A., Zhang Z. Y., Lawrence D. S., Burley S. K. (2007) Structural genomics of protein phosphatases. J. Struct. Funct. Genomics 8, 121–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fonda M. L. (1992) Purification and characterization of vitamin B6-phosphate phosphatase from human erythrocytes. J. Biol. Chem. 267, 15978–15983 [PubMed] [Google Scholar]
- 11. Jang Y. M., Kim D. W., Kang T. C., Won M. H., Baek N. I., Moon B. J., Choi S. Y., Kwon O. S. (2003) Human pyridoxal phosphatase. Molecular cloning, functional expression, and tissue distribution. J. Biol. Chem. 278, 50040–50046 [DOI] [PubMed] [Google Scholar]
- 12. Gohla A., Birkenfeld J., Bokoch G. M. (2005) Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat. Cell Biol. 7, 21–29 [DOI] [PubMed] [Google Scholar]
- 13. Zoudilova M., Kumar P., Ge L., Wang P., Bokoch G. M., DeFea K. A. (2007) β-Arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J. Biol. Chem. 282, 20634–20646 [DOI] [PubMed] [Google Scholar]
- 14. Sun C. X., Magalhães M. A., Glogauer M. (2007) Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J. Cell Biol. 179, 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang T. Y., Minamide L. S., Bamburg J. R., Bokoch G. M. (2008) Chronophin mediates an ATP-sensing mechanism for cofilin dephosphorylation and neuronal cofilin-actin rod formation. Dev. Cell 15, 691–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ivanovska J., Tregubova A., Mahadevan V., Chakilam S., Gandesiri M., Benderska N., Ettle B., Hartmann A., Söder S., Ziesché E., Fischer T., Lautscham L., Fabry B., Segerer G., Gohla A., Schneider-Stock R. (2013) Identification of DAPK as a scaffold protein for the LIMK/cofilin complex in TNF-induced apoptosis. Int. J. Biochem. Cell Biol. 45, 1720–1729 [DOI] [PubMed] [Google Scholar]
- 17. Li C., Liang Y. Y., Feng X. H., Tsai S. Y., Tsai M. J., O'Malley B. W. (2008) Essential phosphatases and a phospho-degron are critical for regulation of SRC-3/AIB1 coactivator function and turnover. Mol. Cell 31, 835–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowling F. G. (2011) Pyridoxine supply in human development. Semin. Cell Dev. Biol. 22, 611–618 [DOI] [PubMed] [Google Scholar]
- 19. di Salvo M. L., Safo M. K., Contestabile R. (2012) Biomedical aspects of pyridoxal 5′-phosphate availability. Front. Biosci. 4, 897–913 [DOI] [PubMed] [Google Scholar]
- 20. Kim J. E., Kim D. W., Kwak S. E., Kwon O. S., Choi S. Y., Kang T. C. (2008) Potential role of pyridoxal-5′-phosphate phosphatase/chronopin in epilepsy. Exp. Neurol. 211, 128–140 [DOI] [PubMed] [Google Scholar]
- 21. Kim J. E., Ryu H. J., Kim M. J., Kim D. W., Kwon O. S., Choi S. Y., Kang T. C. (2010) Pyridoxal-5′-phosphate phosphatase/chronophin induces astroglial apoptosis via actin-depolymerizing factor/cofilin system in the rat brain following status epilepticus. Glia 58, 1937–1948 [DOI] [PubMed] [Google Scholar]
- 22. Pandya C., Brown S., Pieper U., Sali A., Dunaway-Mariano D., Babbitt P. C., Xia Y., Allen K. N. (2013) Consequences of domain insertion on sequence-structure divergence in a superfold. Proc. Natl. Acad. Sci. U.S.A. 110, E3381–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 24. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roth H. M., Römer J., Grundler V., Van Houten B., Kisker C., Tessmer I. (2012) XPB helicase regulates DNA incision by the Thermoplasma acidophilum endonuclease Bax1. DNA Repair 11, 286–293 [DOI] [PubMed] [Google Scholar]
- 26. Battye T. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 28. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Afonine P. V., Grosse-Kunstleve R. W., Adams P. D., Lunin V. Y., Urzhumtsev A. (2007) On macromolecular refinement at subatomic resolution with interatomic scatterers. Acta Crystallogr. D Biol. Crystallogr. 63, 1194–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cho H., Wang W., Kim R., Yokota H., Damo S., Kim S. H., Wemmer D., Kustu S., Yan D. (2001) BeF3− acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF3− complex with phosphoserine phosphatase. Proc. Natl. Acad. Sci. U.S.A 98, 8525–8530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roberts A., Lee S. Y., McCullagh E., Silversmith R. E., Wemmer D. E. (2005) YbiV from Escherichia coli K12 is a HAD phosphatase. Proteins 58, 790–801 [DOI] [PubMed] [Google Scholar]
- 32. Wang W., Cho H. S., Kim R., Jancarik J., Yokota H., Nguyen H. H., Grigoriev I. V., Wemmer D. E., Kim S. H. (2002) Structural characterization of the reaction pathway in phosphoserine phosphatase: crystallographic “snapshots” of intermediate states. J. Mol. Biol. 319, 421–431 [DOI] [PubMed] [Google Scholar]
- 33. Rinaldo-Matthis A., Rampazzo C., Reichard P., Bianchi V., Nordlund P. (2002) Crystal structure of a human mitochondrial deoxyribonucleotidase. Nat. Struct. Biol. 9, 779–787 [DOI] [PubMed] [Google Scholar]
- 34. Jung S. K., Jeong D. G., Chung S. J., Kim J. H., Park B. C., Tonks N. K., Ryu S. E., Kim S. J. (2010) Crystal structure of ED-Eya2: insight into dual roles as a protein tyrosine phosphatase and a transcription factor. FASEB J. 24, 560–569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ghosh A., Shuman S., Lima C. D. (2008) The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol. Cell 32, 478–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim Y., Gentry M. S., Harris T. E., Wiley S. E., Lawrence J. C., Jr., Dixon J. E. (2007) A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc. Natl. Acad. Sci. U.S.A 104, 6596–6601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Felts R. L., Ou Z., Reilly T. J., Tanner J. J. (2007) Structure of recombinant Haemophilus influenzae e (P4) acid phosphatase reveals a new member of the haloacid dehalogenase superfamily. Biochemistry 46, 11110–11119 [DOI] [PubMed] [Google Scholar]
- 38. Spychala J., Chen V., Oka J., Mitchell B. S. (1999) ATP and phosphate reciprocally affect subunit association of human recombinant High Km 5′-nucleotidase. Role for the C-terminal polyglutamic acid tract in subunit association and catalytic activity. Eur. J. Biochem. 259, 851–858 [DOI] [PubMed] [Google Scholar]
- 39. Pesi R., Allegrini S., Careddu M. G., Filoni D. N., Camici M., Tozzi M. G. (2010) Active and regulatory sites of cytosolic 5′-nucleotidase. FEBS J. 277, 4863–4872 [DOI] [PubMed] [Google Scholar]
- 40. Biswas T., Yi L., Aggarwal P., Wu J., Rubin J. R., Stuckey J. A., Woodard R. W., Tsodikov O. V. (2009) The tail of KdsC: conformational changes control the activity of a haloacid dehalogenase superfamily phosphatase. J. Biol. Chem. 284, 30594–30603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lu Z., Wang L., Dunaway-Mariano D., Allen K. N. (2009) Structure-function analysis of 2-keto-3-deoxy-d-glycero-d-galactonononate-9-phosphate phosphatase defines specificity elements in type C0 haloalkanoate dehalogenase family members. J. Biol. Chem. 284, 1224–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu H., Smith P., Wang L. K., Shuman S. (2007) Structure-function analysis of the 3′ phosphatase component of T4 polynucleotide kinase/phosphatase. Virology 366, 126–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Marianayagam N. J., Sunde M., Matthews J. M. (2004) The power of two: protein dimerization in biology. Trends Biochem. Sci. 29, 618–625 [DOI] [PubMed] [Google Scholar]
- 44. Neet K. E., Timm D. E. (1994) Conformational stability of dimeric proteins: quantitative studies by equilibrium denaturation. Protein Sci. 3, 2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Boulanger R. R., Jr., Kantrowitz E. R. (2003) Characterization of a monomeric Escherichia coli alkaline phosphatase formed upon a single amino acid substitution. J. Biol. Chem. 278, 23497–23501 [DOI] [PubMed] [Google Scholar]
- 46. Hashimoto K., Nishi H., Bryant S., Panchenko A. R. (2011) Caught in self-interaction: evolutionary and functional mechanisms of protein homooligomerization. Phys. Biol. 8, 035007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mei G., Di Venere A., Rosato N., Finazzi-Agrò A. (2005) The importance of being dimeric. FEBS J. 272, 16–27 [DOI] [PubMed] [Google Scholar]
- 48. Mazurek S. (2011) Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int. J. Biochem. Cell Biol. 43, 969–980 [DOI] [PubMed] [Google Scholar]
- 49. Tonks N. K. (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7, 833–846 [DOI] [PubMed] [Google Scholar]
- 50. Bilwes A. M., den Hertog J., Hunter T., Noel J. P. (1996) Structural basis for inhibition of receptor protein-tyrosine phosphatase-α by dimerization. Nature 382, 555–559 [DOI] [PubMed] [Google Scholar]
- 51. van der Wijk T., Blanchetot C., Overvoorde J., den Hertog J. (2003) Redox-regulated rotational coupling of receptor protein-tyrosine phosphatase α dimers. J. Biol. Chem. 278, 13968–13974 [DOI] [PubMed] [Google Scholar]
- 52. Chin C. N., Sachs J. N., Engelman D. M. (2005) Transmembrane homodimerization of receptor-like protein tyrosine phosphatases. FEBS Lett. 579, 3855–3858 [DOI] [PubMed] [Google Scholar]
- 53. Wälchli S., Espanel X., Hooft van Huijsduijnen R. (2005) Sap-1/PTPRH activity is regulated by reversible dimerization. Biochem. Biophys. Res. Commun. 331, 497–502 [DOI] [PubMed] [Google Scholar]
- 54. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]