

Abstract
Human immunodeficiency virus (HIV) afflicts an estimated 30 million people globally, making it a continuing pandemic. Despite major research efforts, the rate of new infections has remained relatively static over time. This article reviews an emerging strategy for the treatment of HIV, the inhibition of the co-receptors necessary for HIV entry, CCR5 and CXCR4. The aim of this article is to highlight potential therapeutics derived from peptides and proteins that show particular promise in HIV treatment. Molecules that act on CCR5, CXCR4 or on both receptors will be discussed herein.
Keywords: HIV, CCR5, CXCR4
Introduction
Human immunodeficiency virus (HIV) remains a global pandemic despite monumental advances in the treatment and management of acute infections. The severity of the problem is underscored by the estimated two million new infections annually, and the approximately 30 million people living with HIV.1 The urgent nature of the pandemic has led to the development and implementation of therapeutics targeting all phases of the HIV life cycle. The infection cycle of HIV is characterized by five steps: entry, reverse transcription, integration, assembly and budding.2 All steps of infection have served as therapeutic targets; however, the majority of drugs target the reverse transcription step of the HIV life cycle. These are characterized by their mechanism of action as either nucleotide or non-nucleotide reverse transcriptase inhibitors. Additionally, therapies have been approved that inhibit HIV integrase, protease processing and the viral fusion process. An underdeveloped class of anti-HIV compounds is that targeting the viral entry process, especially that targeted to the co-receptors necessary for viral fusion. There are currently two FDA approved entry/fusion inhibitors, Fuzeon and Maraviroc, which bind to gp41 on the viral surface and CCR5 on the cell surface, respectively.3,4
In the early 1990s several research groups discovered that HIV required two components for infection: CD4 as a targeting factor and an, as then, unidentified co-factor for entry. These factors were subsequently elucidated as either CXCR45 or CCR5,6–8 part of a growing family of C-C and CXC-motif chemokine receptors (Figure 1). The critical nature of this co-receptor in HIV infection was further confirmed by the observation of human populations homozygous for a deletion mutation in CCR5, who had a high resistance to HIV infection.9–11 To date, no human population with a corresponding CXCR4 mutation has been found. The identification of these co-receptors led to an explosion in research surrounding their role in infection and the opportunity to interfere with infection using drugs ranging from natural ligands and derivatives to small molecule therapeutics.
Figure 1.
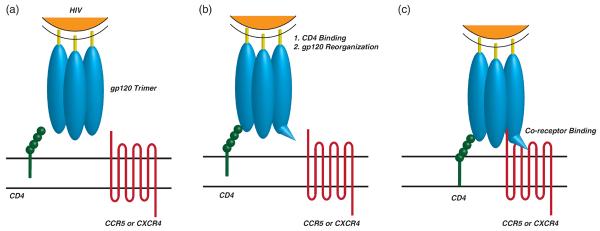
Initial steps in HIV entry. (a) Viral entry into cells is controlled by the envelope glycoproteins on the virus surface, gp41 (rods) and gp120 (ovals). The active complex consists of trimers of both gp120 and gp41. (b) HIV entry is initiated by binding of gp120 to CD4 on the cell surface. This binding event results in a rearrangement of the gp120 protein allowing it to bind to either CXCR4 or CCR5 as a co-receptor. (c) HIV binds to its cognate co-receptor either CCR5 or CXCR4. Co-receptor binding is a novel point of therapeutic intervention and the focus of this review article.
Depending on the co-receptor usage for infection, either CCR5 or CXCR4, HIV tropism can be defined as either R5 or X4, respectively. Typically, infection and early disease progression is dominated by R5 tropic viral strains, making inhibition of CCR5 a viable strategy for pre-exposure prophylaxis and treatment of new infections.12 Alternatively, X4 tropic strains are associated with negative patient outcomes and rapid transition from HIV to AIDS.13 It is unclear whether this relationship is correlative or causative. Nevertheless, CXCR4 inhibition is an important strategy moving forward for the treatment of late stage HIV infection or as a complementary microbicide for the rare X4 tropic infection. Extensive research has gone into the development of small molecules that act on both of these co-receptors, with one drug gaining FDA approval (Maraviroc) and others in late stage clinical trials. One concern of small molecule drugs though, is the propensity for the development of resistant HIV strains.14–17 Macromolecular co-receptor inhibitors, by contrast, require HIV to undergo more difficult and arduous evolutionary paths to either utilize the original receptor in a new way or to switch tropism, particularly in going from R5 to X4 receptor usage.18,19 The development of macromolecular inhibitors of these co-receptors, such as peptides and proteins, presents a potentially innovative means to develop new anti-HIV compounds. The small molecule inhibitors of HIV co-receptors have been extensively reviewed elsewhere, and thus will not be discussed further in this review.20,21 The present article will focus on protein- and peptide-based inhibitors of CCR5, CXCR4, and hybrid inhibitors thereof.
CCR5 inhibitors
As R5 tropic viruses are by far the most prevalent in person-to-person transmission and early stages of infection, inhibitors of the HIV:CCR5 interaction are critical to prevent the spread of the disease. The natural ligands for CCR5 were identified as RANTES, MIP-1α and MIP-1β.22 The first Anti-HIV therapeutic derived from one of these natural ligands, published only months after identification of CCR5 as co-receptor, was RANTES(9–68) which had a deletion of the first eight amino acids on the N-terminus.23 Several concerns of using natural chemokines were that these are inherently pro-inflammatory molecules, that early work showed native ligand had little ability to block infection8,24, and that activation of T-cells could actually lead to enhanced susceptibility to infection.25 One advantage of RANTES(9–68) was that it functioned as an antagonist, perhaps avoiding unwanted downstream pathway activation. However, its binding was lower than that of native RANTES, owing perhaps to the deletion of residues at the N-terminus which later work would show was critical for optimal CCR5 binding.26 Early work in the engineering of protein inhibitors of HIVentry was well reviewed by Hartley and Offord.27 Later work on the use of peptides to mimic parts of RANTES function was the subject of a mini-review by Vangelista et al..28 However, recent work on peptides discovered through phage display libraries and fully recombinant versions of RANTES analogues have yet to be reviewed and are covered herein.
The initial discovery that the N-terminus plays a critical role in RANTES binding to CCR5 was fortuitously found through studies of the recombinant version of the protein, produced in E. coli, which still retained its N-terminal methionine residue.29 The so-named Met-RANTES showed that a single residue could turn the agonist into an antagonist. It was speculated that the hydrophobic nature of the methionine residue played the greatest role in this conversion. To test this, semi-synthetic derivatives of RANTES were generated with chemical modifications of the N-terminus meant to mimic the hydrophobicity of methionine, but improve upon its activity as an HIV inhibitor. The first such derivative was named `Aminooxypentane' or AOP-RANTES.30,31 Further rounds of chemical optimization resulted in PSC-RANTES, an analogue 50 times more potent than AOP-RANTES.32 Use of PSC-RANTES as a topical microbicide was shown to be sufficient to protect against vaginal transmission of R5-tropic virus in a single dose in a macaque model.33 PSC-RANTES was further demonstrated to be capable of preventing viral replication in Langerhans cells. This represented a significant discovery as Langerhans cells are critical in early infection, but frequently thought to be CCR5 negative and thus resistant to CCR5 blocking strategies.34
While PSC-RANTES showed promise as an HIV entry inhibitor, it has two limitations. Firstly, as an agonist it still suffered limitations previously identified for RANTES agonists, namely inflammatory activation. Secondly, due to the fully synthetic route, production would always be costly, limiting its use in developing countries. To circumvent this, a panel of fully recombinant chemokine analogues was generated and tested for agonistic or antagonistic ability. Several rounds of optimization produced three noteworthy analogues: 6P4-RANTES is a strong agonist with prolonged intracellular CCR5 sequestration; 5P12-RANTES and 5P14-RANTES were both complete antagonists, with 5P12-RANTES showing no CCR5 internalization, and 5P14-RANTES only partial internalization.35 5P12-RANTES and 6P4-RANTES were further validated for stability in vaginal pH and in the presence of semen. Stability was only examined at 24 h, and with a cell fusion assay, but was beyond that of PSC-RANTES.36
An alternative route to generating economically viable versions of PSC-RANTES was the study of various semi-synthetic routes of drug production, where the protein could be produced by fermentation and the N-terminus modified by a one-step linkage. A couple viable candidates were identified with moderate activities, but none rivaled that of PSC-RANTES.37 Based on this work, other semisynthetic strategies could yield more successes, such as use of non-natural amino acids and `click' chemistry for high yield reactions.38–40
The first RANTES analogue that demonstrated receptor antagonist activity without receptor activation and internalization was discovered through a different means. Nardese et al.41 explored various N-terminal substitution mutants, the most memorable of which was a serineto-cysteine double mutant C1C5-RANTES, in which an intramolecular loop formed through a disulphide bridge. Antagonistic ability was ascribed to the profound structural change resulting from this N-terminal modification. Hartley et al.44 explored a different strategy, namely the use of phage display libraries to generate mutated N-terminal sequences for full-length RANTES analogues in an effort to identify analogues capable of CCR5 internalization but with antagonistic properties (i.e. failure to activate inflammation). Some putative candidates were identified, but did not warrant further exploration.
A number of other research groups examined the use of peptides, often short sequence fragments of RANTES, to mimic the binding function of RANTES but without activating the signaling of CCR5. Nardese et al.41 identified that the extracellular loops of CCR5 formed a hydrophobic patch crucial for receptor identification and binding. This was a critical discovery in the development of small molecule hydrophobic drugs, but was also crucial in the advancement of peptide mimics of RANTES binding. The sequence with the best identified activity corresponded to two hydrophobic stretches, one in the N-loop (aa 11–16) and one in the β1-strand (aa 27–29) of RANTES.41 The resulting peptide (RANTES aa 11–29) was demonstrated to have potent anti-HIV activity; however, initial modifications with hydrophobic residues did not result in higher efficacy.42 Although recent work, particularly in the use of tryptophan substitutions resulted in peptides with better activity and pharmacologically comparable to other peptides (e.g. T20 a gp41 binding peptide) and to maraviroc (the only FDA approved CCR5 blocking molecule).43
Phage display techniques, originally used to help optimize fully recombinant RANTES analogues,44 were later used to pan for peptides capable of mimicking RANTES binding without activation.45 Initial peptides identified showed modest results (~5 μM affinity) but neither agonistic nor antagonistic activity. Subsequent work used either cells expressing CCR546 or purified CCR5 and analogues47 in phage display selection. While a number of peptides have been identified with moderate enrichment, as yet these techniques have not yielded more viable candidates.
Patel et al.48 identified another research direction for identifying potential peptide based HIV entry inhibitors, namely repurposing a peptide identified in the blocking of other G-Protein Coupled Receptors, such as [D-Lys3]-Growth Hormone Releasing Peptide-6 (DLS) which is used as a selective ghrelin receptor antagonist.48 In these studies they showed it also had the capacity to block HIV binding to CCR5. However, a shortcoming of this approach was the non-specificity, and resultant interference with other ubiquitous GPCRs.
An advantage in using the full-length proteins over shorter peptides is the capacity to take advantage of other internal sequences in the protein. For example, RANTES, similar to many other cytokines, has been demonstrated to have internal glycosaminoglycan (GAG) binding sequences.49 Initial concern was that these sites, responsible for protein aggregation,50 would result in increased viral activation.51 However this has not been observed in vivo. Conversely, Wang et al. exploited the GAG binding capability in an advantageous fashion by using these molecular associations to control the rate of protein delivery, and to prolong the release of RANTES analogues beyond that of diffusion alone.52,53 This strategy is known as `affinity-based drug delivery'.54
Clearly, progress has been made in the development of macromolecular inhibitors of CCR5 with anti-HIV activity. The RANTES-derived proteins show the most promise, advancing to successful in vivo studies. To make translation of these molecules a clinical reality delivery options must be developed to make administration viable in resource-limited settings, most likely as preexposure prophylactic microbicides. Due to high production cost, peptide or protein availability can be augmented through polymer conjugation (such as was demonstrated by PEGylation of RANTES analogue),55 or by better drug delivery strategies.
CXCR4 inhibitors
CXCR4 is a chemokine receptor whose activation is responsible for myriad biological functions, including chemotaxis and cellular differentiation. CXCR4 is a G-protein coupled receptor that is activated by the C-X-C chemokine CXCL12, more commonly named SDF-1α. Due to its diverse biological function, CXCR4 has been targeted as a therapeutic option in a number of disease states besides HIV, the most prevalent of which are cardiovascular disease and cancer. Like CCR5, HIV infection is attenuated by the natural chemokine, however the same concerns of receptor activation and recycling remain. Unlike CCR5 inhibitors, however, development based on alteration of the natural chemokine is just beginning to emerge.56 Owing to its diverse role in many disease states, much more is known about the structural requirements for X4 inhibitor design. Included in this knowledge base is the high-resolution structure of the receptor bound to an antagonist,57 which has already been employed as a resource for computational design of new inhibitors.58
Analogous to CCR5 inhibition, early work in the development of CXCR4 inhibitors focused on developing analogues of the natural chemokine, SDF1-α.59,60 The approach suffered from the same challenges that applied to RANTES development, namely receptor activation and recycling. Engineering productive antagonists from SDF1-α for anti-HIV applications is further complicated by the critical role that the CXCR4-SDF1-α axis plays in the body. Thus, development of macromolecular antagonists centred on peptide fragments derived from components that were able to interact selectively with the receptor, including SDF1-α, gp120, and the viral macrophage inflammatory protein-II (vMIP-II).67 Early work in this field has been reviewed elsewhere,61 and the remainder of the discussion will focus on peptide and protein inhibitors discovered since 2008. A number of recent examples highlight the importance of CXCR4 inhibition in HIV treatment.
The growing body of knowledge regarding CXCR4 and its interactions with inhibitors has enabled the generation of novel structural motifs in both peptides and peptidomimetics with improved antiviral activities. An example from the Camarero laboratory describes grafting the linear CVX15 peptide into a cyclotide framework to improve its potency.62 Cyclotides are a special class of globular mini-proteins (27 to 38 amino acids) that cyclize from head to tail and contain three disulphide bonds to form a complex cystine knot topology.63,64 These molecular architectures are an emerging class of molecules well suited for medicinal applications, owing to their increased serum half lives, compact and rigid molecular structures (important for binding affinity) and near limitless substitution without detriment to structural integrity. Similar scaffolds have already been utilized to template high-affinity peptides for the medicinally relevant integrin receptors.65,66 In this example, researchers adapted a known inhibitor of CXCR4, for which structural information had been recently determined, and grafted it in various positions within a cyclotide scaffold derived from the horseshoe crab peptides polyphemusin I/II. The resultant peptide showed improved activity over the previously reported cyclic CVX15 peptide in HIV inhibition assays, exhibiting EC50 values of ~2 nM. Further promising properties of this peptide inhibitor include improved serum stability relative to linear and simple cyclic analogues, and a lower affinity for serum proteins that would likely be responsible for opsonization and clearance prior to reaching their targets. Taken together, these data indicate that cyclotide scaffolds are promising macromolecular architectures for the discovery of `drug-gable' receptor-specific antagonists.
The T140 peptide, a peptide derived from polyphemusin II, was one of the first X4 inhibitors reported by Fujii and coworkers in 2000, and has served as a scaffold for CXCR4 inhibitor development.68 Shortly thereafter, the Fujii group identified the pharmacophore associated with T140, a linear four amino acid sequence that includes several basic residues and the non-natural amino acid naphthylalanine (Figure 2). The active motif was mapped onto a pentapeptide scaffold to enforce a preferred conformation and thus enhanced specificity, an approach that was already well known to yield active peptides such as cyclic RGD motifs. The approach originally allowed the researchers to identify the peptide FC131, a low nanomolar inhibitor of CXCR4 and likewise active against X4-tropic HIV strains.69 Recent years have seen the iteration of the FC131 scaffold to new compounds with variable potency against X4-tropic HIV strains. For instance, modifications to the peptide backbone that replaced the traditional amide bond with alkene isosteres70 or reduced secondary amines71 resulted in a significant decrease in binding affinity and anti-HIV activity. One of the more potent inhibitors was the FC122 peptide, a derivative of FC131 that had a single N-methyl substituent, and had an approximate three-fold enhancement of activity over the parent peptide.72 Finally, introducing a basic amidine residue into the backbone in place of one amide resulted in a ten-fold improvement in binding affinity over the best previously identified scaffolds, which translated to a similar increase in anti-HIV activity (Figure 2).73
Figure 2.
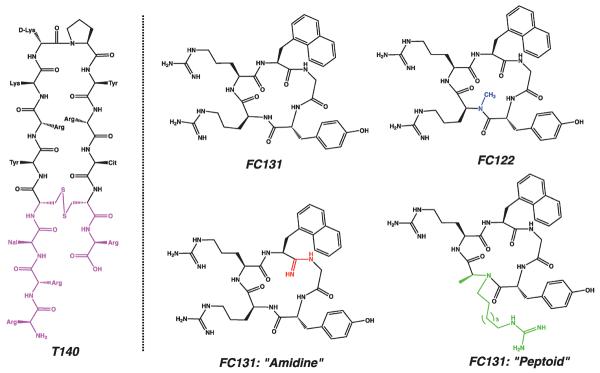
Chemical structures of peptide-based CXCR4 inhibitors. The pharmacophore of T140 is shaded (left). Shaded fragments indicate changes to the promising FC131 peptide scaffold: N-methylated position, amidine, and peptoid.
Kessler and coworkers74 optimized FC131 using structural information garnered from NMR experiments, which indicated a major and minor conformation in solution.74 The researchers were able to modify the peptide scaffold to freeze out its bioactive conformation. This was accomplished by introducing a peptoid residue at the key dynamic bond, thus favouring a trans conformation over the previous cis:trans equilibrium state. This new peptide:peptoid hybrid molecule exhibited exceptionally high affinity for the CXCR4 receptor, with a picomolar IC50 value. The improvement represents a two order of magnitude enhancement of binding affinity over the parent peptide. As expected, this improvement in binding affinity translated to improved efficacy against HIV infection, resulting in nanomolar inhibition (EC50~30 nM) of an X4-tropic HIV-1 strain (LAI). The reported cyclic peptide:peptoid hybrid has a number of desirable features when considered as a candidate for clinical development. Inhibition of HIV-1 infection is an order of magnitude more effective than the clinically approved AMD-3100. The hybrid structure contains both D-amino acids and peptoid residues, likely making the compound resistant to protease degradation and enhancing cell permeability.75–77 It will be of interest in coming years to follow the development of new inhibitors derived from the class of molecules, as structural information regarding the receptor is already leading to an increased understanding of molecular interactions that are critical to producing potent CXCR4 inhibitors.78
An alternative approach to traditional medicinal chemistry is the use of directed evolution to obtain high-affinity ligands for molecular targets. Once limited to phage display panning experiments to identify short peptides (still a highly valuable technique), molecular evolution has advanced to include in vivo binding experiments, identifying novel or improved catalytic activity of enzymes and antibodies, and the identification of so-called nanobodies. Nanobodies are typically engineered from the variable regions of antibodies and can be evolved to exhibit high binding affinity for a molecular target, or enzyme-like catalytic activity. Recently, Smit and coworkers developed an anti-CXCR4 nanobody derived from VHH, immunoglobulin single variable domains found in the Camelidae family.79 In a clever method for generation of high-affinity nanobodies, antibodies were generated in llamas by immunizing animals with CXCR4-expressing cells. PBMCs were then isolated and the VHH fragments cloned into phage vectors for traditional panning experiments. The isolated clones exhibited potent affinities for the CXCR4 receptor in a CXCL12 displacement assay, however when the two most potent nanobodies were joined via a flexible linker to form heterodimers, receptor affinity increased approximately three fold (Ki~0.3 nM) and the resulting proteins acted as inverse receptor agonists. More importantly, the dimeric nanobodies were able to inhibit X4-tropic HIV infection in multiple cell types and under challenge by several HIV strains. The IC50 values represent some of the most potent values reported to date, inhibiting HIV infection at high picomolar concentrations.
Chimeric inhibitors targeting both CCR5 and CXCR4
The potential for mutations in the HIV genome that leads to receptor switching cannot be discounted, thus a new class of entry inhibitor has focused on chimeric molecules that act to block both CCR5 and CXCR4. These types of molecules should be active against a wider breadth of viral strains than single receptor antagonists and potentially offer broad activity in a single therapeutic unit. A recent report by Liwang and coworkers80 describes the genetic modification of RANTES variants with terminal extensions comprised of CXCR4 blockers. The use of protein-based therapies is especially convenient for this class of anti-HIV compounds, as recombinant DNA technology makes the fusion of multiple protein/peptide components a routine laboratory exercise. In this example, the researchers fused the CXCR4 antagonist peptide, C37, to the C-termini of 5P12 and 5P14 RANTES. The growing body of structural information makes approaches such as these likely to succeed. In this instance, it is known that the N-terminus of RANTES is primarily responsible for receptor binding, leaving the C-terminus available for further modification. The chimeric inhibitor strategy has shown promising results, as the resulting protein was able to inhibit both X4 and R5 strains with equal or, in most cases, improved potency over the single acting parent compounds. In experiments with R5 tropic strains, the fusion inhibitor was typically 1–2 orders of magnitude more potent than RANTES alone. Equally impressive was the ability of these inhibitors to block X4 tropic infection, especially in cell lines expressing both CCR5 and CXCR4, a situation aligned with typical T-cell infection. In Tzm-B1 cells, low picomolar inhibition of the X4 tropic HXB2 strain of HIV was observed, showing a synergistic effect between R5 and X4 activity. This effect is presumably due to cell-surface localization enabled by the high affinity interaction between RANTES and CCR5, which subsequently creates a high-local concentration of the chimeric molecule, thus enhancing inhibition of the CXCR4 pathway.
Discussion
The global pandemic of HIV infection clearly necessitates new and improved strategies to combat the disease. Emerging methods for HIV prevention that extend beyond behavioural modification are those of pre-exposure prophylaxis. It is in this realm where macromolecular agents that block co-receptor binding are most likely to find impact as microbicides. There are several advantages associated with using peptide and protein-based molecules in this context. First, peptides and proteins can have extremely high affinity and specificity for their molecular targets, potentially making them highly potent anti-viral agents, as illustrated above. Second, there is evidence that macromolecular agents are more difficult to evade through viral mutation. Thus, employing these agents limits the evolution of more potent and drug-resistant strains of HIV. This is a potential problem for small-molecule therapeutics used for both treatment and prevention, as infected individuals could develop resistant strains that are not susceptible to the prophylactic treatment, furthering the spread of drug-resistant viral strains.
The prospects for the development of peptide and protein inhibitors of HIV co-receptor entry are increasingly promising. RANTES derivatives have been validated in vivo and are especially promising for early stage infection and person-to-person transmission of R5 tropic viruses. Likewise, new and more potent CXCR4 inhibitors are rapidly being identified and tested, showing impressive receptor-binding affinities and inhibition of X4-tropic infection. The recent developments highlighted in this review article, show that the future is bright for these types of molecules. The next step in translating these advances to the clinic is the development of scalable production of these macromolecules and efficient delivery to sites of infection.
ACKNOWLEDGEMENTS
JKP would like to acknowledge the NIH for a pathway to independence award (R00EB011530).
Footnotes
Author contributions: HAV and JKP designed and wrote the manuscript. JKP had primary responsibility for its final content.
REFERENCES
- 1.Joint United Nations Programme on HIV/AIDS. UNAIDS; 2010. Global report: UNAIDS report on the global AIDS epidemic 2010; pp. 1–364. [Google Scholar]
- 2.Adamson CS, Freed EO. Novel approaches to inhibiting HIV-1 replication. Antivir Res. 2010;85:119–41. doi: 10.1016/j.antiviral.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashkenazi A, Wexler-Cohen Y, Shai Y. Multifaceted action of Fuzeon as virus–cell membrane fusion inhibitor. BBA – Biomembranes. 2010;1808:1–7. doi: 10.1016/j.bbamem.2011.06.020. [DOI] [PubMed] [Google Scholar]
- 4.Gilliam BL, Riedel DJ, Redfield RR. Clinical use of CCR5 inhibitors in HIV and beyond. J Transl Med. 2011;9:S9. doi: 10.1186/1479-5876-9-S1-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng Y, Broder CC, Kennedy PE, Berger EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–7. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzi P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–6. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 7.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272:1955–8. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 8.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 9.Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–5. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 10.Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, Goedert JJ, Buchbinder SP, Vittinghoff E, Gomperts E, Donfield S, Vlahov D, Kaslow R, Saah A, Rinaldo C, Detels R, O'Brien SJ. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856–62. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 11.Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–77. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 12.Pope M, Haase AT. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat Med. 2003;9:847–52. doi: 10.1038/nm0703-847. [DOI] [PubMed] [Google Scholar]
- 13.Brumme ZL, Goodrich J, Mayer HB, Brumme CJ, Henrick BM, Wynhoven B, Asselin JJ, Cheung PK, Hogg RS, Montaner JSG. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J Infect Dis. 2005;192:466–74. doi: 10.1086/431519. [DOI] [PubMed] [Google Scholar]
- 14.Nel AM, Coplan P, Smythe SC, McCord K, Mitchnick M, Kaptur PE, Romano J. Pharmacokinetic assessment of dapivirine vaginal microbicide gel in healthy, HIV-negative women. AIDS Res Hum Retroviruses. 2010;26:1181–90. doi: 10.1089/aid.2009.0227. [DOI] [PubMed] [Google Scholar]
- 15.Nel AM, Smythe SC, Habibi S, Kaptur PE, Romano JW. Pharmacokinetics of 2 dapivirine vaginal microbicide gels and their safety vs. hydroxyethyl cellulose-based universal placebo gel. J Acquir Immune Defic Syndr. 2010;55:161–9. doi: 10.1097/QAI.0b013e3181e3293a. [DOI] [PubMed] [Google Scholar]
- 16.Nel A, Smythe S, Young K, Malcolm K, McCoy C, Rosenberg Z, Romano J. Safety and pharmacokinetics of dapivirine delivery from matrix and reservoir intravaginal rings to HIV-negative women. J Acquir Immune Defic Syndr. 2009;51:416–23. doi: 10.1097/qai.0b013e3181acb536. [DOI] [PubMed] [Google Scholar]
- 17.Selhorst P, Vazquez AC, Terrazas-Aranda K, Michiels J, Vereecken K, Heyndrickx L, Weber J, Quiñones-Mateu ME, Ariën KK, Vanham G. Human immunodeficiency virus type 1 resistance or cross-resistance to nonnucleoside reverse transcriptase inhibitors currently under development as microbicides. Antimicrob Agents Chemother. 2011;55:1403–13. doi: 10.1128/AAC.01426-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nedellec R, Coetzer M, Lederman MM, Offord RE, Hartley O, Mosier DE. “Resistance” to PSC-RANTES revisited: two mutations in human immunodeficiency virus type 1 HIV-1 SF162 or simian-human immunodeficiency virus SHIV SF162-p3 do not confer resistance. J Virol. 2010;84:5842–5. doi: 10.1128/JVI.01907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedellec R, Coetzer M, Lederman MM, Offord RE, Hartley O, Mosier DE. Resistance to the CCR5 inhibitor 5P12-RANTES requires a difficult evolution from CCR5 to CXCR4 coreceptor use. PLoS ONE. 2011;6:e22020. doi: 10.1371/journal.pone.0022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson RM, Arts EJ. Past, present, and future of entry inhibitors as HIV microbicides. Curr HIV Res. 2012;10:19–26. doi: 10.2174/157016212799304616. [DOI] [PubMed] [Google Scholar]
- 21.Henrich TJ, Kuritzkes DR. HIV-1 entry inhibitors: recent development and clinical use. Curr Opin Virol. 2013;3:51–57. doi: 10.1016/j.coviro.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–15. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- 23.Arenzana-Seisdedos F, Virelizier JL, Rousset D, Clark-Lewis I, Loetscher P, Moser B, Baggiolini M. HIV blocked by chemokine antagonist. Nature. 1996;383:400. doi: 10.1038/383400a0. [DOI] [PubMed] [Google Scholar]
- 24.Moriuchi H, Moriuchi M, Combadiere C, Murphy PM, Fauci AS. CD8+ T-cell-derived soluble factor(s), but not beta-chemokines RANTES, MIP-1 alpha, and MIP-1 beta, suppress HIV-1 replication in monocyte/macrophages. Proc Natl Acad Sci USA. 1996;93:15341–5. doi: 10.1073/pnas.93.26.15341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinter A, Catanzaro A, Monaco J, Ruiz M, Justement J, Moir S, Arthos J, Oliva A, Ehler L, Mizell S, Jackson R, Ostrowski M, Hoxie J, Offord R, Fauci AS. CC-chemokines enhance the replication of T-tropic strains of HIV-1 in CD4(+) T cells: role of signal transduction. Proc Natl Acad Sci USA. 1998;95:11880–5. doi: 10.1073/pnas.95.20.11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi WT, Nedellec R, Coetzer M, Colin P, Lagane B, Offord RE, Hartley O, Mosier DE. CCR5 Mutations Distinguish N-terminal Modifications of RANTES (CCL5) with Agonist versus Antagonist Activity. J Virol. 2012;86:10218–10220. doi: 10.1128/JVI.00353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartley O, Offord RE. Engineering chemokines to develop optimized HIV inhibitors. Curr Protein Pept Sci. 2005;6:207–19. doi: 10.2174/1389203054065400. [DOI] [PubMed] [Google Scholar]
- 28.Vangelista L, Secchi M, Lusso P. Rational design of novel HIV-1 entry inhibitors by RANTES engineering. Vaccine. 2008;26:3008–15. doi: 10.1016/j.vaccine.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Proudfoot AE, Power CA, Hoogewerf AJ, Montjovent MO, Borlat F, Offord RE, Wells TN. Extension of recombinant human RANTES by the retention of the initiating methionine produces a potent antagonist. J Biol Chem. 1996;271:2599–603. doi: 10.1074/jbc.271.5.2599. [DOI] [PubMed] [Google Scholar]
- 30.Mack M, Luckow B, Nelson PJ, Cihak J, Simmons G, Clapham PR, Signoret N, Marsh M, Stangassinger M, Borlat F, Wells TN, Schlöndorff D, Proudfoot AE. Aminooxypentane-RANTES induces CCR5 internalization but inhibits recycling: a novel inhibitory mechanism of HIV infectivity. J Exp Med. 1998;187:1215–24. doi: 10.1084/jem.187.8.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilken J, Hoover D, Thompson DA, Barlow PN, McSparron H, Picard L, Wlodawer A, Lubkowski J, Kent SB. Total chemical synthesis and high-resolution crystal structure of the potent anti-HIV protein AOP-RANTES. Chem Biol. 1999;6:43–51. doi: 10.1016/S1074-5521(99)80019-2. [DOI] [PubMed] [Google Scholar]
- 32.Hartley O, Gaertner H, Wilken J, Thompson D, Fish R, Ramos A, Pastore C, Dufour B, Cerini F, Melotti A, Heveker N, Picard L, Alizon M, Mosier D, Kent S, Offord R. Medicinal chemistry applied to a synthetic protein: development of highly potent HIV entry inhibitors. Proc Natl Acad Sci USA. 2004;101:16460–5. doi: 10.1073/pnas.0404802101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lederman MM, Veazey RS, Offord R, Mosier DE, Dufour J, Mefford M, Piatak M, Lifson JD, Salkowitz JR, Rodriguez B, Blauvelt A, Hartley O. Prevention of vaginal SHIV transmission in rhesus macaques through inhibition of CCR5. Science. 2004;306:485–7. doi: 10.1126/science.1099288. [DOI] [PubMed] [Google Scholar]
- 34.Kawamura T, Bruse SE, Abraha A, Sugaya M, Hartley O, Offord RE, Arts EJ, Zimmerman PA, Blauvelt A. PSC-RANTES blocks R5 human immunodeficiency virus infection of Langerhans cells isolated from individuals with a variety of CCR5 diplotypes. J Virol. 2004;78:7602–9. doi: 10.1128/JVI.78.14.7602-7609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaertner H, Cerini F, Escola J-M, Kuenzi G, Melotti A, Offord R, Rossitto-Borlat I, Nedellec R, Salkowitz J, Gorochov G, Mosier D, Hartley O. Highly potent, fully recombinant anti-HIV chemokines: reengineering a low-cost microbicide. Proc Natl Acad Sci USA. 2008;105:17706–11. doi: 10.1073/pnas.0805098105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerini F, Landay A, Gichinga C, Lederman MM, Flyckt R, Starks D, Offord RE, Le Gal F, Hartley O. Chemokine analogues show suitable stability for development as microbicides. J Acquir Immune Defic Syndr. 2008;49:472–6. doi: 10.1097/QAI.0b013e31818c953f. [DOI] [PubMed] [Google Scholar]
- 37.Gaertner H, Offord R, Botti P, Kuenzi G, Hartley O. Semisynthetic Analogues of PSC-RANTES, a Potent Anti-HIV Protein. Bioconjug Chem. 2008;19:480–9. doi: 10.1021/bc7003044. [DOI] [PubMed] [Google Scholar]
- 38.Pokorski JK, Hovlid ML, Finn MG. Cell targeting with hybrid Qβ virus-like particles displaying epidermal growth factor. ChemBioChem. 2011;12:2441–7. doi: 10.1002/cbic.201100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc Natl Acad Sci USA. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strable E, Prasuhn DE, Udit AK, Brown S, Link AJ, Ngo JT, Lander G, Quispe J, Potter CS, Carragher B, Tirrell DA, Finn MG. Unnatural amino acid incorporation into virus-like particles. Bioconjug Chem. 2008;19:866–75. doi: 10.1021/bc700390r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nardese V, Longhi R, Polo S, Sironi F, Arcelloni C, Paroni R, DeSantis C, Sarmientos P, Rizzi M, Bolognesi M, Pavone V, Lusso P. Structural determinants of CCR5 recognition and HIV-1 blockade in RANTES. Nat Struct Mol Biol. 2001;8:611–15. doi: 10.1038/89653. [DOI] [PubMed] [Google Scholar]
- 42.Vangelista L, Longhi R, Sironi F, Pavone V, Lusso P. Critical role of the N-loop and beta1-strand hydrophobic clusters of RANTES-derived peptides in anti-HIV activity. Biochem Biophys Res Commun. 2006;351:664–8. doi: 10.1016/j.bbrc.2006.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Secchi M, Longhi R, Vassena L, Sironi F, Grzesiek S, Lusso P, Vangelista L. Enhancement of anti-HIV-1 activity by hot spot evolution of RANTES-derived peptides. Chem Biol. 2012;19:1579–88. doi: 10.1016/j.chembiol.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 44.Hartley O, Dorgham K, Perez-Bercoff D, Cerini F, Heimann A, Gaertner H, Offord RE, Pancino G, Debre P, Gorochov G. Human immunodeficiency virus type 1 entry inhibitors selected on living cells from a library of phage chemokines. J Virol. 2003;77:6637–44. doi: 10.1128/JVI.77.12.6637-6644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vyroubalova EC, Hartley O, Mermod N, Fisch I. Identification of peptide ligands to the chemokine receptor CCR5 and their maturation by gene shuffling. Mol Immunol. 2006;43:1573–8. doi: 10.1016/j.molimm.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 46.Wang FY, Zhang TY, Luo JX, He GA, Gu QL, Xiao F. Selection of CC chemokine receptor 5-binding peptide from a phage display peptide library. Biosci Biotechnol Biochem. 2006;70:2035–41. doi: 10.1271/bbb.50654. [DOI] [PubMed] [Google Scholar]
- 47.Wu Y, Deng R, Wu W. Study on CCR5 analogs and affinity peptides. Protein Eng Des Sel. 2012;25:97–105. doi: 10.1093/protein/gzr062. [DOI] [PubMed] [Google Scholar]
- 48.Patel K, Dixit VD, Lee JH, Kim JW, Schaffer EM, Nguyen D, Taub DD. The GHS-R blocker D-[Lys3] GHRP-6 serves as CCR5 chemokine receptor antagonist. Int J Med Sci. 2012;9:51–8. doi: 10.7150/ijms.9.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Proudfoot AE, Fritchley S, Borlat F, Shaw JP, Vilbois F, Zwahlen C, Trkola A, Marchant D, Clapham PR, Wells TN. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J Biol Chem. 2001;276:10620–6. doi: 10.1074/jbc.M010867200. [DOI] [PubMed] [Google Scholar]
- 50.Czaplewski LG, McKeating J, Craven CJ, Higgins LD, Appay V, Brown A, Dudgeon T, Howard LA, Meyers T, Owen J, Palan SR, Tan P, Wilson G, Woods NR, Heyworth CM, Lord BI, Brotherton D, Christison R, Craig S, Cribbes S, Edwards RM, Evans SJ, Gilbert R, Morgan P, Randle E, Schofield N, Varley PG, Fisher J, Waltho JP, Hunter MG. Identification of amino acid residues critical for aggregation of human CC chemokines macrophage inflammatory protein (MIP)-1alpha, MIP-1beta, and RANTES. Characterization of active disaggregated chemokine variants. J Biol Chem. 1999;274:16077–84. doi: 10.1074/jbc.274.23.16077. [DOI] [PubMed] [Google Scholar]
- 51.Trkola A, Gordon C, Matthews J, Ketas T, Czaplewski L, Proudfoot AE, Moore JP. The CC-chemokine RANTES increases the attachment of human immunodeficiency virus type 1 to target cells via glycosaminoglycans and also activates a signal transduction pathway that enhances viral infectivity. J Virol. 1999;73:6370–9. doi: 10.1128/jvi.73.8.6370-6379.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ham AS, Cost MR, Sassi AB, Dezzutti CS, Rohan LC. Targeted delivery of PSC-RANTES for HIV-1 prevention using biodegradable nanoparticles. Pharm Res. 2009;26:502–11. doi: 10.1007/s11095-008-9765-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang NX, Offord R, Hartley O, Lederman M, Recum von HA. Controlled release of RANTES analogues using heparin affinity. Mol Pharm. in press. [Google Scholar]
- 54.Wang NX, Recum von HA. Affinity-based drug delivery. Macromol Biosci. 2010;11:321–32. doi: 10.1002/mabi.201000206. [DOI] [PubMed] [Google Scholar]
- 55.Shao H, Crnogorac MM, Kong T, Chen S-Y, Williams JM, Tack JM, Gueriguian V, Cagle EN, Carnevali M, Tumelty D, Paliard X, Miranda LP, Bradburne JA, Kochendoerfer GG. Site-specific polymer attachment to a CCL-5 (RANTES) analogue by oxime exchange. JAm Chem Soc. 2005;127:1350–1. doi: 10.1021/ja043096w. [DOI] [PubMed] [Google Scholar]
- 56.Dong CZ, Tian S, Madani N, Choi WT, Kumar S, Liu D, Sodroski JG, Huang Z. Role of CXCR4 internalization in the anti-HIV activity of stromal cell-derived factor-1 probed by a novel synthetically and modularly modified-chemokine analog. Exp Biol Med. 2011;236:1413–19. doi: 10.1258/ebm.2011.011260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu B, Chien EYT, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC, Hamel DJ, Kuhn P, Handel TM, Cherezov V, Stevens RC. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–71. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Planesas JM, Pérez-Nueno VI, Borrell JI, Teixidó J. Impact of the CXCR4 structure on docking-based virtual screening of HIV entry inhibitors. J Mol Graph Model. 2012;38:123–36. doi: 10.1016/j.jmgm.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 59.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, Loetscher M, Baggiolini M, Moser B. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833–5. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 60.Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J, Springer TA. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829–33. doi: 10.1038/382829a0. [DOI] [PubMed] [Google Scholar]
- 61.Liang X. CXCR4, inhibitors and mechanisms of action. Chem Biol Drug Des. 2008;72:97–110. doi: 10.1111/j.1747-0285.2008.00681.x. [DOI] [PubMed] [Google Scholar]
- 62.Aboye TL, Ha H, Majumder S, Christ F, Debyser Z, Shekhtman A, Neamati N, Camarero JA. Design of a novel cyclotide-based CXCR4 antagonist with anti-human immunodeficiency virus (HIV)-1 activity. J Med Chem. 2012;55:10729–34. doi: 10.1021/jm301468k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia AE, Camarero JA. Biological activities of natural and engineered cyclotides, a novel molecular scaffold for peptide-based therapeutics. Curr Mol Pharmacol. 2010;3:153–63. doi: 10.2174/1874467211003030153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Craik DJ, Conibear AC. The chemistry of cyclotides. J Org Chem. 2011;76:4805–17. doi: 10.1021/jo200520v. [DOI] [PubMed] [Google Scholar]
- 65.Kimura RH, Miao Z, Cheng Z, Gambhir SS, Cochran JR. A dual-labeled knottin peptide for pet and near-infrared fluorescence imaging of integrin expression in living subjects. Bioconjug Chem. 2010;21:436–44. doi: 10.1021/bc9003102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kimura RH, Levin AM, Cochran FV, Cochran JR. Engineered cystine knot peptides that bind alphavbeta3, alphavbeta5, and alpha5beta1 integrins with low-nanomolar affinity. Proteins. 2009;77:359–69. doi: 10.1002/prot.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tamamura H, Omagari A, Oishi S, Kanamoto T, Yamamoto N, Peiper SC, Nakashima H, Otaka A, Fujii N. Pharmacophore identification of a specific CXCR4 inhibitor, T140, leads to development of effective anti-HIV agents with very high selectivity indexes. Bioorg Med Chem Lett. 2000;10:2633–2637. doi: 10.1016/s0960-894x(00)00535-7. [DOI] [PubMed] [Google Scholar]
- 68.Tamamura H, Sugioka M, Odagaki Y, Kan Y, Oishi S, Nakashima H, Yamamoto N, Peiper SC, Hamanaka N. Conformational study of a highly specific CXCR4 inhibitor, T140, disclosing the close proximity of its intrinsic pharmacophores associated with strong anti-HIV activity. Bioorg Med Chem Lett. 2001;11:359–62. doi: 10.1016/s0960-894x(00)00664-8. [DOI] [PubMed] [Google Scholar]
- 69.Fujii N, Oishi S, Hiramatsu K, Araki T, Ueda S, Tamamura H, Otaka A, Kusano S, Terakubo S, Nakashima H, Broach JA, Trent JO, Wang Z-X, Peiper SC. Molecular-size reduction of a potent CXCR4-chemokine antagonist using orthogonal combination of conformation- and sequence-based libraries. Angew Chem Int Edit. 2003;42:3251–3. doi: 10.1002/anie.200351024. [DOI] [PubMed] [Google Scholar]
- 70.Narumi T, Hayashi R, Tomita K, Kobayashi K, Tanahara N, Ohno H, Naito T, Kodama E, Matsuoka M, Oishi S, Fujii N. Synthesis and biological evaluation of selective CXCR4 antagonists containing alkene dipeptide isosteres. Org Biomol Chem. 2010;8:616. doi: 10.1039/b917236j. [DOI] [PubMed] [Google Scholar]
- 71.Tamamura H, Araki T, Ueda S, Wang Z, Oishi S, Esaka A, Trent JO, Nakashima H, Yamamoto N, Peiper SC, Otaka A, Fujii N. Identification of novel low molecular weight CXCR4 antagonists by structural tuning of cyclic tetrapeptide scaffolds. J Med Chem. 2005;48:3280–9. doi: 10.1021/jm050009h. [DOI] [PubMed] [Google Scholar]
- 72.Ueda S, Oishi S, Wang Z-X, Araki T, Tamamura H, Cluzeau J, Ohno H, Kusano S, Nakashima H, Trent JO, Peiper SC, Fujii N. Structure-activity relationships of cyclic peptide-based chemokine receptor CXCR4 antagonists: disclosing the importance of side-chain and backbone functionalities. J Med Chem. 2007;50:192–8. doi: 10.1021/jm0607350. [DOI] [PubMed] [Google Scholar]
- 73.Inokuchi E, Oishi S, Kubo T, Ohno H, Shimura K, Matsuoka M, Fujii N. Potent CXCR4 antagonists containing amidine type peptide bond isosteres. ACS Med Chem Lett. 2011;2:477–80. doi: 10.1021/ml200047e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Demmer O, Frank AO, Hagn F, Schottelius M, Marinelli L, Cosconati S, Brack-Werner R, Kremb S, Wester H-J, Kessler HA. A conformationally frozen peptoid boosts CXCR4 affinity and anti-HIV activity. Angew Chem Int Edit. 2012;51:8110–13. doi: 10.1002/anie.201202090. [DOI] [PubMed] [Google Scholar]
- 75.Tan NC, Yu P, Kwon Y-U, Kodadek T. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg Med Chem. 2008;16:5853–61. doi: 10.1016/j.bmc.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kwon Y-U, Kodadek T. Quantitative evaluation of the relative cell permeability of peptoids and peptides. J Am Chem Soc. 2007;129:1508. doi: 10.1021/ja0668623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller S, Simon R, Ng S, Zuckermann R, Kerr J, Moos W. Comparison of the proteolytic susceptibilities of homologous L-amino acid, D-amino acid, and N-substituted glycine peptide and peptoid oligomers. Drug Dev Res. 1995;35:20–32. [Google Scholar]
- 78.Yoshikawa Y, Kobayashi K, Oishi S, Fujii N, Furuya T. Molecular modeling study of cyclic pentapeptide CXCR4 antagonists: new insight into CXCR4-FC131 interactions. Bioorg Med Chem Lett. 2012;22:2146–50. doi: 10.1016/j.bmcl.2012.01.134. [DOI] [PubMed] [Google Scholar]
- 79.Jaähnichen S, Blanchetot C, Maussang D, Gonzalez-Pajuelo M, Chow KY, Bosch L, De Vrieze S, Serruys B, Ulrichts H, Vandevelde W. CXCR4 nanobodies (VHH-based single variable domains) potently inhibit chemotaxis and HIV-1 replication and mobilize stem cells. Proc Natl Acad Sci USA. 2010;107:20565–70. doi: 10.1073/pnas.1012865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao B, Mankowski MK, Snyder BA, Ptak RG, Liwang PJ. Highly potent chimeric inhibitors targeting two steps of HIV cell entry. J Biol Chem. 2011;286:28370–81. doi: 10.1074/jbc.M111.234799. [DOI] [PMC free article] [PubMed] [Google Scholar]