

Abstract
We report that, in the rat, administering insulin-like growth factor II (IGF-II) significantly enhances memory retention and prevents forgetting. Inhibitory avoidance learning leads to an increase in hippocampal expression of IGF-II, which requires the transcription factor CCAAT enhancer binding protein β and is essential for memory consolidation. Furthermore, injections of recombinant IGF-II into the hippocampus after either training or memory retrieval significantly enhance memory retention and prevent forgetting. To be effective, IGF-II needs to be administered within a sensitive period of memory consolidation. IGF-II-dependent memory enhancement requires IGF-II receptors, new protein synthesis, the function of activity-regulated cytoskeletal-associated protein and glycogensynthase kinase 3 (GSK3). Moreover, it correlates with a significant activation of synaptic GSK3β and expression of GluR1 a-amino-3-hydroxy-5-methyl-4-isoxasoleproprionic acid receptor subunits. In hippocampal slices, IGF-II promotes IGF-II receptor-dependent, persistent long-term potentiation after weak synaptic stimulation. Thus, IGF-II may represent a novel target for cognitive enhancement therapies.
Elucidating the mechanisms of memory enhancement is critical for the development of cognitive enhancement therapies. Memory strengthening and persistence depend on consolidation, a process whereby newly learned information, which is initially labile, becomes stronger and resilient to disruption1. This process recruits evolutionarily conserved de novo RNA and protein syntheses, the function of members of the cAMP response element binding protein (CREB) and CCAAT enhancer binding protein (C/EBP) transcription factor families2-4, and correlates with synaptic structural changes2,5. Stable memories can again become fragile if retrieved, and undergo a process of reconsolidation that, like the initial consolidation, requires de novo RNA and protein synthesis, CREB and C/EBP6,7 to re-stabilize8. The identity of the target genes regulated by CREB and C/EBP is still largely unknown. Studies in liver and other tissues show that C/EBP binding sites are present in tissue-specific promoters of IGF-II9, a growth factor that is expressed in the brain but still is poorly characterized.
IGF-II is a mitogenic polypeptide, which together with insulin and insulin-like growth factor 1 (IGF-I) belongs to the IGF/IGFBP (IGF/IGF Binding Protein) system. This system is important in normal somatic growth and development, tissue repair and regeneration throughout the lifespan10,11. IGF-II, the less characterized member of the family, is expressed in the brain both during development and in adulthood and declines with aging12. In the adult brain, it is the most abundantly expressed of the IGFs10, and its relative concentration is highest in the hippocampus13. Given that IGF-II is a putative C/EBP target gene, we investigated its expression and functional role in memory formation.
C/EBPβ-dependent IGF-II expression is regulated by training
In previous studies, we showed that IA training induces a significant increase in hippocampal C/EBPβ, which starts between 6 and 9h after training, lasts for at least 28h and returns to baseline by 48h after training20 (supplementary Fig. S1). Here we tested whether C/EBPβ regulates the expression of the putative target gene IGF-II. The numeric values, and number of animals/group (n) of all experiments are shown in Supplementary Tables. Northern blot analysis showed that, compared to controls that were exposed to the box without footshock and either euthanized immediately after (0h−) or at paired timepoints (no shock, −), the hippocampal expression of IGF-II mRNA did not change at 6 and 9h but significantly increased at 20h and had a strong trend toward an increase at 36h after training (Fig. 1a).
Figure 1. C/EBPβ-dependent IGF-II expression significantly increases following training.

a, Northern blot examples and densitometric analyses of IGF-II (cyclophilin-normalized). Data are expressed as mean %±s.e.m. of 0h− (one-way ANOVA comparing all groups F8,59=2.46, P=0.0249, post-hoc t-test *P<0.05). b, Real-time PCR of hippocampal IGF-II and IGF-I mRNA (18S RNA-normalized). Data are expressed as mean fold change± s.e.m. of 20h−/0h− (Student’s t-test ***P< 0.0001). c, Western blot analyses of hippocampal IGF-II from 0h−, unpaired (Un) and trained (+) rats euthanized 20, 72 or 96h later (actin-normalized). Data are expressed as mean %± s.e.m. of 0h− (one-way ANOVA comparing 0h−, 20un, and 20h+ F2,29=4.69, P=0.0172, Newman-Keuls post-hoc test, *P<0.05). d, Western blot analysis of hippocampal IGF-II from trained or Un rats injected (↓) with either SC-ODN or β-ODN 5h post-training and euthanized 24h post-training (actin-normalized). Data are expressed as mean %± s.e.m. of SC-ODN-Un (two-way ANOVA F1,19=4.62, P=0.0447 for interaction, F1,19=1.45, P=0.2434 for ODN-treatment, F1,19=6.46, P=0.0199 for training-paradigm, Bonferroni post-hoc **P<0.01;*P<0.05).
Quantitative RT-PCR analyses of mRNA extracts confirmed the significant increase of IGF-II mRNA 20h after training compared to no-shock and 0h− controls, whereas, in the same extracts, IGF-I mRNA remained unchanged (Fig. 1b).
Quantitative western blot analyses with an anti-IGF-II antibody that specifically recognizes IGF-II and not IGF-I (supplementary Fig. S2), showed that, hippocampal levels of IGF-II protein significantly increased at 20 but not at 72 or 96h after training, compared to both time-matched unpaired and 0h− controls (Fig. 1c). The unpaired control protocol temporally dissociates, within subject, context and footshock exposure by one hour, and does not produce long-term IA memory (supplementary Fig. S3). Thus, IA training leads to an increase in IGF-II that temporally overlaps that of C/EBPβ20. We next investigated whether the IGF-II increase requires C/EBPβ. Taubenfeld et al.15 have established that hippocampal bilateral injection of C/EBPβ antisense oligodeoxynucleotide (β-ODN), 5h after IA training, blocks the training-dependent C/EBPβ induction and completely disrupts memory consolidation. Using this injection protocol and quantitative western blot analyses we found that, compared to control scrambled ODN (SC-ODN), β-ODN completely disrupted the training-induced IGF-II increase without changing the IGF-II expression in unpaired control rats 24h after training (Fig. 1d). Chromatin immunoprecipitation of hippocampal extracts confirmed that C/EBPβ binds in vivo to a C/EBPβ consensus sequence in the promoter region of the rat IGF-II exon 1 (supplementary Fig. S4). Thus, IA training leads to an increase in hippocampal C/EBPβ that regulates a downstream increase in IGF-II.
Temporal requirement of IGF-II during memory consolidation
We then investigated the functional kinetic of hippocampal IGF-II during IA memory consolidation. Bilateral injections of IGF-II ODN antisense (IGF-II-ODN) were used to selectively knock-down the IGF-II expression in the dorsal hippocampus. Injection either immediately- or 8h after training, or at both timepoints, showed that double, but not single, injections of IGF-II-ODN significantly disrupted memory retention at 24h after training, compared to SC-ODN (Fig. 2a, supplementary Fig. S5). Quantitative RT-PCR confirmed that, compared to SC-ODN, double injections of IGF-II-ODN selectively and significantly decreased the levels of IGF-II, but not of IGF-I mRNA, 16h after training (supplementary Fig. S6). IGF-II-ODN doubly injected at 24 and 32h after training, compared to SC-ODN, significantly disrupted memory retention at 48h after training (Fig. 2a) and re-training of the amnesic rats resulted in normal memory retention 24h after re-training (Fig. 2a), thus excluding hippocampal damage or non-specific effects. However, IGF-II-ODN doubly injected at 96 and 104h after training did not affect memory retention 24h later (Fig. 2a). The amnesia caused by IGF-II-ODN double injections was rescued by the co-administration of recombinant IGF-II but not IGF-I (Fig. 2b), further proving that IGF-II expression is essential for IA memory consolidation. Furthermore, while IGF-I had no effect (compare SC-ODN/IGF-I from Fig. 2b and SC-ODN from Fig. 2a), IGF-II seemed to enhance memory retention, although the effect was not significant, possibly because the testing latency was cut-off at 540 sec.
Figure 2. Hippocampal IGF-II is required for memory consolidation.

Schedules shown above figures. a, Mean latency± s.e.m. of rats given double hippocampal injections (↓) of SC-ODN or IGF-II-ODN (one-way ANOVA for treatment F5,47= 2.54, P=0.043, post-hoc Student’s t-test * P<0.05 for 0h/8h and 24h/32h). b, Mean latency± s.e.m. of rats given double hippocampal injections (↓) of SC-ODN or IGF-II-ODN with either IGF-II or IGF-I (two-way ANOVA F1,33=4.29, P=0.0468 for interaction, F1,31=6.34, P=0.173 for ODN-treatment, F1,31=11.38, P<0.0021 for IGF-treatment, Bonferroni post-hoc **P<0.01)
We concluded that hippocampal IGF-II has a critical role for IA memory consolidation during a limited time window that lasts for more than one but less than 4 days.
IGF-II significantly enhances memory and prevents forgetting
Because of the tendency toward memory enhancement in our IGF-II rescue experiment (Fig. 2c), we tested whether exogenously administered IGF-II into the hippocampus immediately after training modulates memory strength. The latency cut-off time was raised to 900 sec. Bilateral injections of IGF-II immediately after training significantly and persistently enhanced memory retention at 24h and 7 days, compared to IGF-I or vehicle (Fig. 3a). This enhancement was not due to a non-specific locomotor effect (supplementary Fig. S7).
Figure 3. Hippocampal post-training IGF-II administration enhances memory and prevents forgetting.

Schedules shown above figures. a, Mean latency± s.e.m. of trained rats given hippocampal injection (↓) of vehicle, IGF-II or IGF-I and tested 24h and 7-days later (two-way ANOVA F2,38=0.44, P=0.6463 for interaction, F2,38=26.7, P<0.0001 for treatment, F1,38=4.24, P=0.0466 for test, Bonferroni post-hoc test **P<0.01, ***P<0.001). b, Mean latency± s.e.m. of trained rats given an hippocampal injection (↓) of vehicle or IGF-II (Student’s t-test *P=0.0261). c, Mean % freezing of trained rats injected with vehicle or IGF-II (Student’s t-test *P<0.0434). d, Mean latency± s.e.m. of trained rats given bilateral amygdala injection (↓) of vehicle or IGF-II.
The IGF-II-mediated memory enhancement was dose-dependent (supplementary Fig. S8): hippocampal injections of 25 or 2.5 ng, like 250 ng, immediately after training incrementally enhanced memory retention at 24h.
Finally, hippocampal injection of IGF-II immediately after training significantly enhanced memory retention tested 3 weeks later when the latency of vehicle-injected rats was not significantly different from acquisition, indicating that IGF-II prevents forgetting (Fig. 3b).
The IGF-II effect generalized to another memory task, contextual fear conditioning. Bilateral hippocampal injection of IGF-II immediately after contextual-auditory fear conditioning training significantly enhanced contextual fear conditioning retention 24h later, without affecting auditory fear conditioning tested 48h after training (Fig. 3c). No difference in baseline freezing was found between groups prior to footshock delivery (Fig. 3c).
Finally, because IA consolidation also critically involves the amygdala21, we tested the effect of bilateral IGF-II injections into the amygdala immediately after training, but found no effect at testing 24h later (Fig. 3d).
Hence, IGF-II in the hippocampus acts as a strong memory enhancer and also prevents forgetting.
IGF-II-mediated enhancement: effect on reconsolidation
An established memory, resilient to disruption, becomes again labile and undergoes another protein-synthesis-dependent reconsolidation process if retrieved22,23. Bilateral hippocampal injection of IGF-II 24h after training had no effect on memory retention tested at 48h (Fig. 4a). However, if 24h after training IGF-II was given after memory retrieval, (Test1), memory retention was significantly enhanced 24h later (Final Test, Fig. 4a).
Figure 4. Post-retrieval IGF-II administration enhances memory and the effect is temporally limited.
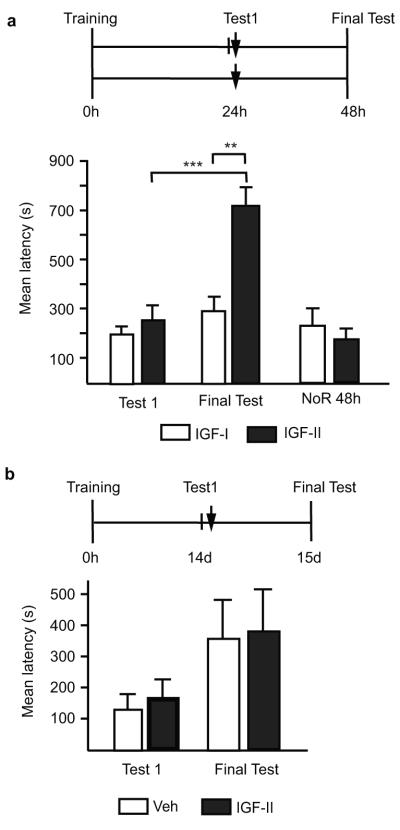
Schedules shown above figures. a, Mean latency± s.e.m. of trained rats, tested 24h post-training and, immediately after, injected (↓) with IGF-II or IGF-I. Non-reactivated rats (NoR) were trained and injected (↓) without testing. Rats were tested 48h post-training (two-way ANOVA F1,26=5.67, P=0.0249 for interaction, F1,26=9.82, P=0.0042 for treatment, F1,26=13.67, P=0.0001 for test, Bonferroni post-hoc **P<0.01, *P<0.05). b, Mean latency± s.e.m.of trained rats, tested 14d post-training and, immediately after, injected (↓) with vehicle or IGF-II; memory was tested 15d after training.
Studies in IA and other types of learning24-27, but not all8,28, have shown that reconsolidation is temporally limited. IA memory undergoes protein-synthesis-dependent reconsolidation if retrieved 2 or 7 days after training but not 2 or 4 weeks after training25. Hence, we asked whether the enhancing effect of IGF-II is also temporally restricted, and coincides with the reconsolidation-sensitive temporal window. Bilateral hippocampal injection of IGF-II immediately after retrieval (Test1), two weeks after training, did not change memory retention tested one day later, compared to vehicle (Final Test, Fig. 4b). Hence, hippocampal IGF-II-mediated memory enhancement occurs only within the sensitive temporal window during which IA memory undergoes reconsolidation.
Mechanisms underlying IGF-II-mediated memory enhancement
IGF-II activates both IGF-I and IGF-II receptors but with different affinity29. To determine whether IGF-II-mediated memory enhancement recruits one or both of these receptors, we tested the effect of IGF-I and IGF-II receptor (R) selective inhibitors. Specific inhibitors of IGF-IIR (anti-IGF-IIR antibody) but not IGF-IR (JB1) co-injected with IGF-II completely abolished the memory enhancement compared to respective controls (Fig. 5a). The inhibitors alone did not affect memory retention (Fig. 5a). Similarly to the antisense experiments, compared to control IgG, a single bilateral hippocampal injection of anti-IGF-2R antibody immediately after training did not affect memory retention (Fig. 5a), whereas double injections, immediately- and 8 hours after training, caused a complete amnesia 24h after training (Fig. 5b).
Figure 5. The role of IGF-II receptors, de novo protein synthesis, and Arc in memory consolidation and IGF-II-mediated enhancement.

Schedules shown above figures. a, Mean latency± s.e.m. of trained rats injected (↓) with vehicle, IGF-II, IGF-II/Anti-IGF-2R, IGF-II/JB1, Anti-IGF-2R, or JB1 (one-way ANOVA F5,40=3.82, P=0.0023, Newman-Keuls post-hoc test *P<0.05 **P<0.01). b, Mean latency± s.e.m. of trained rats given double injections of IgG or anti-IGF-2R antibody (Student’s t-test **P<0.0041). c, Mean latency± s.e.m. of rats trained, tested then injected (↓) with vehicle, IGF-II or IGF-II+anisomycin (two-way ANOVA F2,34=5.25, P=0.0103 for interaction, F2,34=4.68, P=0.0161 for treatment, F1,34=13.7, P=0.0008 for test, Bonferroni post-hoc **P<0.01,***P<0.001). d, Mean latency± s.e.m. of rats trained, tested and injected (↓) with vehicle+SC-ODN, vehicle+Arc-ODN, IGF-II+SC-ODN, or IGF-II+Arc-ODN (two-way ANOVA F1,18=7.8, P=0.0119 for interaction, F1,18=17.3, P=0.0006 for ODN-treatment, F1,18=12.3, P=0.0025 for veh/IGF-II-treatment, Bonferroni post-hoc ***P<0.001).
We next asked whether IGF-II-mediated memory enhancement recruits new protein synthesis. Since memory consolidation per se requires new protein synthesis in the hippocampus, blocking protein synthesis in IGF-II-injected rats after training would not be informative. However, because new protein synthesis is not required in the hippocampus for IA reconsolidation15, we tested the effect of protein synthesis inhibition on retrieval-dependent IGF-II-mediated enhancement.
Bilateral hippocampal co-injection of IGF-II and the protein synthesis inhibitor anisomycin immediately after Test1, 24h after training, showed that anisomycin, compared to vehicle, completely disrupted the IGF-II-mediated memory enhancement tested 24h later (Fig. 5c) without changing the training-induced retention levels. Hence, memory enhancement, but not reconsolidation, requires hippocampal de novo protein synthesis.
To begin identifying which proteins are required for the memory enhancement, we investigated the role of C/EBPβ. Bilateral hippocampal injection of β-ODN 5h after retrieval (Test1) did not affect the IGF-II-mediated memory enhancement tested 48h after training (supplementary Fig. S9). The timing of the ODN injections was based on previous kinetics studies showing maximal disruptive effect of β-ODN7,15. To test whether a prolonged β-ODN treatment could affect the post-retrieval IGF-II-mediated memory enhancement we injected β-ODNs at both 1h before and 5h after reactivation. This treatment, compared to control SC-ODN, also failed to affect the IGF-II-evoked memory enhancement (supplementary Fig. S9), suggesting that, although de novo protein synthesis is critical for memory enhancement, C/EBPβ is not.
We therefore hypothesized that the protein synthesis-mediated enhancement may recruit synaptic rather than cell-wide, transcriptional mechanisms. One rapidly regulated translation known to occur at activated synapses and critical for long-term plasticity and memory is that of activity-regulated cytoskeletal-associated protein (Arc)30. Bilateral hippocampal injection of Arc antisense (Arc-ODN), compared to relative SC-ODN, 1h before retrieval (Test1), completely blocked the post-retrieval IGF-II-mediated memory enhancement, without affecting the basal level of the memory 2 days after training (Fig. 5d).
Thus, IGF-II-mediated enhancement requires IGF-II- but not IGF-I receptors. Furthermore, retrieval-dependent IGF-II-mediated enhancement requires de novo protein synthesis and Arc but not C/EBPβ, suggesting that it may use synaptic rather than cell-wide-regulatory mechanisms.
Memory consolidation requires the CREB-C/EBP-dependent gene cascade2. In IA, both CREB phosphorylation in ser133 (pCREB) and the expression of C/EBPβ are significantly increased in the hippocampus for more than 20 hours after training20. Here, we examined whether IGF-II mediated memory enhancement following training correlates with an enhanced hippocampal activation of the CREB-C/EBP pathway. Quantitative western blot analyses confirmed that training significantly increased both pCREB and C/EBPβ in the hippocampus 20 h later20 (trained-vehicle vs naïve-vehicle, Fig. 6a). Compared to vehicle, IGF-II treatment immediately after training resulted in only a tendency toward a further increase in both markers (Fig. 6a). Thus, IGF-II-mediated memory enhancement does not correlate with significant enhancement in the activation of the CREB-C/EBP cascade, strengthening our hypothesis that IGF-II-regulated mechanisms may be synaptic rather than cell-wide. We therefore investigated the synaptic expression levels of GluR1 and GluR2 AMPA receptor subunits. Synaptic GluR1 AMPA receptors have been shown to rapidly increase following IA training and play a critical role in consolidation31,32. Furthermore, AMPA receptor subunit trafficking is known to accompany long-term plasticity in LTP and long-term depression (LTD)33. Quantitative western blot analyses of synaptoneurosomal extract (see supplementary Fig. S10 for the biochemical characterization), revealed that there was an increased expression of synaptic GluR1 30 minutes after training compared to naive, which, however, was not significant, probably due to the relatively low shock intensity used (Fig. 6b). Importantly, synaptic GluR1 levels were significantly increased in trained rats treated with IGF-II compared to vehicle. This increase was completely abolished by anti-IGF-IIR antibody. On the other hand, GluR2 levels remained unchanged across groups.
Figure 6. Mechanisms of IGF-II-mediated memory enhancement. IGF-II promotes LTP.

a, Western blot analysis of hippocampal pCREB and C/EBPβ from naïve or trained rats injected (↓) with vehicle or IGF-II and euthanized 20h later (actin-normalized). Data are expressed as mean %± s.e.m. of naïve-Veh (one-way ANOVA, pCREB: F2,20=4.3, P=0.0287, C/EBPβ: F2,19=5.7, P=0.0117, Newman-Keuls post-hoc test, *P<0.05). b, Western blot analysis of hippocampal GluR1 and GluR2 from naïve or trained rats injected (↓) with vehicle, IGF-II, IGF-II+Anti-IGF-IIR antibody (actin-normalized). Data are expressed as mean%± s.e.m. of naïve-Veh (one way ANOVA F3,19=4.24, P=0.0188, Newman-Keuls post hoc test * P<0.05). c, Western blot analysis of hippocampal pGSK3β and GSK3β from the same extracts as in (B) (actin normalized). Data are expressed as mean%± s.e.m. of naïve-veh (one-way ANOVA F3,19=4.93, P=0.130, Newman-Keuls post hoc test * P<0.05,**P<0.01). d, Mean latency± s.e.m. of rats trained, tested and injected (↓) with vehicle, IGF-II, SB216763 (SB) or IGF-II+SB (two-way ANOVA F3,56=4.44, P=0.0072 for interaction, F3,56=5.07, P=0.0035 for treatment, F1,56=9.12, P=0.0038 for test, Bonferroni post-hoc **P<0.01,***P<0.001) e, Time-courses of fEPSPs in area CA1 stratum radiatum are shown with sample traces obtained during the baseline period, 2 min after high frequency stimulation (HFS) and 100 min after HFS (gray traces: no HFS; black traces: HFS). Scale bars: 0.5 mV, 5 ms. Left panel: Weak HFS induced only transient potentiation that returned to baseline levels within 100 min. Middle panel: In the presence of IGF-II, the same protocol induced stable LTP (Student’s t-test P<0.05). Right panel: In slices pretreated with antibodies against the IGFII receptor, IGF-II failed to facilitate the induction of stable LTP.
Previous studies reported that AMPA receptor trafficking and dendritic expression of GluR1 in neurons are regulated by GSK334 and, interestingly, IGF-II has been implicated in GSK3 regulation35. As depicted in Fig. 6c, the IGF-II-mediated significant increase of GluR1 was paralleled by a significant synaptic activation of GSK3β (measured by its dephosphorylation at ser936), which was also completely abolished by anti-IGF-IIR antibody. Furthermore, while blocking hippocampal GSK3 function with pretraining injection of the inhibitor SB216763 completely disrupted IA memory (data not shown), the same treatment delivered immediately after retrieval (Test1) selectively eliminated the IGF-II-mediated enhancement tested 2d after training (Test2) without affecting memory reconsolidation (Fig. 6d).
Thus, IGF-II-dependent memory enhancement requires the activation of GSK3β and correlates with increased synaptic expression of GluR1.
IGF-II enables long-term potentiation (LTP)
To determine whether the effect of IGF-II was generalized to long-term synaptic plasticity, we tested the effect of IGF-II on hippocampal LTP, which is widely regarded as a cellular correlate of long-term memory37. IGF-II was applied to acute hippocampal slices and both LTP in the Schaffer collateral pathway and basal synaptic transmission were investigated. As shown in Fig. 6e, a weak tetanus elicited a transient synaptic potentiation that decayed to baseline within 100 minutes after induction (slope=109.4±9.7%, calculated as the average of the final 10 min of recording normalized to the full baseline period for each slice). When this weak stimulus was delivered in the presence of IGF-II, stable LTP was expressed (slope=135.2±6.6% of baseline). This enabling effect was completely blocked in slices that were pretreated with anti-IGF-IIR antibody (112.05±4.7%) (Fig. 6E). Neither IGF-II nor the anti-IGF-IIR antibody affected basal synaptic transmission (supplementary Figs. S11 and S12).
Discussion
Our study shows that memory retention can be enhanced, LTP promoted and forgetting prevented by the administration of IGF-II, a growth factor physiologically regulated following learning. IGF-II is endogenously upregulated following learning as a C/EBPβ2 target gene and required in the hippocampus for memory consolidation during the first 1-2 days after training but not at later times, extending previous conclusions that the transcription- and translation-dependent phase of IA consolidation in the dorsal hippocampus lasts for more than one, but less than 4 days.
The effect of IGF-II as memory enhancer is temporally restricted to active phases induced by either learning or memory retrieval, generalized to different types of hippocampal-dependent memories and occurs with an acute treatment in low doses. The training-related IGF-II-dependent memory enhancement is restricted to a temporal window that lasts less than a day. However, at later times, the enhancing effect reemerges if IGF-II is given in combination with memory retrieval, which is known to reactivate the memory and induce reconsolidation22. The IGF-II effect following retrieval is also temporally limited, and, in fact, restricted to a temporal window that overlaps with the reconsolidation sensitive period of IA7,15,25. Both retrieval-induced memory fragility and IGF-II-dependent enhancement require new protein synthesis but in different brain regions: the former in the amygdala, the latter in the hippocampus. Hence, during the first 1-2 weeks after training, IA memory is in a sensitive period, during which, if in an active state, it can be either significantly weakened or enhanced. These findings strengthen our previously proposed hypothesis that reconsolidation is a phase of a lingering consolidation process22,38. We speculate that the retrieval-induced memory fragility mediated by amygdala mechanisms may be critical for promoting memory enhancement mediated by hippocampal mechanisms and that the temporal boundary of the sensitive period may reflect the hippocampal-cortical redistribution of memory storage39,40 or the multiple trace distribution of memory41.
Intriguingly, the effect of IGF-II as memory enhancer is selectively mediated by IGF-II and not IGF-I receptors. IGF-IIR is identical to the mannose 6 phosphate (M6P) receptor11 and plays a role in lysosomal enzyme trafficking, clearance and endocytosis-mediated degradation of IGF-II and possibly in transmembrane-receptor-mediated signal transduction11 but, in general, little is known about its function in the brain.
IGF-II-mediated memory enhancement is not paralleled by significant activation of pCREB or C/EBPβ and does not functionally require C/EBPβ, but critically depends on GSK3 and Arc and is accompanied by a significant increase in synaptic GSK3activation and GluR1 expression. Since C/EBPβ is significantly upregulated for more than 28h after training20, it is possible that this induction is sufficient to also mediate the memory enhancement. Alternatively, the enhancement may use mechanisms either downstream of C/EBPβ or distinct from those mediating consolidation. Thus, the IGF-II-dependent enhancement might not recruit the activation of new cells, but rather uses those that have been transcriptionally “marked” by training and target synaptic mechanisms, possibly those at activated synapses. One of these mechanisms might be GSK3-regulated GluR1 synaptic mobilization, a hypothesis in line with previous reports of functional links between GSK3 activity and dendritic clustering of GluR134, and of Arc expression and membrane trafficking of GluR1, synaptic plasticity and memory consolidation30. We cannot exclude that IGF-II-dependent memory enhancement may occur via recruitment of new cell activation, which however would be independently from the activation and function of CREB-C/EBPβ.
Thus, IGF-II may be a novel target for cognitive enhancement therapies.
Methods
Animals
Long Evans adult male rats (Harlan, Indianapolis, IN) weighing between 200 and 250 grams at the beginning of the experiments were used. Rats were housed individually on a 12 hour light-dark cycle with ad libitum access to food and water. All experiments were done during the light cycle between 9 AM and 6 PM. All rats were handled for 2-3 min per day for 5 days before any behavioral procedure. All protocols complied with the National Institute of Health Guide for the Care and Use of Laboratory Animals and were approved by the Mount Sinai School of Medicine Animal Care Committees.
Inhibitory Avoidance (IA)
IA was carried out as previously described14,15. The IA chamber (Med Associates. Inc., St. Albans, VT) consisted of a rectangular Perspex box divided into a safe compartment and a shock compartment. The safe compartment was white and illuminated; while the shock compartment was black and dark. Foot shocks were delivered to the grid floor of the shock chamber via a constant current scrambler circuit. The apparatus was located in a sound-attenuated, nonilluminated room. During training sessions, each rat was placed in the safe compartment with its head facing away from the door. After 10 sec, the door separating the compartments was automatically opened, allowing the rat access to the shock compartment; the rats usually enter the shock (dark) compartment within 10-20 sec of the door opening. The door closed 1 sec after the rat entered the shock compartment, and a brief foot shock (0.6mA for 2 sec for all experiments except for those of Fig. 6a which was done at 0.9mA) was administered. Latency to enter the shock compartment was taken in seconds as acquisition. The rat was then returned to its home cage and tested for memory retention at the designated time-point(s). Retention tests were done by placing the rat back in the safe compartment and measuring its latency to enter the shock compartment. Foot shocks were not administered on the retention tests, and testing was terminated at 540 sec or 900 sec as indicated in the figures.
Controls consisted of rats that remained in their home cage (Naive), rats exposed to the training apparatus without foot shock (−). or rats exposed to the training apparatus and to foot shock 1 h later (unpaired, Un).
In reactivation (reconsolidation) experiments, rats were trained as described, and, at the indicated time points, were tested. This test reactivated the memory. Immediately after or at the designated time-points, rats were injected with the indicated compounds and subsequently tested again for retention.
During testing, the experimenter was blind to the treatments given.
Contextual and auditory fear conditioning
Fear conditioning was carried out as previously described16. Rats were conditioned in contextual fear conditioning chamber (CFC), which consisted of a rectangular Plexiglass box (30.5 cm × 24.1 cm × 21.0 cm) with a metal grid floor (Model ENV-008 Med Associates). All rats were pre-exposed to this chamber for 5 min. On the next day, rats were placed in CFC chamber for 120 sec and then presented with 30 sec of the auditory cue consisting of a 5 kHz 75 dB tone that co-terminated with a 0.6 mA 2-sec footshock. One hundred twenty sec after the first footshock, another 30 sec auditory cue was presented that also co-terminated with another 0.6 mA 2-sec footshock. Rats were returned to their homecage 120 sec after the second footshock. Freezing levels during the first 148 sec (before the presentation of the first footshock) was recorded, scored and reported as baseline freezing. Freezing was defined as lack of movement except for breathing. Twenty-four hour later, rats were placed back in the CFC chamber and their freezing levels recorded for 5 min and scored. Twenty-four hours after CFC test, rats were placed in a different context (the illuminated IA box) for 120 sec before being presented with three 30 sec auditory cues. The three 30 sec auditory cues were separated by 120 sec. Freezing levels during the cue presentations was recorded and scored by an experimenter who was blind to the treatment conditions.
Cannulae implants and hippocampal and amgydala injections
Hippocampal and amygdala injections were given as previously described14,15,17. Rats were anesthetized with ketamine (65 mg/kg, i.p.) and xylazine (7.5mg/kg, i.p.), and stainless-steel guide cannulae (22-gauge) were stereotactically implanted to bilaterally target the hippocampus (4.0 mm posterior to the bregma; 2.6 mm lateral from midline; and 2.0 mm ventral). For amygdala injections, 26-gauge guide cannuale were implanted to bilaterally target the basolateral amygdala (2.8 mm posterior to bregma; 5.3 mm lateral from midline; and 6.25 mm ventral). The rats were returned to their home cages and allowed to recover from surgery for 7 days. At the indicated time points before or after training or retrieval, rats received bilateral injections of compounds as specified. All injections are indicated by arrow in the experimental schedule. All hippocampal injections were carried out in 1 μl per side, whereas all amygdala injections were done in 0.5 μl per side. Hippocampal injections used a 28-gauge needle and amygdala injections used 33-gauge needles that extended 1.5 mm beyond the tip of the guide cannula and connected via polyethylene tubing to a Hamilton syringe. The infusions were delivered at a rate of 0.33 μl/min using an infusion pump. The injection needle was left in place for two min after the injection to allow complete dispersion of the solution.
To verify proper placement of cannula implants, at the end of the behavioral experiments, rats were anesthetized and perfused with 4% paraformaldehyde in PBS. Their brains were postfixed overnight in the same fixative with 30% sucrose. Forty-micrometer coronal sections were cut through the hippocampus, stained with cresyl violet, and examined under a light microscope. Rats with incorrect cannula placement were discarded from the study.
Antisense ODNs and relative scrambled sequences (SC-ODNs) were injected at 2 nmol/μl in all antisense experiments. Sequences: C/EBPβ antisense (β-ODN: 5′-CCAGCAGGCGGTGCATGAAC-3′), C/EBPβ scrambled (SC-ODN: 5′-TCGGAGACTAAGCGCGGCAC-3′); IGF-II antisense (IGF-II-ODN: 5′-CCCATTGGTACCTGAAGTTG-3′); IGF-II scrambled (SC-ODN: 5′-CGCCTTGTGATACGACTTAG-3′); Arc antisense (Arc-ODN: 5′-GTCCAGCTCCATCTGCTCGC-3′); and Arc scrambled (SC-ODN: 5′-CGTGCCCTCTCGCAGCTTC-3′). Vehicle: phosphate-buffered saline (PBS, pH 7.4). The antisense for C/EBPβ has been previously shown to knock-down C/EBPβ in the hippocampus15. The antisense for IGF-II mRNA was specific for the sequence that includes the translational start site and was previously used successfully to knockdown IGF-II in other tissues42. The antisense for Arc has been previously shown to block Arc protein expression and long-term memory consolidation when injected into the hippocampus43. The respective SC-ODNs, which served as control, contained the same base composition but in a random order and show no homology to sequences in the GenBank database. All ODNs were phosphorothioated on the three terminal bases of both 5′ and 3′ ends to produce increased stability. Both ODNs were RPC (Reverse-Phased-Cartridge)-purified and obtained from Gene Link (Hawthorne, NY). Recombinant IGF-I and IGF-II were purchased from R&D Systems (Minneapolis, MN) and were dissolved in 0.1% bovine serum albumin (BSA) in 1xPBS. All experiments with recombinant IGF-II or IGF-I were carried out with 250 ng/injection, except for the dose-response curve (250, 25 or 2.5 ng) and for those in Fig. 5a where 25 ng was used. The IGF-I receptor (IGF-1R) antagonist JB1 (Bachem Biosciences, Torrance, CA) was dissolved in PBS. JB1 was injected at 20 ng/μl, a concentration that has been used successfully to block IGF-1 activity in various tissues including the brain44,45. Anti-IGF-II receptor antibody (anti-IGF2R, R&D Systems, Minneapolis, MN) was dissolved in 1xPBS and injected at 5ng/μl. This concentration blocked 95% of IGF-II receptor in an in-vitro binding assay (R&D).
Anisomycin (Sigma Aldrich, St. Louis, MO) was dissolved in 0.9% saline pH 7.4. and injected at 125μg/μl. This dose blocks more than 80% of protein synthesis in the dorsal hippocampus for up to 6h46.
GSK3 inhibitor SB216763 was purchased from Sigma and was dissolved in 1% DMSO in PBS and injected at 1ng/μl. This dose has been shown to block GSK3β activity (as measured by its dephosphorylation levels) in the brain47.
Synaptoneurosomal preparation and Western blot analysis
Synaptoneurosomal preparation was adapted from Elkobi et al.48. Briefly, dorsal hippocampi were rapidly dissected in ice-cold cortical dissection buffer followed by homogenization in buffer containing 10mM HEPES, 2mM EDTA, 2mM EGTA, 0.5mM TT, phosphatase and protease inhibitor cocktails (Sigma). Glass-teflon homogenizer was used and homogenates were filtered through 100 μm nylon mesh filter and 5 μm nitrocellulose filters sequentially. Synaptoneurosomes were obtained by centrifugating the filtrate at 1000g for 10 min. Synaptoneurosomal fraction was enriched for PSD-95 and N-methyl-D-aspartic acid (NMDA) receptor subunit NR-1 (Supplementary Fig. S10). Western blot analysis was done as previously reported 14,15. Hippocampal total extracts from rat were obtained by polytron homogenization in cold lysis buffer with protease and phosphatase inhibitors (0.2 M NaCl, 0.1 M HEPES, 10% glycerol, 2 mM NaF, 2 mM Na4P2O7, aprotonin 4U/ml, DTT 2mM, EGTA 1mM, microcystin, 1μM, benzamidine, 1mM). Protein concentrations were determined using the BioRad protein assay (BioRad Laboratories, Hercules, CA.). Equal amounts of total protein (10-20 μg/lane) were resolved on denaturing SDS-PAGE gels and transferred to Hybond-P membranes (Millipore, Billenca, MA) by electroblotting. Membranes were dried and then reactivated in methanol for 5 min and then washed with 3 changes of water. The membrane was then blocked in 3% milk/PBS or according to manufacturers’ instruction for 1 h at room temperature, then incubated with either anti-IGF-II (1/500, Millipore, Billerica, MA), or anti-actin (1/5000, Santa Cruz Biotechnology, Santa Cruz, CA) antisera in PBS overnight at 4°C. Anti-phospho-CREB (1/1000), anti-GluR1 (1/2000), anti-GluR2 (1/2000), anti-PSD95 (1/5000) and anti-NR1 (1/1000) antibodies were purchased from Millipore. Anti-C/EBPβ antibody was purchased from Santa Cruz Biotechnology (1/1000, Santa Cruz, CA). pGSK3β and GSK3β antibodies were purchased from Cell Signaling (1/1000, Danvers, MA.) pGSK3β was normalized to actin and GSK3β. The colloidal gold total protein stain was purchased from BioRad. The membranes were washed, treated with a secondary HRP-labeled donkey anti-rabbit antibody (1/4000, GE Healthcare, Waukesha, WI) for 1 h, washed again and incubated with HRP-streptavidin complex and ECL detection reagents (GE healthcare, Waukesha, WI). Membranes were exposed to Denville Scientific HyBlotCL (Denville Scientific, Metuchen, NJ), and quantitative densitometric analysis was performed using NIH ImageJ.
Real time quantitative RT-PCR
Hippocampal total RNA was extracted with TRizol (Invitrogen, Carlsbad, CA) and reverse transcribed using SuperScript II RNAse H minus RT (Invitrogen). Real-time PCR was done with an ABI Prism 7900HT (Applied Biosystems, Foster City CA). Five hundred pg of the first-strand cDNA was subjected to PCR amplification using a Quantitect SYBR Green PCR kit (Qiagen, Valencia, CA). IGF-II primers (forward: CCCAGCGAGACTCTGTGCGGA; reverse, GGAAGTACGGCCTGAGAGGTA); IGFI primers (forward: CCTGGGCTTTGTTTTCACTTCGG; reverse: CACAGCTCCGGAAGCAACACTCA). Forty cycles of PCR amplification were performed as follows: denature at 95°C for 30 sec, anneal at 55°C for 30 sec and extend for 30 sec at 72°C. Three PCR assays with triplicates were performed for each cDNA sample. 18S rRNA (forward, CGCCGCTAGAGGTGAAATTCT; reverse, CAGACCTCCGACTTTCGTTCT) was used as internal controls. Data were analyzed with Sequence Detector System version 2.0 software (Applied Biosystems, Foster City CA). The cycle threshold method (CT, see Applied Biosystems User Bulletin Number 2, P/N 4303859) was chosen to determine the relative quantification of gene expression in trained and control rats.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as described in Tsankova et al.18. The rat hippocampi were dissected and minced into ~1mm pieces, and immediately cross-linked in 1% formaldehyde for 17 min at room temperature rotating. The cross-linking reaction was stopped by adding glycine to a final concentration of 0.125 M and incubated for 7 min. The tissue was washed five times in cold PBS containing protease inhibitor (Roche Applied Sciences, Indianapolis, IN.) and then frozen on dry ice. The chromatin was solubilized and extracted by adding 500μl of lysis buffer (1% SDS, 50mM Tris-HCl pH8.1, 10mM EDTA), followed by sonication. The homogenate was diluted in 1.1ml ChIP dilution buffer (1.1% Triton X-100, 167mM NaCl, 16.7mM Tris-HCL pH8.1, 1.2mM EDTA, 0.01% SDS). The homogenate was used for C/EBPβ ChIP. 30μl of Magnetic Protein A beads (EZ-Magna ChIP A kit, Millipore) and 5μg of C/EBPβ antibody was added to the homogenate. The mixture was incubated rotating overnight in 4°C. The wash, elution, and reverse cross-link to free DNA were all performed according to the manufacturer’s protocol (EZ-Magna ChIP A kit).
Specific primers were designed to amplify the proximal promoter region of approximately 150 bp 5′ of exon one of rat IGF-II (GenBank: X17012.1), which contains a putative C/EBP binding site. Putative C/EBP binding site was predicted using an online program AliBaba 2.1. Similar C/EBP binding sites have been identified in other species49,50. Primer sequences used: forward GGTTCCCCACGTTAGGCTTGGAT; reverse TTGCGGCCCTGGGAATGAGTG. A standard thirty-five cycle PCR was performed as followed: denature at 95°C for 30 sec, anneal at 58°C for 30 sec and extend for 30 sec at 72°C. The PCR reaction was resolved on a 2% agarose gel and sequenced. Sequencing confirmed the identity of the fragment. DNA sequencing was performed by W.M. Keck Facility at Yale University, New Haven, CT.
Northern blot analysis
Northern blot analyses were performed as previously described 20. The rat IGF-II probe consisted of a 224 bp fragment corresponding to nucleotides 1145-1368 of the IGF-II sequence in GenBank accession number NM-031511. The same membrane was stripped and rehybridized with a full-length rat cyclophilin probe that was used as a loading control. Probes were labeled with random oligonucleotide primers (Prime-It II kit, Stratagene) and [a-32P]dCTP (Amersham). Quantitative densitometry analysis was performed using NIH Image J. Data were expressed as mean percentage ± s.e.m. of the 0h− (100%) control mean values.
Electrophysiology methods
Brains were removed from isoflurane-anesthetized male Long-Evans rats (6-8 weeks old), and chilled in ice-cold ACSF (in mM: 1.25 NaH2PO4,1.3 mM MgSO4, 2.5 mM CaCl2, 3.5 KCl, 15 glucose, 24 NaHCO3, and 118 NaCl) bubbled with 95% O2 / 5% CO2 (pH = 7.35). Acute transverse slices of dorsal hippocampus (400 μm thick) were recovered in an interface chamber at room temperature, as described previously 19. Slices were individually transferred to a recirculating submersion recording chamber and superfused with ACSF at 30-32 °C. Field EPSPs (fEPSPs) were recorded with a ACSF-filled pipette (2-4 MΩ) positioned in stratum radiatum of area CA1, and Schaffer collateral inputs were stimulated with 50 μs monophasic pulses using a bipolar concentric electrode placed in area CA3. Weak HFS, which normally induces only transient synaptic potentiation 19, consisted of two 1-s trains of 100 Hz pulses, delivered 20 s apart, with stimulus intensity set at 20% of the spike threshold.
IGF-II, freshly prepared from stock to a final concentration of 1 nM, was introduced 20 min before HFS was delivered and was present for the remainder of experiment. The antibody against the IGF-II receptor was used at a final concentration of 16 or 50 μg / mL, prepared fresh before use. As these two concentrations produced indistinguishable results, the data were pooled for analysis and presentation. The antibody was introduced at least 30 min before HFS was delivered, and remained in the superfusate for the rest of the experiment. The n’s for the electrophysiology experiment are: Veh (wLTP : n=4; no stim.: n=4), IGF-II (wLTP: n=5; no stim.: n=6), IGF-II+anti-IGF-IIR antibody (wLTP: n=5; no stim.: n=5).
Statistical Analysis
One- or two-way analysis of variance (ANOVA) followed by either the Newman-Keuls, or Bonferroni post-hoc test, or Student’s t-test was used for statistical analyses. For the electrophysiology experiments, EPSP slopes were compared by a Student’s t-test.
Supplementary Material
Acknowledgments
This work was supported by grants R01-MH065635, R01-MH074736, NARSAD, the Hirschl Foundation and Philoctetes Foundation awarded to C.M.A, F31-MH816213 to D.Y.C., and T32-MH087004 to S.A.S. R21-DA29298, R01-GM054508 to R.D.B. We thank Mark Baxter for assistance with statistical analyses. We thank Jian Feng, Ja-Wook Koo and Chia-Ying Lu for technical assistance. We thank Akinobu Suzuki and Amy Arguello for comments on the manuscript. We thank Reginald Miller and the Center for Comparative Medicine and Surgery Facility at Mount Sinai School of Medicine for technical support.
Footnotes
Reprints and permission information are available at www.nature.com/reprints.
The authors declare competing financial interests: details accompany the full-text HTML version of the paper at (url of journal website).
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
Author Contributions D.Y.C., A.G.-O. and C.M.A. designed and developed this study. D.Y.C., S.A.S., A.G.-O. carried out the behavioral studies. D.Y.C., A.G.-O. G.P. and D.B.-M. carried out the biochemical studies and analyses. R.D.B. and B.S.-R. designed and conducted the electrophysiology experiments. D.Y.C. and C.M.A. wrote the manuscript.
References
- 1.McGaugh JL. Memory--a century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- 2.Alberini CM. Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev. 2009;89:121–145. doi: 10.1152/physrev.00017.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kandel ER. The molecular biology of memory storage: A dialog between genes and synapses. Biosci. Rep. 2001;21:565–611. doi: 10.1023/a:1014775008533. [DOI] [PubMed] [Google Scholar]
- 4.Silva AJ, Kogan JH, Frankland PW, Kida S. Creb and memory. Annu. Rev. Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 5.Bailey CH, Kandel ER. Structural changes accompanying memory storage. Annu. Rev. Physiol. 1993;55:397–426. doi: 10.1146/annurev.ph.55.030193.002145. [DOI] [PubMed] [Google Scholar]
- 6.Kida S, et al. Creb required for the stability of new and reactivated fear memories. Nat. Neurosci. 2002;5:348–355. doi: 10.1038/nn819. [DOI] [PubMed] [Google Scholar]
- 7.Milekic MH, Pollonini G, Alberini CM. Temporal requirement of c/ebpbeta in the amygdala following reactivation but not acquisition of inhibitory avoidance. Learn. Mem. 2007;14:504–511. doi: 10.1101/lm.598307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nader K, Schafe GE, Le Doux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 9.Shamblott MJ, Leung S, Greene MW, Chen TT. Characterization of a teleost insulin-like growth factor ii (igf-ii) gene: Evidence for promoter ccaat/enhancer-binding protein (c/ebp) sites, and the presence of hepatic c/ebp. Mol. Mar. Biol. Biotechnol. 1998;7:181–190. [PubMed] [Google Scholar]
- 10.Russo VC, Gluckman PD, Feldman EL, Werther GA. The insulin-like growth factor system and its pleiotropic functions in brain. Endocr. Rev. 2005;26:916–943. doi: 10.1210/er.2004-0024. [DOI] [PubMed] [Google Scholar]
- 11.Hawkes C, Kar S. The insulin-like growth factor-ii/mannose-6-phosphate receptor: Structure, distribution and function in the central nervous system. Brain Res. Brain Res. Rev. 2004;44:117–140. doi: 10.1016/j.brainresrev.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 12.Kitraki E, Bozas E, Philippidis H, Stylianopoulou F. Aging-related changes in igf-ii and c-fos gene expression in the rat brain. Int. J. Dev. Neurosci. 1993;11:1–9. doi: 10.1016/0736-5748(93)90029-d. [DOI] [PubMed] [Google Scholar]
- 13.Kar S, Chabot JG, Quirion R. Quantitative autoradiographic localization of [125i]insulin-like growth factor i, [125i]insulin-like growth factor ii, and [125i]insulin receptor binding sites in developing and adult rat brain. J. Comp. Neurol. 1993;333:375–397. doi: 10.1002/cne.903330306. [DOI] [PubMed] [Google Scholar]
- 14.Garcia-Osta A, et al. Musk expressed in the brain mediates cholinergic responses, synaptic plasticity, and memory formation. J. Neurosci. 2006;26:7919–7932. doi: 10.1523/JNEUROSCI.1674-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taubenfeld SM, Milekic MH, Monti B, Alberini CM. The consolidation of new but not reactivated memory requires hippocampal c/ebpbeta. Nat. Neurosci. 2001;4:813–818. doi: 10.1038/90520. [DOI] [PubMed] [Google Scholar]
- 16.Muravieva EV, Alberini CM. Limited efficacy of propranolol on the reconsolidation of fear memories. Learn. Mem. 2010;17:306–313. doi: 10.1101/lm.1794710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tronel S, Alberini CM. Persistent disruption of a traumatic memory by postretrieval inactivation of glucocorticoid receptors in the amygdala. Biol. Psychiatry. 2007;62:33–39. doi: 10.1016/j.biopsych.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsankova NM, Kumar A, Nestler EJ. Histone modifications at gene promoter regions in rat hippocampus after acute and chronic electroconvulsive seizures. J. Neurosci. 2004;24:5603–5610. doi: 10.1523/JNEUROSCI.0589-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsokas P, et al. Local protein synthesis mediates a rapid increase in dendritic elongation factor 1a after induction of late long-term potentiation. J. Neurosci. 2005;25:5833–5843. doi: 10.1523/JNEUROSCI.0599-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taubenfeld SM, et al. Fornix-dependent induction of hippocampal ccaat enhancer-binding protein [beta] and [delta] co-localizes with phosphorylated camp response element-binding protein and accompanies long-term memory consolidation. J. Neurosci. 2001;21:84–91. doi: 10.1523/JNEUROSCI.21-01-00084.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang KC, et al. Post-training amygdaloid lesions impair retention of an inhibitory avoidance response. Behav. Brain. Res. 1982;4:237–249. doi: 10.1016/0166-4328(82)90002-x. [DOI] [PubMed] [Google Scholar]
- 22.Alberini CM. Mechanisms of memory stabilization: Are consolidation and reconsolidation similar or distinct processes? Trends Neurosci. 2005;28:51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Nader K, Schafe GE, LeDoux JE. The labile nature of consolidation theory. Nat. Rev. Neurosci. 2000;1:216–219. doi: 10.1038/35044580. [DOI] [PubMed] [Google Scholar]
- 24.Eisenberg M, Dudai Y. Reconsolidation of fresh, remote, and extinguished fear memory in medaka: Old fears don’t die. Eur. J. Neurosci. 2004;20:3397–3403. doi: 10.1111/j.1460-9568.2004.03818.x. [DOI] [PubMed] [Google Scholar]
- 25.Milekic MH, Alberini CM. Temporally graded requirement for protein synthesis following memory reactivation. Neuron. 2002;36:521–525. doi: 10.1016/s0896-6273(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 26.Suzuki A, et al. Memory reconsolidation and extinction have distinct temporal and biochemical signatures. J. Neurosci. 2004;24:4787–4795. doi: 10.1523/JNEUROSCI.5491-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Litvin OO, Anokhin KV. Mechanisms of memory reorganization during retrieval of acquired behavioral experience in chicks: The effects of protein synthesis inhibition in the brain. Neurosci. Behav. Physiol. 2000;30:671–678. doi: 10.1023/a:1026698700139. [DOI] [PubMed] [Google Scholar]
- 28.Debiec J, LeDoux JE, Nader K. Cellular and systems reconsolidation in the hippocampus. Neuron. 2002;36:527–538. doi: 10.1016/s0896-6273(02)01001-2. [DOI] [PubMed] [Google Scholar]
- 29.Nissley SP, Rechler MM. Somatomedin/insulin-like growth factor tissue receptors. Clin. Endocrinol. Metab. 1984;13:43–67. doi: 10.1016/s0300-595x(84)80008-0. [DOI] [PubMed] [Google Scholar]
- 30.Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: Regulation, mechanisms, and function. J. Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- 32.Slipczuk L, et al. Bdnf activates mtor to regulate glur1 expression required for memory formation. PLoS One. 2009;4:e6007. doi: 10.1371/journal.pone.0006007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kessels HW, Malinow R. Synaptic ampa receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei J, Liu W, Yan Z. Regulation of ampa receptor trafficking and function by glycogen synthase kinase 3. J. Biol. Chem. 285:26369–26376. doi: 10.1074/jbc.M110.121376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scalia P, et al. Regulation of the akt/glycogen synthase kinase-3 axis by insulin-like growth factor-ii via activation of the human insulin receptor isoform-a. J. Cell Biochem. 2001;82:610–618. doi: 10.1002/jcb.1196. [DOI] [PubMed] [Google Scholar]
- 36.Dajani R, et al. Crystal structure of glycogen synthase kinase 3 beta: Structural basis for phosphate-primed substrate specificity and autoinhibition. Cell. 2001;105:721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 37.Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain. 2006;129:1659–1673. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- 38.Alberini CM, Milekic MH, Tronel S. Mechanisms of memory stabilization and de-stabilization. Cell Mol. Life Sci. 2006;63:999–1008. doi: 10.1007/s00018-006-6025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- 40.Maviel T, Durkin TP, Menzaghi F, Bontempi B. Sites of neocortical reorganization critical for remote spatial memory. Science. 2004;305:96–99. doi: 10.1126/science.1098180. [DOI] [PubMed] [Google Scholar]
- 41.Nadel L, Land C. Memory traces revisited. Nat. Rev. Neurosci. 2000;1:209–212. doi: 10.1038/35044572. [DOI] [PubMed] [Google Scholar]
- 42.Elizalde PV, et al. Involvement of insulin-like growth factors-i and -ii and their receptors in medroxyprogesterone acetate-induced growth of mouse mammary adenocarcinomas. J. Steroid Biochem. Mol. Biol. 1998;67:305–317. doi: 10.1016/s0960-0760(98)00123-x. [DOI] [PubMed] [Google Scholar]
- 43.Guzowski JF, et al. Inhibition of activity-dependent arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J. Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pietrzkowski Z, et al. Inhibition of cellular proliferation by peptide analogues of insulin-like growth factor 1. Cancer Res. 1992;52:6447–6451. [PubMed] [Google Scholar]
- 45.Quesada A, Micevych PE. Estrogen interacts with the igf-1 system to protect nigrostriatal dopamine and maintain motoric behavior after 6-hydroxdopamine lesions. J. Neurosci. Res. 2004;75:107–116. doi: 10.1002/jnr.10833. [DOI] [PubMed] [Google Scholar]
- 46.Milekic MH, Brown SD, Castellini C, Alberini CM. Persistent disruption of an established morphine conditioned place preference. J. Neurosci. 2006;26:3010–3020. doi: 10.1523/JNEUROSCI.4818-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu CM, et al. Glycogen synthase kinase 3beta in the nucleus accumbens core mediates cocaine-induced behavioral sensitization. J. Neurochem. 2009;111:1357–1368. doi: 10.1111/j.1471-4159.2009.06414.x. [DOI] [PubMed] [Google Scholar]
- 48.Elkobi A, et al. Erk-dependent psd-95 induction in the gustatory cortex is necessary for taste learning, but not retrieval. Nat. Neurosci. 2008;11:1149–1151. doi: 10.1038/nn.2190. [DOI] [PubMed] [Google Scholar]
- 49.Lo JH, Chen TT. Ccaat/enhancer binding protein beta2 is involved in growth hormone-regulated insulin-like growth factor-ii gene expression in the liver of rainbow trout (oncorhynchus mykiss) Endocrinology. 151:2128–2139. doi: 10.1210/en.2009-0960. [DOI] [PubMed] [Google Scholar]
- 50.van Dijk MA, Rodenburg RJ, Holthuizen P, Sussenbach JS. The liver-specific promoter of the human insulin-like growth factor ii gene is activated by ccaat/enhancer binding protein (c/ebp) Nucleic Acids Res. 1992;20:3099–3104. doi: 10.1093/nar/20.12.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.