

Abstract
Objective:
To determine the histopathologic bases for the observed incidence of parkinsonism in families with C9ORF72 expansions, which typically cause amyotrophic lateral sclerosis (ALS) and/or frontotemporal dementia.
Methods:
DNA was extracted from 377 brains with the histopathologic diagnosis of idiopathic Parkinson disease or related disorders and analyzed for C9ORF72 expansions. α-Synuclein and p62 immunohistochemistry of the substantia nigra (SN) was undertaken in brains of 17 ALS cases with (C9ORF72+) and 51 without (C9ORF72−) the C9ORF72 expansion.
Results:
Only 1 of 338 cases with pathologically confirmed idiopathic Parkinson disease had a C9ORF72 expansion. Similarly, only 1 of 17 C9ORF72+ brains displayed features suggestive of α-synucleinopathy. In contrast, p62-positive, TDP-43–negative neuronal cytoplasmic inclusions within the SN were considerably more frequent in C9ORF72+ brain tissue than in the C9ORF72− brains (p = 0.005). Furthermore, there was a more marked loss of dopaminergic neurons in the SN of C9ORF72+ ALS brains than C9ORF72− ALS brains (p = 0.029).
Conclusions:
SN involvement is common in C9ORF72+ ALS but can be clearly distinguished from Parkinson disease–related mechanisms by the presence of p62-positive inclusions and the absence of α-synuclein–positive Lewy bodies or Lewy neurites.
Substantia nigra (SN) involvement in amyotrophic lateral sclerosis (ALS) has previously been noted clinically1 and neuropathologically.2,3 Expansions of C9ORF72 with >30 repeats (C9ORF72+) are the most common identifiable genetic cause of ALS and frontotemporal dementia (FTD).3,4 We and others have reported parkinsonian phenotypes at a greater frequency within C9ORF72+ families3,5 and sporadic cases,3,6,7 than in those with ALS/FTD who did not have a C9ORF72 expansion (C9ORF72−). Thus, it seems likely that intronic expansions of C9ORF72 explain at least in part the observed association between ALS and parkinsonism. However, it is currently unclear whether C9ORF72+ mutation carriers develop parkinsonism due to C9ORF72+ causing an α-synucleinopathy—as observed in idiopathic Parkinson disease (iPD)—or whether the underlying pathology in these patients is more in keeping with typical C9ORF72+ extramotor pathology with p62-positive, TDP-43–negative, ubiquitylated neuronal and glial cytoplasmic inclusions.3 Of note, the 9p21 locus has not been implicated in genetic association studies of iPD.8
To further clarify these crucial issues, we have genotyped a large number of brain tissue samples with the histopathologically confirmed diagnosis of iPD or related disorders for C9ORF72 expansions. We also hypothesized that subclinical involvement of the SN may be more common in C9ORF72+ than in C9ORF72− brains of patients who clinically presented only with features in keeping with ALS/FTD.
METHODS
Standard protocol approvals, registrations, and patient consents.
Ethical approval was obtained from the respective local and national ethics committees. Postmortem histology reports were provided by the Parkinson’s UK Brain Bank and the Queen Square Brain Bank for Neurological Disorders.
Subjects.
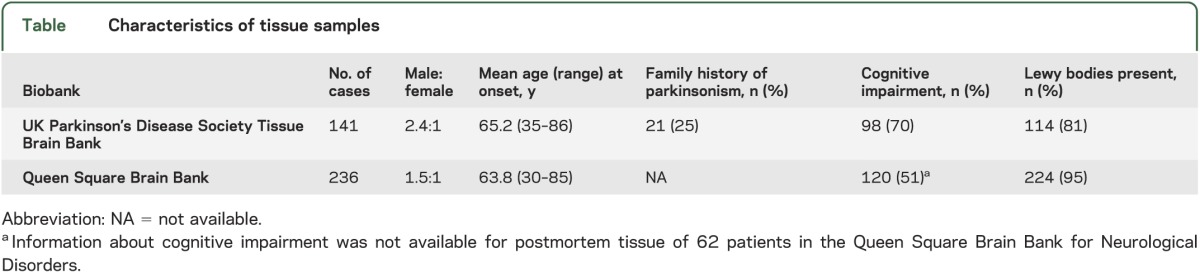
A total of 377 cases with a clinical diagnosis of PD were included; Lewy-body–positive, α-synucleinopathy was pathologically confirmed in 338 (90%) of cases (see table). Thus, a sufficient number of cases was obtained to allow comparison with the known frequency of expanded C9ORF72 in controls.3
Table.
Characteristics of tissue samples
Genotyping and immunohistochemistry.
DNA was extracted from brain tissue using standard methods and analyzed for C9ORF72 expansions as previously described.3 α-Synuclein and p62 immunohistochemistry was performed on 17 C9ORF72+ cases of ALS, including one case known to have autopsy-confirmed iPD as well as ALS.3 Immunohistochemistry for p62 was also performed on an additional 51 C9ORF72− ALS cases. The SN was examined on one side of each brain. Seven-micron-thick tissue sections from selected blocks were subjected to immunohistochemistry using antibodies to α-synuclein (Novocastra, Milton Keynes, UK) and p62 (BD Transduction Laboratories, Oxford, UK). α-Synuclein pathology was assessed as present or absent in C9ORF72+ cases. The number of p62-positive inclusions was classified as high (≥10 positive neuronal cytoplasmic inclusions), intermediate (5–9 inclusions), or low (≤4 inclusions). The number of cases with and without the C9ORF72 mutation with high, intermediate, and low numbers of p62-positive neuronal cytoplasmic was compared by χ2. To determine whether the ubiquitylated neuronal cytoplasmic inclusion pathology of the SN was associated with neuronal loss, both C9ORF72+ and C9ORF72− brains were semiquantitatively assessed as having no, mild, or severe neuronal loss. The extent of the neuronal cell loss was compared by χ2.
RESULTS
Only one of the brain bank iPD cases had a C9ORF72 expansion containing >30 repeats (1/377 = 0.2% of the total number of cases screened, and 1/338 = 0.3% of the Lewy-body–positive cases). This C9ORF72+ patient presented with clinically typical PD at the age of 67 years. His father had died of ALS. Neuropathologic assessment revealed features of 1) classic PD with Braak stage 6, diffuse neocortical Lewy-body pathology; 2) classic TDP-43 pathology with frontotemporal lobar degeneration (FTLD)-TDP type-A features9; and 3) C9ORF72-ALS/FTLD pathology with numerous p62-positive, TDP-43–negative neuronal cytoplasmic inclusions of star-shaped morphology in the hippocampus, and smaller cytoplasmic inclusions in cerebellar granule cells. Unfortunately, his spinal cord was not available.
All but one of the 17 C9ORF72+ ALS brains were devoid of α-synuclein–positive neuronal cytoplasmic inclusions in the SN. The single case with α-synuclein pathology was known to have coincident PD-ALS and has been discussed elsewhere.3
The 17 C9ORF72+ ALS brains had a considerably higher number of p62-positive, TDP-43–negative neuronal cytoplasmic inclusions in the SN (7 cases with >10, 2 cases with 5–9, and 8 cases with ≤4 p62-positive inclusions) than the 51 C9ORF72− ALS cases (4 cases >10, 6 cases with 5–9, and 41 cases with ≤4 p62-positive inclusions; χ2 = 10.724, df = 2, p = 0.005). No/moderate/severe neuronal cell loss was observed in 3/9/5 cases with C9ORF72 mutations and 22/17/4 cases without this mutation (χ2 = 7.074, df = 2, p = 0.029). Thus, the burden of p62-positive disease is much greater than that seen on α-synuclein immunohistochemistry and was associated with a variable degree of neuronal loss in the SN (figure).
Figure. Substantia nigra histopathology in C9ORF72−and C9ORF72+ cases.
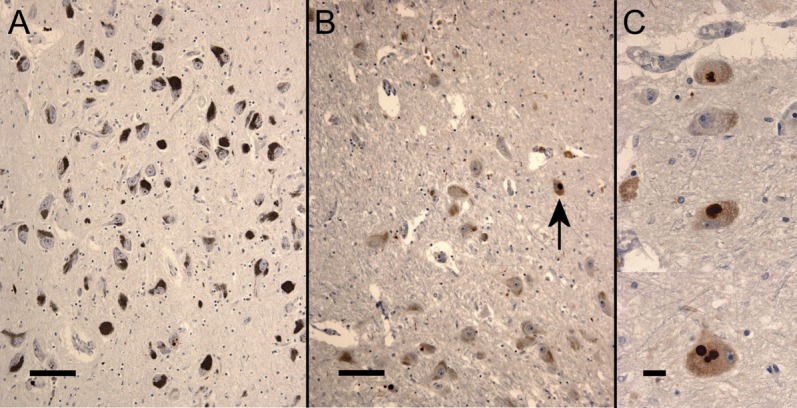
Photomicrographs of the substantia nigra after immunohistochemistry for p62 in C9ORF72− brains (A) and C9ORF72+ brains (B, low power, and C, high power) of patients with the clinical diagnosis of amyotrophic lateral sclerosis showing neuronal loss and p62-positive cytoplasmic inclusions (arrow) in the C9ORF72+ brain. Bar = 100 μm (A and B), and 20 μm (C).
DISCUSSION
Previous studies investigating a possible association among C9ORF72 expansions, parkinsonism, and iPD have concentrated on patients with the clinical rather than the pathologic diagnosis of iPD.e1–e7 These studies have all concluded that C9ORF72 expansions are not a common cause of iPD; however, given that clinical diagnosis has a higher false-positive rate, we chose to conduct a study of pathologically confirmed iPD. Furthermore, by focusing on neuropathology, we also investigated the pathologic basis of parkinsonian presentations in C9ORF72+ ALS patients.
We identified a single C9ORF72+ patient with clinically typical iPD of 377 tested; this frequency is similar to that in controls3 and thus we conclude that C9ORF72 expansions are not a major cause of iPD. Notably, this patient had a family history of ALS and neuropathology consistent with C9ORF72+ disease; although it was not possible to investigate motor neuron loss in the spinal cord, we suspect this patient had subclinical FTD. The presence of type-A FTLD pathology suggests that motor neuron pathology was unlikely.
It is noteworthy that of the 2 cases that were known to have coincident PD-ALS in the Sheffield brain bank, one did3 and one did not have the C9ORF72 repeat expansion.
The absence of α-synuclein pathology in the SN of the vast majority of C9ORF72+ brains further strengthens our assumption that the intracellular mechanisms leading to neuronal cell loss in ALS/FTD and those causing α-synuclein pathology in iPD are distinct. In contrast, p62-positive, TDP-43–negative inclusions in combination with neuronal loss are considerably more common in the SN of C9ORF72+ ALS patients than in C9ORF72− ALS. This p62-positive extrapyramidal pathology is therefore the likely cause of the previously reported increased incidence of parkinsonian features in C9ORF72-related ALS. One could therefore consider C9ORF72-related neurodegeneration as a clinically and pathologically heterogeneous syndrome characterized by a combination of TDP-43 proteinopathy with superimposed extramotor p62-positive, TDP-43–negative pathology. The distribution and severity of this latter pathology is likely to govern the presence of cognitive impairment (in the presence of hippocampal and neocortical pathology) or parkinsonism (in the presence of basal ganglia pathology).
Until the pathogenesis of C9ORF72 disease is fully understood, it remains impossible to exclude C9ORF72 expansions as a very rare cause of α-synucleinopathy and clinical iPD. However, our observation of an alternative pathologic basis for the observed incidence of parkinsonism in C9ORF72+ patients, significantly strengthens the case that C9ORF72 disease and α-synucleinopathy represent distinct pathologic entities.
Understanding that C9ORF72 expansions are a cause of both ALS and a parkinsonian phenocopy is likely to be crucial to the counseling and management of patients with ALS presenting with parkinsonian features, particularly if they have a family history of ALS/PD. Genetic testing for expansions of C9ORF72 will help to differentiate patients with C9ORF72 neurodegeneration from those who have developed more typical PD; a similar suggestion has been made for the use of C9ORF72 genotyping in the differentiation of true Alzheimer disease from FTD caused by mutation of C9ORF72.10
Supplementary Material
ACKNOWLEDGMENT
The authors are grateful to all of the patients with ALS and PD who donated biosamples for research purposes.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- C9ORF72
chromosome 9 open reading frame 72
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobar degeneration
- iPD
idiopathic Parkinson disease
- PD
Parkinson disease
- SN
substantia nigra
- TDP-43
TAR DNA-binding protein 43
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
The study was conceived and designed by authors J.C.-K., J.R.H., G.C., P.J.S., N.W.W., and O.B. Data acquisition was performed by authors J.C.-K., A.F., J.R.H., G.C., J.K., A.M., J.H., S.B.W., P.G.I., C.M.D., T.L., and T.R. Data analysis and interpretation were performed by J.C.-K., A.F., J.R.H., G.C., T.L., T.R., N.W.W., and O.B. The manuscript was critically revised by J.C.-K., J.R.H., P.J.S., and O.B. The study was supervised by T.R., P.J.S., N.W.W., and O.B.
STUDY FUNDING
The biosample collection for the ALS cases was supported by the MND Association and the Wellcome Trust (P.J.S.). Genetic testing was supported by National Institute for Health Research (NIHR) CLAHRC for South Yorkshire and by a Wellcome Trust/MRC joint strategic award (WT089698/Z/09/Z) (N.W.W.). The Queen Square Brain Bank for Neurological Disorders is supported by the Reta Lila Weston Institute for Neurological Studies, the MRC, and the PSP (Europe) Association. The research was, in part, supported by the NIHR Biomedical Research Unit in Dementia based at University College London Hospitals, University College London (UCL).
DISCLOSURE
J. Cooper-Knock is supported by MND Association/Medical Research Council Lady Edith Wolfson Fellowship awards (G0 800380) and (R/132205). A. Frolov reports no disclosures. J. Highley is supported by MND Association/Medical Research Council Lady Edith Wolfson Fellowship awards (G0 800380) and (R/132205), and by Joint Programme for Neurodegenerative Disease (JPND) grant SOPHIA. G. Charlesworth reports no disclosures. J. Kirby is supported by JPND grant SOPHIA and an FP7 grant EuroMOTOR (no. 259867). A. Milano, J. Hartley, P. Ince, and C. McDermott report no disclosures. T. Lashley is supported by an Alzheimer's Research UK fellowship award (ARUK-RF2012-1). T. Revesz received research support from Alzheimer's Research UK, Parkinson's UK, and the Multiple System Atrophy Trust, received honoraria for lectures from Novartis, serves as editorial board member of Acta Neuropathologica, as editorial advisory board member of Neuropathology and Applied Neurobiology, and as Associate Editor of the Journal of Parkinson's Disease. He also received funding for 2 trips from Merck Serono. P. Shaw is supported by JPND grant SOPHIA and an FP7 grant EuroMOTOR (no. 259867). N. Wood is supported by a Wellcome Trust/MRC joint strategic award (WT089698/Z/09/Z). O. Bandmann is supported by Parkinson's UK (G-1007). He received a speaker honorarium from GlaxoSmithKline in 2012. He is a member of the Editorial Board of Neurology®.
REFERENCES
- 1.Lim YM, Park HK, Kim JS, et al. Clinical and neuroimaging characteristics in neurodegenerative overlap syndrome. Neurol Sci 2013;34:875–881 [DOI] [PubMed] [Google Scholar]
- 2.Nishihira Y, Tan CF, Onodera O, et al. Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 2008;116:169–182 [DOI] [PubMed] [Google Scholar]
- 3.Cooper-Knock J, Hewitt C, Highley JR, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012;135:751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majounie E, Renton AE, Mok K. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Dowd S, Curtin D, Waite AJ, et al. C9ORF72 expansion in amyotrophic lateral sclerosis/frontotemporal dementia also causes parkinsonism. Mov Disord 2012;27:1072–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lill CM, Roehr JT, McQueen MB, et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson's disease genetics: the PDGene database. PLoS Genet 2012;8:e1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mackenzie IR, Neumann M, Baborie A, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol 2011;122:111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majounie E, Abramzon Y, Renton AE, et al. Repeat expansion in C9ORF72 in Alzheimer's disease. N Engl J Med 2012;366:283–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.