

Abstract
Purpose:
Fabry disease is a rare, X-linked, inherited lysosomal storage disorder that can be treated with the enzymes agalsidase alfa (Replagal) and agalsidase beta (Fabrazyme). Currently, there is a global shortage of agalsidase beta, and this has increased global demand for agalsidase alfa. We assess the feasibility of switching patients on agalsidase beta treatment to agalsidase alfa instead.
Methods:
This analysis is part of an ongoing observational study involving 11 patients with Fabry disease in whom the treatment was switched from agalsidase beta (1 mg/kg every other week) to agalsidase alfa (0.2 mg/kg every other week). Data were collected for a minimum of 36 months: 24 months before and 12 months after the switch. Serial data were evaluated with respect to renal function, cardiac mass, pain, quality of life, and tolerability/safety.
Results:
Indexes of renal function (estimated glomerular filtration rate) and cardiac mass (left-ventricular mass index), pain (Brief Pain Inventory), and quality of life (EuroQoL-Dimensions) clearly showed that, in patients switched to agalsidase alfa, Fabry disease stabilized during the 12 months of follow-up.
Conclusion:
Despite the limitations of this preliminary observational study, it was found that all the patients maintained disease stability when treated with agalsidase alfa, as evidenced by estimated glomerular filtration rate, left-ventricular mass index, pain scores, and quality-of-life indexes, throughout 12 months of follow-up.
Keywords: agalsidase alfa, agalsidase beta, Fabry disease, Fabryzyme, Replagal
Introduction
Fabry disease is an X-linked, inherited, lysosomal storage disorder caused by a deficiency of the enzyme α-galactosidase A.1 α-Galactosidase deficiency results in the accumulation of globotriaosylceramide (Gb3 or ceramide trihexoside) and other neutral glycolipids in many tissues and cell types throughout the body, ultimately leading to cellular abnormalities and triggering inflammation and fibrosis.2 The consequences of these biochemical changes can be wide-ranging, and include acroparesthesias, hypohidrosis, angiokeratomas, corneal opacities, cerebrovascular lesions, cardiac disorders, and renal dysfunction.1,3 Organ dysfunction and pain are usually the first clinical manifestations of Fabry disease and, at this stage, some degree of irreversible damage may already have occurred. The onset of symptoms generally starts during childhood and, by middle-age, life-threatening complications often develop in untreated patients.2
Fabry disease is rare, with an incidence estimated to be between 1 in 40,000 and 1 in 117,000 worldwide.4 There are no known ethnic predispositions, although regional pockets with increased prevalence can occur because of founder effects, as documented in Nova Scotia (Canada) and West Virginia (USA).2 Life expectancy is reduced by ~20 years in untreated men and by ~15 years in untreated women.3,5 The cause of mortality in female patients is usually cardiac failure, whereas renal failure is the most frequent cause of death in male patients.3 In the recent past, organ transplantation and dialysis have increased the longevity of patients with Fabry disease, and this has resulted in an increased prevalence of cardiac and cerebrovascular events in these patients.6,7
A milestone in the treatment of Fabry disease occurred with the introduction of enzyme replacement therapy (ERT) in 2001.8 Two preparations of the enzyme α-galactosidase A are now available: agalsidase alfa (Replagal; Shire Human Genetic Therapies, Lexington, MA) and agalsidase beta (Fabrazyme; Genzyme, Cambridge, MA). To date, >2,000 patients with Fabry disease have been treated with ERT.9 However, since June 2009, there has been a worldwide shortage of agalsidase beta as a result of viral contamination in the manufacturer's production facility. This shortage has led to a series of recommendations including dosage reductions (i.e., spreading the usual dose over a longer time period). The recommendation was made in an attempt to maintain current users on the limited supplies of agalsidase beta that are available, in view of the fact that full-scale production is not anticipated to be restored before the first quarter of 2012 at the earliest. However, these new guidelines were reviewed by a consensus group of treating physicians and reported by the European Medicines Agency in October 2010.10,11,12 At this meeting, it was noted that there was a steady increase in the number of adverse events reported, presumably because of clinical deterioration in patients receiving agalsidase beta at less than the licensed dose. Currently, the general consensus is that patients should receive only licensed, approved doses of agalsidase beta (1 mg/kg once every 2 weeks), and if this is not possible, then switching to treatment with agalsidase alfa (0.2 mg/kg once every 2 weeks) is a possible option.10 However, as noted by the European Medicines Agency, very limited clinical data are available regarding patients in whom the treatment is switched between the different forms of α-galactosidase A enzyme.10,11,12
In Japan, from the year 2009, we started switching treatment from agalsidase beta to agalsidase alfa for patients with Fabry disease, because of the worldwide shortage of agalsidase beta. In this report, we review clinical findings in 11 patients who have been receiving the new treatment (after the switch) for at least 12 months. The aim of this study was to ascertain the safety and effects on disease stability when treatment was switched from agalsidase beta to agalsidase alfa. We investigated the potential clinical changes by evaluating 12-month prospective data and comparing them with retrospective data recorded in the patients' medical charts for at least 24 months before the switch.
Methods
Study design and plan
This is an ongoing observational study involving male and female patients with Fabry disease in whom treatment was switched from agalsidase beta (Fabrazyme) to agalsidase alfa (Replagal). Before the supply shortage of agalsidase beta occurred, 13 Fabry patients in Japan were being treated with agalsidase beta 1 mg/ kg every other week in our hospital. Of these, 11 agreed to switch to agalsidase alfa 0.2 mg/kg every other week without any interval, whereas the other 2 patients preferred to remain on agalsidase beta at the decreased dose of 0.3 mg/kg every other week. The first patient for the switched treatment regimen was recruited on 5 November 2009, and the final patient on 24 November 2009. All data were collected from medical records at the medical institutes where the patients were being treated for Fabry disease. The names and identities of the patients have been removed or changed to preserve patient anonymity and to maintain confidentiality.
This study reports the data from these 11 patients with Fabry disease who underwent a switch in therapy, and for whom at least 36 months of continuous data were available, including 24 months before and 12 months after the switch from agalsidase beta to agalsidase alfa. Clinical assessment was performed through evaluation of relevant disease measures, treatment parameters, safety/tolerability, and Fabry disease–related clinical events, as recorded on the medical charts of individual patients. The study was approved by the appropriate institutional review boards and was conducted in accordance with the ethical provisions set out in the Declaration of Helsinki, and all applicable local laws and regulations. All the patients were required to provide written, informed consent before inclusion into the study.
Patients
Patients were eligible for the study if they met the following criteria:
Hemizygous male or heterozygous female with a confirmed diagnosis of genetically acquired Fabry disease;
Currently being treated with agalsidase beta at the recommended dosage of 1 mg/kg every other week for the indication of Fabry disease as previously diagnosed at our institution using Japanese diagnostic criteria including reduced α-galactosidase A activity and genotyping; family history; abnormal Gb3 deposits in biopsy samples; and increased urinary Gb3 excretion;
Received agalsidase alfa 0.2 mg/kg every other week for >12 months after the switch from agalsidase beta; and
The medical chart data included detailed information regarding Fabry disease history, laboratory data, clinical findings relevant to Fabry disease, and relevant safety information.
The main exclusion criteria were
Concomitant administration of agalsidase alfa before the switch;
The switch took place before November 2009;
The switch was initiated for any reason other than the worldwide shortage of agalsidase beta; or
The patient received any investigational drugs, or treatment using investigational medical devices, within the 30 days before commencement of the study.
Treatment
Patients who were treated with agalsidase beta 1 mg/kg every other week were switched immediately (without any interval) to treatment with agalsidase alfa 0.2 mg/kg, administered intravenously every other week, over a period of 40–60 min.
Data collection and study end points
The data collected retrospectively included patient demographics at the time of the switch, such as gender, age, height and body weight, medical history, Fabry disease diagnosis, vital signs, and laboratory data (serum biochemistry, hematology, and urinalysis). Also documented retrospectively were relevant data pertaining to plasma and urinary Gb3 levels, renal function (serum and urinary creatinine levels and/or estimated glomerular filtration rate (eGFR) if available), cardiac mass (left-ventricular mass index (LVM index)) obtained by echocardiography, Fabry disease–related pain and quality of life (QOL), and safety/tolerability data (all adverse events, infusion reactions, and antibody titers of anti-agalsidase alfa and beta antibodies).
The following were determined at the preswitch period (baseline) and prospectively at 6-month intervals:
Plasma Gb3 levels
Renal function (eGFR, using the Chronic Kidney Disease Epidemiology Collaboration equation)13
Cardiac mass (LVM adjusted for height with the Devereux formula)14,15
Pain scores using the Brief Pain Inventory questionnaire (average and worst scores)16
QOL (EuroQoL-Dimensions)17
Severity of signs and symptoms of Fabry disease using the Mainz Severity Score Index18
The safety of agalsidase alfa was evaluated by monitoring the occurrence of adverse events, the use of concomitant drugs, laboratory test results, vital signs, antibody production, and the use of premedication (for infusion reactions).
Statistical methods
Unless stated otherwise, the data are presented as mean values ± s.d., maximum, minimum, median, and 25 and 75% quartiles. Given the small numbers of patients involved in this case series, limited statistical analyses were performed. Paired t-tests or one-sample Wilcoxon tests were used to compare preswitch findings on agalsidase beta with 6-month and 12-month results after switching to agalsidase alfa; P values <0.05 were considered statistically significant.
Results
Demographics
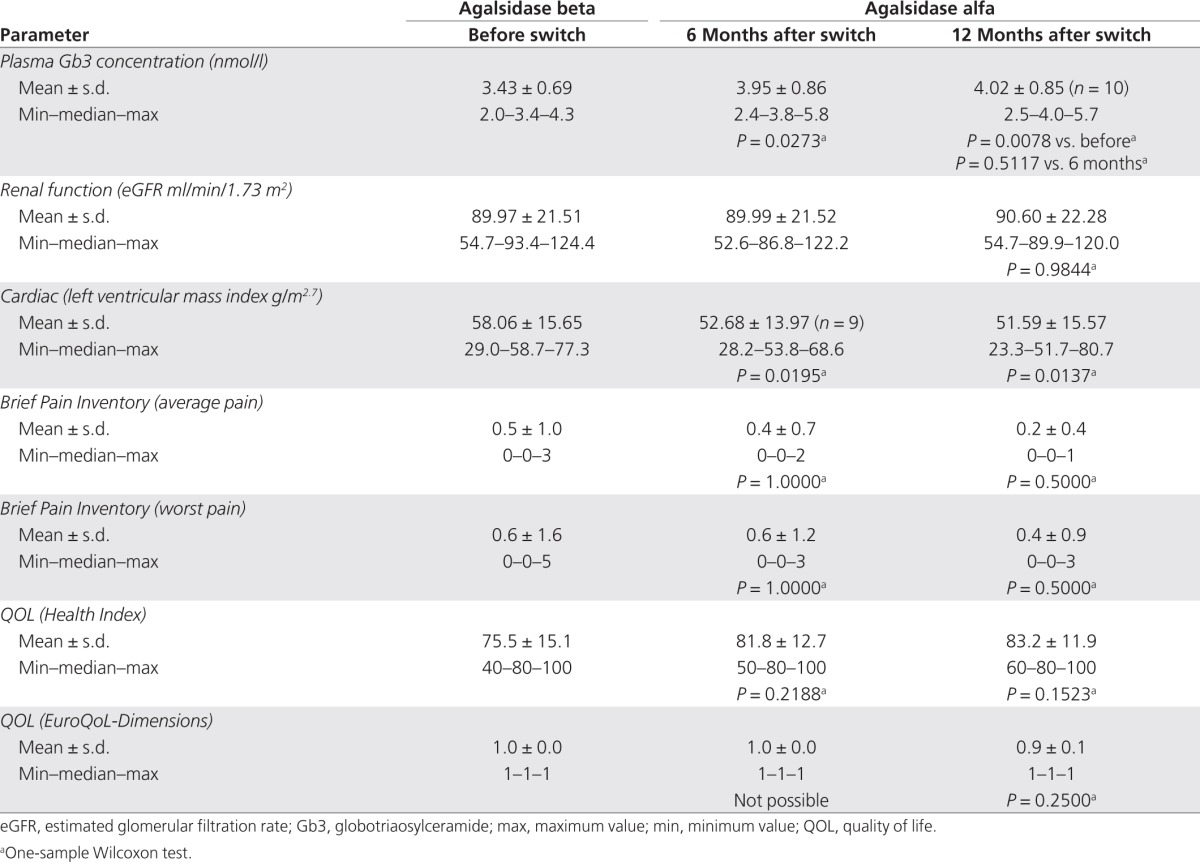
This study is ongoing and the data (retrospective, baseline, and postswitch) reported herein are those of 11 patients with Fabry disease who agreed to switch treatment from agalsidase beta to agalsidase alfa. They were part of a group of 13 patients on agalsidase beta treatment for whom a change in ERT was necessitated by the shortage of supply of the treatment drug; 2 of the 13 patients chose to remain on agalsidase beta treatment at a reduced dosage of 0.3 mg/kg; the data of these 2 patients are not included in this analysis. The 11 patients in whom this preliminary assessment was carried out comprised 7 women (mean age 52 years) and 4 men (mean age 38 years) with Fabry disease who had completed 12 months of postswitch therapy with agalsidase alfa by the end of 2010. Individual patient baseline demographic data, including the date at which treatment was initiated, are shown in Table 1; the mean, median, minimum, and maximum baseline findings relating to renal function, LVM index, pain, and QOL are presented in Table 2. Three patients were receiving concomitant medications: carbamazepine for pain relief (n = 2) and an angiotensin receptor blocker for hypertension (n = 1).
Table 1. Individual demographic and preswitch data for 11 patients with Fabry disease who switched from agalsidase beta to agalsidase alfa.
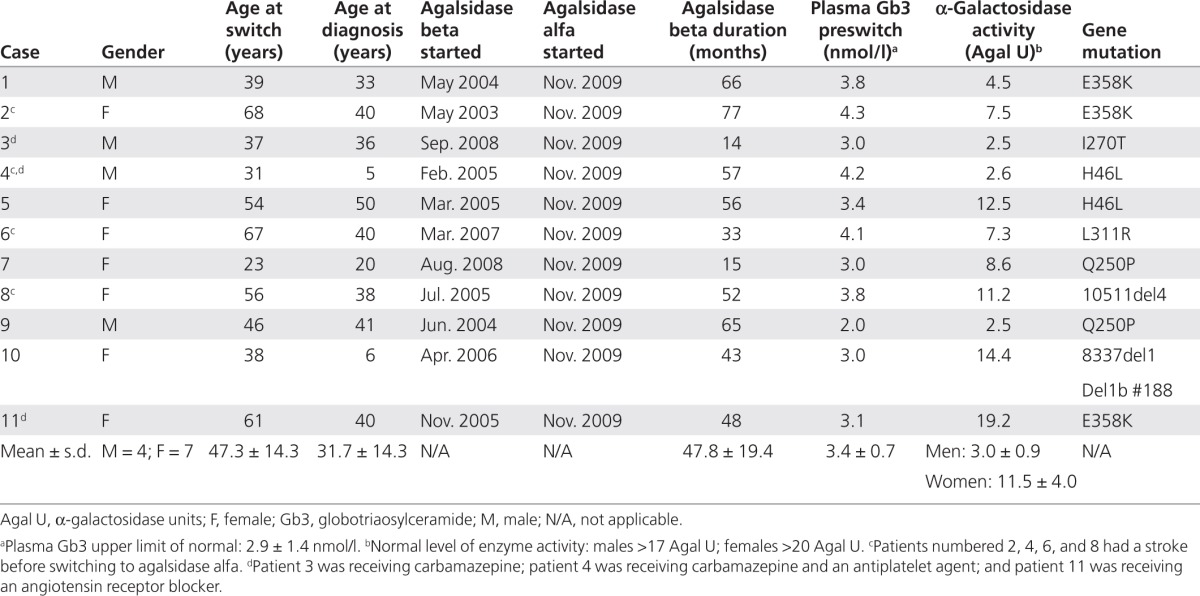
Table 2. Clinical measures at baseline, and 6 and 12 months after switching from agalsidase beta (Fabrazyme) to agalsidase alfa (Replagal) in 11 patients with Fabry disease.
Clinical evaluations after the switch to agalsidase alfa treatment
The main clinical findings in this group of 11 patients with Fabry disease for whom the treatment regimen was switched from agalsidase beta to agalsidase alfa (parameters at 6 and 12 months after the switch versus before the switch) are summarized in Table 2. In terms of clinical outcomes (renal function, cardiac mass, pain, and QOL), there were no statistically or clinically significant changes at follow-up evaluations carried out at 6 and 12 months after the treatment switch as compared with preswitch values. Plasma Gb3 levels increased from a preswitch value of 3.43 ± 0.69 nmol/l to 3.95 ± 0.86 nmol/l (P = 0.0273) at 6 months, and then remained stable, with a mean value of 4.02 ± 0.85 nmol/l at 12 months (P = 0.5117 relative to the 6-month value). In addition, Mainz Severity Score Index scores indicated that all the patients had mild-to-moderate Fabry disease at baseline (Mainz Severity Score Index ≤40)18 and that the disease remained stable at 12 months after switching to agalsidase alfa, with some slight improvements in general and neurological scores recorded in three of the individuals (Table 3). In the period before the switching of the treatment, four patients had a history of stroke. Of these, one has moderate hemiparesis and is receiving antiplatelet medication, whereas the other three are mild cases as confirmed by computed tomography scans. No patient suffered a stroke during the 12 months of follow-up after switching to agalsidase alfa. One patient experienced a marked drop in GFR during the last 12 months of treatment on agalsidase beta (from 75 to 55 ml/min/1.73 m2); after switching to agalsidase alfa treatment, the GFR level in this patient remained relatively stable and was 55 ml/min/1.73 m2 at the last visit (12 months after the switch).
Table 3. Mainz Severity Scores at baseline and 12 months after switching from agalsidase beta (Fabrazyme) to agalsidase alfa (Replagal) in 11 patients with Fabry disease.
Renal function
All indexes of renal function as assessed by eGFR, including mean, median, minimum, and maximum values, clearly remained stable in both male and female patients with Fabry disease who switched from agalsidase beta to agalsidase alfa (Table 2). Individual values confirm this finding, with no patient demonstrating clinically significant changes in eGFR after the ERT switch (Figure 1). As regards renal disease, at 24 months before the switch when the patients were receiving agalsidase beta, five patients were in stage 1 of renal disease (eGFR ≥90 ml/min/1.73 m2), five were in stage 2 (60–89 ml/ min/1.73 m2), and one was in stage 3 (30–59 ml/ min/1.73 m2). Identical results were obtained in these patients after 12 months of treatment with agalsidase alfa (no statistically significant differences were observed using the extended McNemar test).
Figure 1.

Individual eGFR values in 11 patients with Fabry disease treated with agalsidase beta (months −24 to 0) before being switched to treatment with agalsidase alfa (months 6 and 12). eGFR, estimated glomerular filtration rate.
In addition, blood levels of creatinine, sodium, potassium, and β2-microglobulin were determined, and urinalysis was performed to determine the presence of protein, occult blood, and β2-microglobulin. None of the results revealed any statistically significant or clinically relevant abnormalities after the switch from agalsidase beta to agalsidase alfa.
Cardiac status
There was significant improvement in cardiac mass after the switch in treatment, as demonstrated by reductions in LVM index from a preswitch mean ± s.d. value of 58.06 ± 15.65 to 52.68 ± 13.97 (P = 0.0195) and 51.59 ± 15.57 g/m2.7(P = 0.0137) at 6 months and 12 months, respectively (Table 4). The benefits were observed in both female patients (60.43 ± 18.35 preswitch to 54.54 ± 15.59, P = 0.0625 at 6 months and 52.64 ± 18.27 g/ m2.7, P = 0.0469 at 12 months), and male patients (53.93 ± 10.32 preswitch to 50.35 ± 13.53, P = 0.3750 at 6 months and 49.75 ± 11.54 g/m2.7, P = 0.3750 at 12 months). However, the numbers of patients in these subgroups were too small to detect statistically significant differences at all time points.
Table 4. Cardiac measures at baseline, and 6 and 12 months after switching from agalsidase beta (Fabrazyme) to agalsidase alfa (Replagal) in 11 patients with Fabry disease.
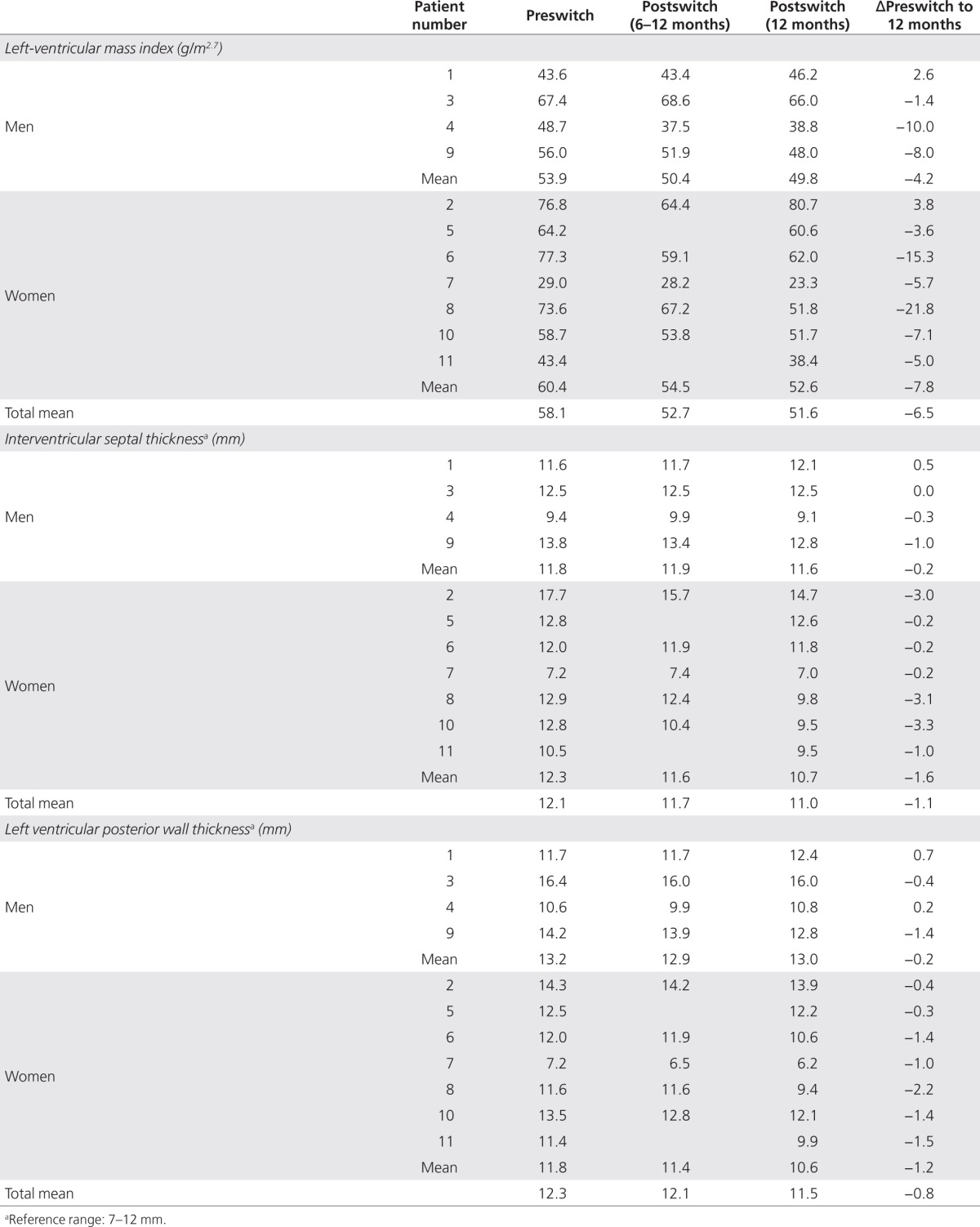
Cardiac ultrasonography was performed during treatment with agalsidase beta, and again at 6 and 12 months after the switch to agalsidase alfa. Before the switch, the mean ± s.d. interventricular septum thickness was 12.1 ± 2.6 mm and the ventricular posterior wall thickness was 12.3 ± 2.4 mm; 12 months after the switch to agalsidase alfa, the interventricular septum thickness was 11.0 ± 2.2 mm and ventricular posterior wall thickness was 11.5 ± 2.6 mm (Table 4). The improvements in these parameters consequent to the switch in treatment, while mirroring the reduction in cardiac mass, did not achieve statistical significance.
In female patients with heterozygous Fabry disease, preswitch values of interventricular septum thickness and ventricular posterior wall thickness were 12.3 ± 3.1 mm and 11.8 ± 2.3 mm, respectively; at 12 months after the switch, these values had decreased to 10.7 ± 2.5 mm and 10.6 ± 2.5 mm, respectively. However, these decreases in interventricular septum thickness (Δmean ± s.d.: −1.6 ± 1.5 mm) and ventricular posterior wall thickness (Δmean ± s.d.: −1.2 ± 0.7 mm), while encouraging, were not statistically significant (Table 4).
Electrocardiography and chest X-ray examination were performed at intervals of 6 months. Electrocardiography findings identified a decrease in RV5 + SV1 in two of the four patients with classic-type Fabry disease, and in three of the seven patients with heterozygous disease. In 10 of the 11 patients, no clinically relevant changes were noted in the electrocardiograph after the switch from agalsidase beta to agalsidase alfa. One patient with classic-type Fabry disease developed atrial fibrillation, but it is not known whether this event was associated with worsening of Fabry disease or with the switch from agalsidase beta to agalsidase alfa. The atrial fibrillation resolved naturally without intervention within 6 months. With regard to chest X-rays, no significant differences in cardiothoracic ratio were found after the switch to agalsidase alfa.
Pain
Pain, measured using the Brief Pain Inventory questionnaire for average and worst scores, showed no increase after the switch to agalsidase alfa (Table 2). Of the 11 patients, 9 had no pain at the time of switching, and they have remained pain free 1 year later. Two patients were experiencing pain when the switch took place, but there has been some improvement in one of them, who has managed to discontinue carbamazepine.
QOL
QOL as assessed by EuroQoL-Dimensions confirmed the stabilization of Fabry disease relative to preswitch values (Table 2).
Safety and tolerability
Throughout the observation period, blood tests were performed monthly, and urinalysis every 3 months. No treatment-related adverse reactions were noted, with the absence of abnormal laboratory test values such as white-cell disturbances, thrombocytopenia, and changes in hepatic or renal function. No treatment-related atrial fibrillation, ventricular premature beats, tachyarrhythmias, cardiac failure, or other events were reported within 24 h of administration of ERT.
One case of a treatment-related allergic reaction (presenting as worsening allergic rhinitis) was noted after administration of agalsidase beta. This patient consistently developed nasal discharge when agalsidase beta was administered, but, after the switch to agalsidase alfa, no treatment-related allergic symptoms occurred in this patient. In the other 10 patients, no allergic findings such as chills, nasal discharge, or urticaria were documented.
One patient developed antibodies against agalsidase beta (enzyme-linked immunosorbent assay method) and the antibody test remained positive until the switch to agalsidase alfa (Immunoglobulin G (IgG) titer 1:3,200). Before the switch in treatment was carried out, and again at 6 months and 12 months after the switch, all the patients were tested for antibodies against agalsidase alfa using the enzyme-linked immunosorbent assay method. The results were negative for all the patients at all times, including in the patient who tested positive for antibodies against agalsidase beta.
Discussion
In this report, we present data from 11 patients with Fabry disease who have received at least 12 months of treatment with agalsidase alfa after switching from agalsidase beta. The switch was necessitated by a worldwide shortage of agalsidase beta as a result of manufacturing problems. There are several important differences between these two products. Agalsidase alfa is produced by a genetically engineered human cell system whereas agalsidase beta is produced using Chinese hamster ovary cells. Additional differences include posttranslational modifications such as glycosylation, sialylation, and phosphorylation of mannose residues, which may affect tissue distribution and antibody production rates for the two enzymes. The composition of the commercial products, including other constituents of the formulation, may also cause differences between the two drugs as regards risk for hypersensitivity. The approved dosage and method of administration of agalsidase alfa involve administering 0.2 mg/kg intravenously over a period of 40 min every other week; agalsidase beta is administered at 1 mg/kg intravenously over a period of 1–2 h every other week. Although both drugs came onto the market ~10 years ago in Europe, there appears to be very little clinical information in terms of efficacy and/or safety profiles of patients who have switched from one enzyme to the other.10,11,12 Since our article was submitted for publication, Smid and colleagues in The Netherlands have reported findings from a study of 35 patients with Fabry disease whose treatment was managed during the worldwide shortage of agalsidase beta.19 Initially, all the patients were given a reduced-dosage regimen with agalsidase beta before two patients were switched to agalsidase alfa. In the final 12 months, another 18 patients switched to agalsidase alfa at different times. The results of this study demonstrated no increase in the incidence of clinical events during the period of enzyme shortage, although lysoGb3 levels increased. The authors attributed these increases to a reduction in agalsidase dosage rather than to the change of enzyme.19
Long-term treatment with agalsidase alfa has been shown to produce substantial and sustained clinical benefits in patients with Fabry disease.20 In this study, we undertook a systematic chart review of retrospective clinical data pertaining to 11 patients who switched ERT from agalsidase beta to agalsidase alfa, and who underwent prospective assessment of renal function, cardiac mass, pain, QOL, and plasma Gb3 levels at 6 and 12 months after switching. The initial findings are encouraging, and patients who switched to agalsidase alfa remained stable as assessed by eGFR, while cardiac mass (as estimated by LVM index) was significantly improved. Furthermore, the 11 patients have remained mostly pain free with no detrimental changes in QOL. After switching to agalsidase alfa, the patients had somewhat higher plasma Gb3 levels: 3.43 ± 0.69 nmol/l preswitch vs. 3.95 ± 0.86 nmol/l (P = 0.0297) after 6 months. However, from 6 months onward the plasma Gb3 level remained relatively constant and was 4.02 ± 0.85 nmol/l after 12 months. The clinical relevance of these changes is questionable because, in the absence of a control group, it is not known whether the level would have increased even if the patients had remained on agalsidase beta treatment. Furthermore, the role of Gb3 as a biological marker for Fabry disease has been questioned.8
The development of neutralizing antibodies is a consideration with respect to recombinant proteins such as agalsidase alfa and agalsidase beta. Cross-reactivity of IgG antibodies has been observed against agalsidase alfa and agalsidase beta in patients with Fabry disease.21 However, the IgE antibody response to ERT, and possible cross-reactivity of IgE antibodies to agalsidase alfa and agalsidase beta, does not appear to have been reported. Of note, a case has been reported recently of a 17-year-old Japanese boy with Fabry disease who developed anti-agalsidase beta antibodies with subsequent eosinophilia and adverse clinical symptoms. After the treatment was switched to agalsidase alfa, no IgE antibody response occurred and the eosinophilia resolved.22
It is important to discuss the inherent limitations of our preliminary investigation of the results of switching ERT in patients with Fabry disease. First and foremost, the data are available only for a small number of patients, but this is of course due to the rare nature of this genetic disorder. This limitation could mean that the sample is not truly representative in terms of baseline characteristics, disease severity, disease duration, level of impairment, or other parameters. It also means that, for some of the analyses, the sample size lacks the necessary power to detect statistically significant differences and increases the likelihood of false positives/negatives. Another potential limitation is the fact that clinical assessments were performed by medical chart review at several different institutions; some of the retrospective data may, therefore, lack consistency because of variations among centers as regards the range and type of investigations performed and the rigor of data entry. However, these limitations highlight the problems facing physicians when treating patients with Fabry disease, given that each case is different. Despite these limitations, the study revealed that all the patients maintained disease stability as evidenced by plasma Gb3 levels, eGFR, LVM index, pain scores, and QOL indexes throughout the 12 months of follow-up. However, longer-term observation is needed to confirm the efficacy and safety of switching therapy from agalsidase beta to agalsidase alfa in patients with Fabry disease. We will continue to monitor clinical efficacy and safety outcomes in all the patients in whom treatment is switched from agalsidase beta to agalsidase alfa.
In conclusion, the clinical findings from this study, although preliminary in nature, provide an encouraging level of support for the idea that patients with Fabry disease who require an alternative to agalsidase beta will find it useful to switch to agalsidase alfa. This switch in therapy raised no safety concerns after 12 months of follow-up.
Disclosure
Shire funded medical writing assistance by Steve Clissold, Content Ed Net.
Acknowledgments
The authors wish to thank all patients who participated in this study, and Steve Clissold PhD, Content Ed Net, for editorial assistance, which was funded by Shire PLC, Japan.
References
- Nagueh SF. Fabry disease. Heart. 2003;89:819–820. doi: 10.1136/heart.89.8.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate YA, Hopkin RJ. Fabry's disease. Lancet. 2008;372:1427–1435. doi: 10.1016/S0140-6736(08)61589-5. [DOI] [PubMed] [Google Scholar]
- Fabry H. Angiokeratoma corporis diffusum–Fabry disease: historical review from the original description to the introduction of enzyme replacement therapy. Acta Paediatr Suppl. 2002;91:3–5. doi: 10.1111/j.1651-2227.2002.tb03102.x. [DOI] [PubMed] [Google Scholar]
- Mehta A, Ricci R, Widmer U.et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey Eur J Clin Invest 200434236–242. [DOI] [PubMed] [Google Scholar]
- MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 98 hemizygous males. J Med Genet. 2001;38:750–760. doi: 10.1136/jmg.38.11.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhart A, Kampmann C, Zamorano JL, European FOS Investigators et al. Cardiac manifestations of Anderson-Fabry disease: results from the international Fabry outcome survey. Eur Heart J. 2007;28:1228–1235. doi: 10.1093/eurheartj/ehm153. [DOI] [PubMed] [Google Scholar]
- Mehta A, Clarke JT, Giugliani R, FOS Investigators et al. Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. J Med Genet. 2009;46:548–552. doi: 10.1136/jmg.2008.065904. [DOI] [PubMed] [Google Scholar]
- Lidove O, Joly D, Barbey F.et al. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature Int J Clin Pract 200761293–302. [DOI] [PubMed] [Google Scholar]
- Hoffmann B. Fabry disease: recent advances in pathology, diagnosis, treatment and monitoring. Orphanet J Rare Dis. 2009;4:21. doi: 10.1186/1750-1172-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMEA Assessment report on the shortage of Fabrazyme. Overview of Shortage Period: Spontaneous Reports from June 2009 through 15 September 2010 and Registry Data from June 2009 through 05 August 2010 . http://www.ema.europa.eu/docs/en_GB/document_library/ Other/2010/11/WC500099241.pdf . Accessed September 2011.
- Sirrs S. The Fabrazyme shortage–a call to action for metabolic physicians. Mol Genet Metab. 2011;102:4–5. doi: 10.1016/j.ymgme.2010.11.002. [DOI] [PubMed] [Google Scholar]
- Linthorst GE, Germain DP, Hollak CE, European Medicines Agency et al. Expert opinion on temporary treatment recommendations for Fabry disease during the shortage of enzyme replacement therapy (ERT) Mol Genet Metab. 2011;102:99–102. doi: 10.1016/j.ymgme.2010.11.155. [DOI] [PubMed] [Google Scholar]
- Levey AS, Stevens LA, Schmid CH, CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devereux RB, Alonso DR, Lutas EM.et al. Echocardiographic assessment of left ventricular hypertrophy: comparison to necropsy findings Am J Cardiol 198657450–458. [DOI] [PubMed] [Google Scholar]
- Lang RM, Bierig M, Devereux RB.et al.; Chamber Quantification Writing Group; American Society of Echocardiography's Guidelines and Standards Committee; European Association of Echocardiography. Recommendations for chamber quantification: a report from the American Society of Echocardiography's Guidelines and Standards Committee and the Chamber Quantification Writing Group, developed in conjunction with the European Association of Echocardiography, a branch of the European Society of Cardiology J Am Soc Echocardiogr 2005181440–1463. [DOI] [PubMed] [Google Scholar]
- Tan G, Jensen MP, Thornby JI, Shanti BF. Validation of the Brief Pain Inventory for chronic nonmalignant pain. J Pain. 2004;5:133–137. doi: 10.1016/j.jpain.2003.12.005. [DOI] [PubMed] [Google Scholar]
- The EuroQoL Group EuroQoL-a new facility for the measurement of health-related quality of life. Health Policy. 1990;16:199–208. doi: 10.1016/0168-8510(90)90421-9. [DOI] [PubMed] [Google Scholar]
- Whybra C, Kampmann C, Krummenauer F.et al. The Mainz Severity Score Index: a new instrument for quantifying the Anderson-Fabry disease phenotype, and the response of patients to enzyme replacement therapy Clin Genet 200465299–307. [DOI] [PubMed] [Google Scholar]
- Smid BE, Rombach SM, Aerts JM.et al. Consequences of a global enzyme shortage of agalsidase beta in adult Dutch Fabry patients Orphanet J Rare Dis 2011669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Beck M, Elliott P, Fabry Outcome Survey investigators et al. Enzyme replacement therapy with agalsidase alfa in patients with Fabry's disease: an analysis of registry data. Lancet. 2009;374:1986–1996. doi: 10.1016/S0140-6736(09)61493-8. [DOI] [PubMed] [Google Scholar]
- Linthorst GE, Hollak CE, Donker-Koopman WE, Strijland A, Aerts JM. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Takeda T, Hoshina T.et al. Enzyme replacement therapy in a patient with Fabry disease and the development of IgE antibodies against agalsidase beta but not agalsidase alpha J Inherit Metab Dis 2010. e-pub ahead of print 22 June 2010. [DOI] [PubMed]