

Abstract
The EGFR-family member HER4 undergoes regulated intramembrane proteolysis (RIP) to generate an intracellular domain (4ICD) that functions as a transcriptional coactivator. Accordingly, 4ICD coactivates the estrogen receptor (ER) and associates with ER at target gene promoters in breast tumor cells. However, the extent of 4ICD coactivation of ER and the functional significance of the 4ICD/ER transcriptional complex is unclear. To identify 4ICD coactivated genes we performed a microarray gene expression analysis of β-estradiol treated cells comparing control MCF-7 breast cancer cells to MCF-7 cells where HER4 expression was stably suppressed using a shRNA. In the MCF-7 cell line, β-estradiol significantly stimulated or repressed by 2-fold or more 726 or 53 genes, respectively. Significantly, HER4/4ICD was an obligate coactivator for 277 or 38% of the β-estradiol stimulated genes. Ingenuity Pathway Analysis of β-estradiol regulated genes identified significant associations with multiple cellular functions regulating cellular growth and proliferation, cell cycle progression, cancer metastasis, decreased hypoplasia, tumor cell migration, apoptotic resistance of tumor cells, and increased transcription. Genes coactivated by 4ICD displayed functional specificity by only significantly contributing to cellular growth and proliferation, cell cycle progression, and decreased hypoplasia. In direct concordance with these in situ results we show that HER4 knockdown in MCF-7 cells results in a loss of estrogen stimulated tumor cell proliferation and cell cycle progression, whereas, estrogen stimulated tumor cell migration was unaffected by loss of HER4 expression. In summary, we demonstrate for the first time that a cell surface receptor functions as an obligate ER coactivator with functional specificity associated with breast tumor cell proliferation and cell cycle progression. Nearly 90% of ER positive tumors coexpress HER4, therefore we predict that the majority of breast cancer patients would benefit from a strategy to therapeutic disengage ER/4ICD coregulated tumor cell proliferation.
Keywords: Breast cancer, EGFR-family, steroid receptors, cell cycle, p160 steroid receptor coactivator, gene regulation
INTRODUCTION
The estrogen receptor (ER) is a nuclear receptor overexpressed in over 70% of primary breast cancers [1]. The biological activity of ER is regulated, in part, through the association with a functionally diverse complex of transcriptional coregulators at target gene promoters. For example, independently suppressed expression of p160 steroid receptor coactivator (SRC) family members results in impaired estrogen stimulated tumor cell proliferation [2,3]. The important contribution of ER associated coactivator complexes to breast tumor cell proliferation is further underscored by the success of tamoxifen therapy for the treatment of ER positive breast cancer. Binding of tamoxifen alters ER structure thereby disrupting ER association with coactivators and arresting tumor cell cycle progression [4,5]. Therefore targeting ER coactivators has been suggested as a potential therapeutic strategy for breast cancer [6].
The epidermal growth factor receptor (EGFR) family member, HER4, is a putative ER coactivator with unique properties. HER4 undergoes proteolytic processing at the cell surface to release an independently signaling HER4 intracellular domain (4ICD) with transcriptional coactivator activity [7,8]. For example, 4ICD interacts with ER in response to estrogen and coactivates specific genes by associating with their promoters in a complex with ER [9,10]. Suppression of HER4 expression abrogates estrogen stimulated breast tumor cell proliferation [10,11,12] raising the possibility that 4ICD coactivation of ER regulates genes associated with tumorigenesis. Accordingly, 4ICD coactivates CXCL12 (SDF-1) [10], an estrogen regulated gene involved in tumor growth and metastasis [13]. Clinically, HER4 expression is significantly associated with ER expression [14] and up to 90% of ER positive tumors coexpress HER4 [15]. These clinical and experimental observations suggest a strong tumor cell selection for establishing a HER4/ER proliferative signal in breast cancer.
Although substantial evidence supports a role for ER coactivators in breast tumorigenesis, the estrogen regulated genes which serve as coactivator targets contributing to tumor cell proliferation remain to be determined. Here we use microarray gene expression analysis to demonstrate that 4ICD is required for the coactivation of 38% of estrogen regulated genes. Using a systems biology approach and biological assays we further show primary functions for 4ICD coactivated genes in cell cycle progression and tumor cell proliferation. Our results represent the first global gene expression functional analysis for an ER coactivator and establish 4ICD as an important ER coactivator required for estrogen stimulated breast tumor cell proliferation.
MATERIALS AND METHODS
Cell Lines
MCF-7 cells were purchased from American Type Culture Collection and cultured according to their instructions. For stable suppression of HER4 to generate the pooled MCF-7/H4Si4 cell line, MCF-7 cells were transfected with the MISSION shRNA plasmid-DNA TRCN0000039690 targeting HER4 (Sigma) using Fugene6 (Roche). The pooled MCF-7/pLKO vector control cell line was generated by transfecting MCF-7 cells with the pLKO.1 plasmid (Sigma) using Fugene6.
Western Blot Analysis
Total cell lysates were prepared in RIPA Buffer (10 mM NaPO4, pH 7.2, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Na-deoxycholate, 1% Nonedit P40) containing Complete EDTA-free Protease Inhibitor Cocktail (Roche) and PhosSTOP phosphatase inhibitor (Roche). Lysates were solubilized in NuPAGE LDS Sample Buffer (4X) (Life Technologies) with NuPAGE Sample Reducing Agent (10X) (Life Technologies) added to 1X. Lysates were separated by electrophoresis with 20 μg of protein per lane in a NuPAGE 4–12% Bis-Tris Gel (Life Technologies) and transferred to an Immobilon-FL 0.45 μm Pore Size Transfer Membrane (Millipore) using a Trans-Blot Semi-Dry Electrophoretic Transfer Cell (Bio-Rad). Membranes were blocked and all antibody dilutions were performed in 5% bovine serum albumin (BSA) (Sigma) in TBST (10 mM Tris, pH 7.5, 150 mM NaCl, 0.1% Tween-20). All washes were performed in TBST. Primary antibodies included ErbB4 (E200) (Abcam) and α-tubulin #05829 (Upstate Biotechnology).
Microarray Gene Expression Profiling
MCF-7/pLKO and MCF-7/H4Si4 cells were incubated in phenol red-free MEM supplemented with 5% CS-FBS for 48 hrs then left untreated or treated with 100 pM 17-β-estradiol (Sigma) for 6 hrs. Total RNA from three independent experiments was purified using the miRVANA RNA Isolation System (Life Technologies) according to the manufacturer's instructions and RNA integrity was confirmed using a Bioanalyzer (Agilent Technologies). Two-color microarray gene expression analysis was performed using the Agilent Technologies DNA Microarray Scanner and associated protocols. Briefly, 100 ng of total RNA was diluted to generate the spike mix using the RNA Spike-In Kit (Agilent Technologies) exactly as described by the manufacturer. Amplified and cyanine 3 (Cy3)- or cyanine 5 (Cy5)-labeled complementary RNA (cRNA) was prepared for each total RNA sample from 2 μl of spike mix and 100 ng of total RNA using the Low Input Quick Amp Labeling Kit (Agilent Technologies) exactly as described by the manufacturer. The cRNA was purified using the RNeasy Mini Kit (Qiagen) and purified cRNA and Cy3 or Cy5 were quantitated using a NanoDrop 2000 (Thermo Scientific). Using the Gene Expression Hybridization Kit (Agilent Technologies) 825 ng each of Cy3- or Cy5-labeled, amplified, and purified cRNA were hybridized to a single microarray of a Whole Human Genome 4 × 44K Microarray G4112F (Agilent Technologies) in a SureHyb chamber (Agilent Technologies) and Agilent G2545A Hybridization Oven (Agilent Technologies) at 65°C for 17 hrs as described by the manufacturer. Washed microarray slides were scanned in a DNA MicroArray Scanner G2505C (Agilent Technologies). Microarray data was extracted and quality controlled using Feature Extraction software (Version 10.7.1.1)(Agilent Technologies). Statistical analysis of gene expression data comparing triplicate untreated to 17-β-estradiol treated samples was performed using GeneSpring GX (Agilent Technologies). Genes in each dataset with a 2-fold or greater change in expression following 17-β-estradiol treatment (p < 0.05) were subjected to Cellular Function Analysis using Ingenuity Pathway Analysis (IPA) software (Version 17199142).
Quantitative RT-PCR
Cells were preincubated in phenol red–free MEM supplemented with 5% charcoal-stripped FBS (CS-FBS) for 48 hrs and were left untreated or treated with 100 pM 17-β-estradiol for 6 hrs. Triplicate total RNA samples were purified using the miRVANA RNA Isolation System according to the manufacturer's instructions and RNA integrity was confirmed using a Bioanalyzer. First-strand complementary DNA (cDNA) was synthesized from 1.0 μg of total RNA in a 20 μl reaction volume using the Superscript III First-Strand Synthesis System (Life Technologies) with random hexamers exactly as described by the manufacturer. Following reverse transcription, 180 μl of DEPC Treated Water (Invitrogen) was added to the cDNA reaction and 2 μl of the diluted cDNA was used in a 20 μl Power SYBR Green PCR Master Mix (Applied Biosystems) with 250 nM of each oligonucleotide primer to amplify GAPDH, TFF1, CXCL12, or PgR described elsewhere [10] or the RASGPR1 oligonucleotide primers 5'-ACATTTAGCCAAAGGAGCCA and 5'-TACTTCGACACAGGTTTCCA. Reactions were amplified in the 7500 Fast Real-Time PCR system (conditions as follows: 55°C for 20 min, 95°C for 10 min and then 40 cycles of 95°C for 15 sec and 60°C for 60 sec), as described by the manufacturer (Applied Biosystems). The CT analysis for each reaction was performed using the supplied 7500 Software v2.0.5 (Applied Biosystems). Gene expression levels were normalized to GAPDH and 17-β-estradiol stimulated expression relative to untreated control was calculated using the 2−ΔΔCT method. Each sample was prepared in triplicate and the data represent the mean and standard error (SE) of at least three independent experiments. Statistically significant differences between data sets were determined using paired Student's t test.
Colony Formation Assay
Cells were plated at 1,000 cells per well in a 6-well plate with phenol red–free MEM supplemented with 5% CS-FBS with or without 10 nM 17-β-estradiol. Media was replaced every two days for 12 days total. Colonies were fixed in 100% methanol and stained with crystal violet. Colony number was calculated using a ColCount Colony Counter (Oxford Optronix) and the supplied statistical software. Each sample was prepared in duplicate and the data represent the mean and SE of at least three independent experiments. Statistically significant differences between data sets were determined using paired Student's t test.
xCELLigence Cell Proliferation Assay
Cell proliferation was determined using the xCELLigence System (Roche) by plating 2000 cells in an E-Plate 16 in the RTCA DP Instrument (Roche) according to the manufacturer's instructions. After 24 hrs media was changed to phenol red–free MEM supplemented with 5% CS-FBS and after an additional 48 hrs cells were left untreated or treated with 10 nM 17-β-estradiol. Cell proliferation as a function of real-time changes in electrical impedance, also referred to as cell index, was monitored by the xCELLigence System for 72 hrs. The slope of the change in cell index over time and the standard deviation of replicates were calculated using the supplied RTCA Software (Roche). Each sample was prepared in triplicate and the data represent the mean and SE of at least three independent experiments. Statistically significant differences between data sets were determined using paired Student's t test.
Cell Cycle Analysis
Cells were preincubated in phenol red–free MEM supplemented with 5% CS-FBS for 48 hrs followed by serum-free phenol red-free MEM for 24 hrs. Cells were returned to phenol red–free MEM and 5% CS-FBS with or without 10 nM 17-β-estradiol. After 24 hrs cells were trypsinized and fixed in 100% ethanol overnight. Fixed cells were stained with Guava Cell Cycle Reagent (Millipore) and cell cycle was analyzed in a Guava Easy Cyte Mini Base System (Millipore) using the supplied statistical software exactly as described previously [16]. The data represent the mean and SE of at least three independent experiments. Statistically significant differences between data sets were determined using paired Student's t test.
xCELLigence Cell Migration Assay
Cell migration was determined using the xCELLigence System (Roche) with the CIM-Plate 16 and RTCA DP Instrument (Roche) according to the manufacturer's instructions. Briefly, Cells were preincubated in phenol red–free MEM supplemented with 5% CS-FBS for 48 hrs and then 40,000 cells were added to the upper CIM-Plate 16 chamber in phenol red–free MEM supplemented with 0.2% CS-FBS with or without 10 nM 17-β-estradiol. Phenol red–free MEM supplemented with 5% CS-FBS with or without 10 nM 17-β-estradiol was added to the lower chamber and the CIM-Plate 16 was incubated in the RTCA DP Instrument for 48 hrs. Cell migration as a function of real-time changes in electrical impedance was monitored by the xCELLigence System. Cell Index (referred to here as Migration Index) and standard deviation of replicates were calculated using the supplied RTCA Software (Roche). Each sample was prepared in triplicate and the data represent the mean and SE of at least three independent experiments. Statistically significant differences between data sets were determined using paired Student's t test.
RESULTS AND DISCUSSION
HER4/4ICD is an Obligate Coactivator of Estrogen Regulated Genes
We have previously shown that the HER4 intracellular domain (4ICD) coactivates and interacts with ER at the promoters of progesterone receptor (PgR) and CXCL12 (also referred to as SDF-1), but 4ICD failed to coactivate the estrogen regulated gene TFF1 (also referred to as pS2) [9,10]. These observations suggest that 4ICD is a selective ER coactivator. To determine the extent of 4ICD coactivation of β-estradiol regulated genes we developed a MCF-7 cell line with stable knockdown of HER4 (MCF-7/H4Si4) using a HER4 targeting shRNA expression construct (Fig. S1). We first identified β-estradiol regulated genes in the vector control, MCF-7/pLKO, cell line by gene expression array. β-estradiol treatment at 100 pM for 6 hrs significantly stimulated 726 MCF-7/pLKO genes by 2-fold or greater and significantly suppressed 53 genes by 2-fold or greater (Table S1). To determine the extent of 4ICD coregulation of the estrogen regulated genes we performed microarray gene expression analysis of the MCF-7/H4Si4 cell line also treated with 100 pM of β-estradiol for 6 hrs. Loss of 4ICD expression failed to significantly alter expression of the 53 MCF-7/pLKO β-estradiol suppressed genes. These results indicate that 4ICD does not function as a β-estradiol-activated ER corepressor in MCF-7 cells. However, 11 β-estradiol regulated genes were significantly repressed by 2-fold or more in the absence of 4ICD (ZKSCAN5, DDX58, BRWD1, ADCK2, ARF1, ZNF148, PRKAA1, HKR1, ZNF627, PRPF48, PAN3) suggesting that 4ICD expression prevents transcriptional repression of these genes in response to β-estradiol (Table S2). Consistent with 4ICD functioning as an important ER coactivator, expression of 277 or 38% of the 726 MCF-7/pLKO estrogen regulated genes was significantly reduced by 2-fold or greater in the absence of 4ICD expression (Table S2). Interestingly, only eight 4ICD coregulated genes exhibited a β-estradiol response of 2-fold or greater in the absence of 4ICD (CXCL12, CYP1B1, FOS, SGK3, MYBL1, FRMD3, NUDCD1, DDX21)(Table S2). Accordingly, the remaining 269 4ICD coregulated genes failed to maintain a significant response to estrogen in the absence of 4ICD, indicating that 4ICD is an obligate coactivator for 37% of the MCF-7/pLKO β-estradiol stimulated genes. Consistent with our previously published results CXCL12 was identified as a 4ICD coactivated gene in the gene array analysis (Table S2) [10]. Another previously identified 4ICD coactivated gene, PgR [9,10], was not represented on the array. We used qRT-PCR to validate 4ICD coactivation of CXCL12 and PgR but not RASGPR1 and confirm that loss of 4ICD results in enhanced TFF1 expression (Fig. 1). Consistent with our previously published results [9,10] and the microarray data presented here (Tables S1 and S2) β-estradiol stimulated TFF1 expression increased in the absence of 4ICD whereas β-estradiol stimulated expression of CXCL12 and PgR was suppressed by greater than two fold in the absence of 4ICD (Fig. 1). Taken together these results further establish 4ICD as a potent ER coactivator and implicate 4ICD as an obligate coactivator of 37% of the β-estradiol stimulated genes in breast tumor cells.
Fig. 1.
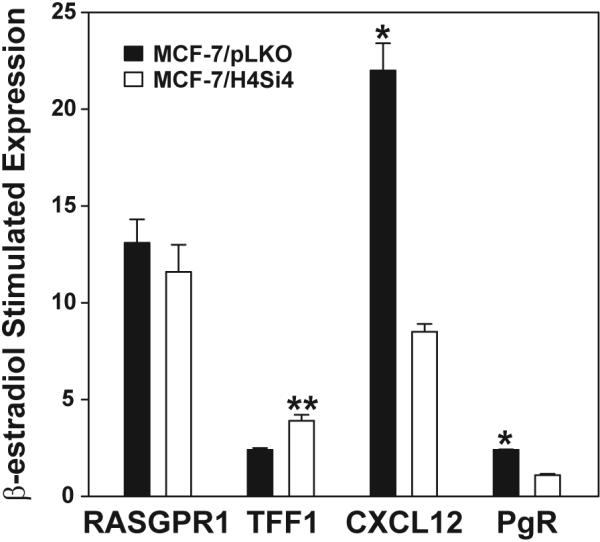
Quantitative RT-PCR validation of microarray gene expression data. MCF-7/pLKO vector control and MCF-7/H4Si4 HER4 knockdown cell lines were preincubated in phenol red–free MEM supplemented with 5% CS-FBS for 48 hrs and were left untreated or treated with 100 pM 17-β-estradiol for 6 hrs. RNA was isolated and expression of the indicated genes was determined by qRT-PCR. The data represents fold change in expression of 17-β-estradiol treated versus untreated. Each sample was prepared in triplicate and the data represent the mean +/− SE of at least three independent experiments. Statistically significant differences between data sets were determined using paired Student's t test. Asterisk and double asterisks represent p < 0.001 and p = 0.011, respectively.
HER4/4ICD Coactivates Estrogen Stimulated Genes That Function in Cell Cycle and Tumor Cell Proliferation
Although 4ICD coactivates a significant percentage of β-estradiol regulated genes, it remained unclear if the 4ICD coactivated genes are associated with specific β-estradiol regulated functions or if 4ICD is a broad-spectrum ER coactivator lacking obvious functional specificity. To determine if 4ICD coactivated genes are enriched for specific β-estradiol regulated functions we performed Ingenuity Pathway Analysis (IPA) and determined the extent of functional overlap between β-estradiol regulated genes and 4ICD coactivated genes. IPA software analyzes gene expression data and predicts functional relationships within the gene set. For each of the more than 1000 IPA cellular functions an activation z-Score is calculated. An activation z-Score greater than 2 indicates a significantly increased cellular function whereas a z-Score less than −2 indicates a significantly decreased cellular function.
β-estradiol stimulation of the MCF-7/pLKO cell line was significantly associated with 36 IPA cellular functions (Table 1). Consistent with the role of β-estradiol driving breast tumor proliferation, the most significant z-Scores associated with β-estradiol regulated gene expression included multiple cellular functions associated with cellular growth and proliferation and cell cycle progression, as well as, decreased hypoplasia (Table 1). Importantly, these same functional categories had the most significant z-Scores associated with 4ICD coactivated genes (Table 2). Nevertheless, 4ICD coactivation of ER appears to have functional specificity. Accordingly, 4ICD coactivated genes fail to contribute to several significantly important β-estradiol regulated functional categories including cellular movement associated with tumor cell migration, cell death and survival associated with apoptotic resistance of tumor cells, small molecule biochemistry, gene expression related to increased transcription, and cancer associated cell transformation and metastasis (compare Tables 1 and 2).
Table 1.
Ingenuity Pathway Analysis reveals cellular functions significantly associated with estrogen regulated genes in breast tumor cells.
| Category | Functions Annotation | Regulation | p-Value | Activation z-Score |
|---|---|---|---|---|
| Cellular Growth and Proliferation | Proliferation of Cells | Increased | 3.80E-04 | 5.553 |
| Proliferation of Tumor Cell Lines | 2.21E-04 | 3.422 | ||
| Proliferation of Breast Cell Lines | 1.01E-03 | 2.590 | ||
| Colony Formation of Cells | 1.08E-02 | 2.222 | ||
| Proliferation of Epithelial Cells | 5.96E-03 | 2.108 | ||
| Proliferation of Breast Cancer Cell Lines | 2.98E-03 | 2.006 | ||
| Developmental Disorder | Hypoplasia | Decreased | 3.70E-03 | −4.931 |
| Cellular Movement | Cell Movement | Increased | 7.43E-03 | 3.817 |
| Migration of Tumor Cell Lines | 1.40E-04 | 3.696 | ||
| Cell Movement of Tumor Cell Lines | 3.12E-05 | 3.025 | ||
| Invasion of Cells | 1.02E-03 | 2.980 | ||
| Cell Movement of Stem Cells | 9.52E-03 | 2 385 | ||
| Migration of Stem Cells | 1.13E-02 | 2.176 | ||
| Cell Cycle | Entry Into Interphase | Increased | 3.93E-04 | 3.352 |
| Entry Into S Phase | 1.33E-03 | 3.073 | ||
| S phase | 1.13E-04 | 2.476 | ||
| Interphase | 9.23E-04 | 2.389 | ||
| Cell Death and Survival | Cell Viability of Tumor Cell Lines | Increased | 5.63E-04 | 3.112 |
| Apoptosis of Tumor Cell Lines | Decreased | 1.67E-05 | −2.169 | |
| Apoptosis | Decreased | 1.31E-03 | −2.063 | |
| Small Molecule Biochemistry | Synthesis of Lipid | Incresaed | 4.21E-03 | 2.813 |
| Oxidation of Lipid | 1.55E-04 | 2.505 | ||
| Uptake of Amino Acids | 1.08E-02 | 2.395 | ||
| Uptake of Alpha-Amino Acid | 2.22E-03 | 2.191 | ||
| Beta-Oxidation of Fatty Acid | 8.66E-04 | 2.190 | ||
| Gene Expression | Transactivation of RNA | Increased | 5.57E-03 | 2.734 |
| Transactivation | 5.66E-03 | 2.593 | ||
| Transcription | 9.12E-05 | 2.426 | ||
| Expression of RNA | 4.48E-04 | 2.254 | ||
| Transcription of RNA | 1.44E-04 | 2.240 | ||
| Cancer | Cell Transformation | Increased | 8.61E-03 | 2.733 |
| Metastasis | Increased | 8.46E-03 | 2.291 | |
| Benign Neoplasia | Decreased | 3.09E-04 | −2.081 | |
| Cellular Development | Differentiation of Cells | Increased | 2.80E-03 | 2.402 |
| DNA Replication, Recombination, and Repair | Breakage of Chromosomes | Decreased | 8.60E-03 | −2.348 |
| Cellular Function and Maintenance | Cellular Homeostasis | Decreased | 8.46E-03 | 2.025 |
Table 2.
Ingenuity Pathway Analysis reveals cellular functions significantly associated with estrogen regulated genes coactivated by 4ICD in breast tumor cells.
| Category | Functions Annotation | Regulation | p-Value | Activation z-Score |
|---|---|---|---|---|
| Developmental Disorder | Hypoplasia | Decreased | 1.64E-03 | −4.067 |
| Cellular Growth and Proliferation | Proliferation of Cells | Increased | 1.35E-02 | 3.371 |
| Proliferation of Tumor Cell Lines | 3.88E-04 | 2.526 | ||
| Cellular Assembly and Organization | Formation of Cellular Protrusions | Increased | 1.76E-02 | 2.911 |
| Organization of Cytoskeleton | 2.58E-02 | 2.671 | ||
| Organization of Cytoplasm | 1.64E-02 | 2.671 | ||
|
| ||||
| Molecular Transport | Transport of Molecule | Increased | 1.42E-03 | 2.635 |
| Cell Cycle | Entry Into S Phase | Increased | 3.37E-03 | 2.592 |
| Interphase | 9.29E-03 | 2.112 | ||
| S Phase | 6.18E-05 | 2.076 | ||
| Energy Production | Consumption of Oxygen | Decreased | 9.58E-03 | −2.415 |
| DNA Replication, Recombination, and Repair | Repair of DNA | Increased | 1.03E-02 | 2.211 |
HER4/4ICD Expression is Required for Estrogen Stimulated Growth of MCF-7 Breast Tumor Cells
We next determined if there was a concordance between the influence of 4ICD coactivation on IPA functional predictions based upon gene expression profiling and the impact of 4ICD expression on MCF-7 cells in biological assays. The IPA analysis predicted a role for 4ICD in β-estradiol stimulated MCF-7 tumor cell proliferation and cell cycle progression. To confirm these results we compared β-estradiol stimulated growth of MCF-7/pLKO cells to MCF-7/H4Si4 cells in a colony formation assay, as well as, a real-time xCELLigence growth assay. As predicted β-estradiol significantly enhanced the growth of the vector control MCF-7/pLKO cell line by 1.7 fold and 1.6 fold in the colony formation and xCELLigence assays, respectively (Fig. 2A). In contrast, the MCF-7/H4Si4 cell line failed to respond to the β-estradiol proliferative signal with an insignificant 1.1 and 1.0 fold change in the colony formation and xCELLigence assays, respectively (Fig. 2A). These results indicate that HER4/4ICD is an obligate mediator of breast tumor cell proliferation in response to β-estradiol. To determine if 4ICD contributes to β-estradiol stimulated cell cycle progression we performed a cell cycle analysis of β-estradiol treated MCF-7/pLKO and MCF-7/H4Si4 cells. In direct concordance with the IPA functional analysis results β-estradiol stimulated cell cycle progression of the MCF-7/pLKO cell line with a significant decrease in G1 phase cells and concomitant significant increase in the percentage of S phase cells (Fig. 2B). In contrast, β-estradiol failed to stimulate cell cycle progression of the MCF-7/H4Si4 cell line (Fig. 2B). Taken together, our biological data confirms the IPA functional data underscoring the obligate role for 4ICD coactivated gene expression in β-estradiol stimulated tumor cell proliferation and cell cycle progression.
Fig. 2.
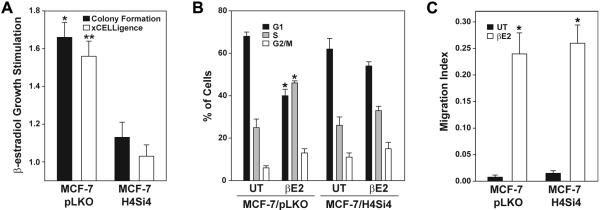
HER4 expression is required for estrogen-stimulated proliferation of breast tumor cells. (A) For colony formation assay MCF-7/pLKO and MCF-7/H4Si4 cells were plated at 1,000 cells per well in a 6-well plate with phenol red–free MEM supplemented with 5% CS-FBS with or without 10 nM 17-β-estradiol. Media was changed every two days and after 12 days the number of fixed and stained colonies was calculated using a ColCount Colony Counter. For the xCELLigence cell growth assay 2000 cells were plated in an E-Plate 16 in the RTCA DP Instrument. After 24 hrs media was changed to phenol red–free MEM supplemented with 5% CS-FBS and after an additional 48 hrs cells were left untreated or treated with 10 nM 17-β-estradiol. The slope of the change in cell index during a 72 hrs treatment was used to determine relative growth stimulation. Data represents 17-β-estradiol stimulated growth relative to untreated cells. (B) Cell cycle analysis of each cell line preincubated in phenol red–free MEM supplemented with 5% CS-FBS for 48 hrs followed by serum-free phenol red-free MEM for 24 hrs. Cells were returned to phenol red–free MEM and 5% CS-FBS with (βE2) or without (UT) 10 nM 17-β-estradiol for 24 hrs and cell cycle analysis was performed on fixed and stained cells in a Guava Easy Cyte Mini Base System. (C) Cell migration assay of each cell line preincubated in phenol red–free MEM supplemented with 5% CS-FBS for 48 hrs and then transferred to the upper CIM-Plate 16 chamber. The upper and lower chambers contained phenol red–free MEM supplemented with 0.2% and 5% CS-FBS, respectively, with or without 10 nM 17-β-estradiol. Using the xCELLigence System cell Index (referred to here as Migration Index) and standard deviation of replicates were calculated after 48 hrs using the RTCA Software. For all experiments samples were prepared in triplicate and the data represents the mean +/− SE of at least three independent experiments. Statistically significant differences between data sets were determined using paired Student's t test. Asterisk and double asterisks in (A) represent p = 0.013 and p = 0.008, respectively. Asterisk in (B) and (C) represent p = 0.001 and p < 0.005, respectively.
Following cellular growth and proliferation, cellular movement associated with migration of tumor cell lines was the second most significant β-estradiol stimulated MCF-7/pLKO IPA functional category (Table 1). In contrast, IPA failed to identify a significant association of 4ICD coactivated genes with the cellular movement functional category, suggesting that 4ICD coactivation does not contribute to β-estradiol-stimulated tumor cell migration (Table 2). To determine the impact of 4ICD loss-of-function on β-estradiol stimulated tumor cell migration we performed an xCELLigence cell migration assay comparing β-estradiol stimulated cell migration between MCF-7/pLKO and MCF-7/H4Si4 cell lines. Consistent with the IPA functional analysis, β-estradiol stimulated a modest but significant increase in MCF-7/pLKO tumor cell migration (Fig. 2C). Likewise, as predicted by the IPA functional analysis, suppression of HER4 expression in the MCF-7/H4Si4 cell line failed to impact β-estradiol stimulated cell migration which was equivalent to the MCF-7/pLKO cell line (Fig. 2C). Taken together, the IPA functional analysis and multiple biological assays indicate that 4ICD is a functionally selective ER coregulator primarily coactivating genes promoting β-estradiol stimulated tumor cell proliferation and cell cycle progression while failing to coactivate genes contributing to β-estradiol stimulated tumor cell migration.
Estrogen regulated gene expression and breast tumor cell growth assays have also been performed where the SRC family of canonical ER coregulators are independently suppressed. Of the three SRC family members, SRC-2 and SRC-3 (amplified in breast cancer 1; AIB1) displayed activities similar to 4ICD by contributing to estrogen stimulated growth [2,3] and cell cycle progression [2] of MCF-7 cells. In addition, both 4ICD and SRC-3 are required for estrogen stimulated PgR expression [2,9,10]. Although there appears to be partial overlap between 4ICD and SRC family coregulated genes, 4ICD has unique activities. For example, all three SRCs bind with ER at the TFF1 promoter and independent suppression of each SRC results in suppressed estrogen induced TFF1 expression [2,17]. In contrast, 4ICD fails to bind with ER at the TFF1 promoter [10] and we consistently observe enhanced estrogen stimulated TFF1 expression when 4ICD expression is suppressed [10]. These results indicate that 4ICD fails to coactivate TFF1 and may function as an indirect TFF1 corepressor. Conversely, 4ICD is required for maximal estrogen induced MYC expression (Table S2); whereas none of the SRC family members appear to influence MYC expression [2]. These observations suggest that although 4ICD and SRC family members have some functional overlap, 4ICD exhibits coregulator activities distinct from SRC family members.
In conclusion, our data provides the first experimental evidence that a cell surface receptor directly functions as an important nuclear receptor transcriptional coactivator. Specifically, we have previously shown that 4ICD coregulates estrogen gene expression by selectively associating with ER at estrogen responsive promoters [9,10]. Here we used a global gene expression microarray approach to show that 4ICD is an obligate coactivator for 37% of the estrogen regulated genes in breast tumor cells. Furthermore, our biological and in situ data indicates that 4ICD selectively coactivates estrogen regulated genes associated with tumor cell proliferationand cell cycle progression. Clinically, nearly 90% of ER positive breast tumors coexpress HER4 [15] suggesting a predominant tumor cell selection for ER/4ICD coregulated gene expression driving tumor cell proliferation. By extension, our evidence that 4ICD coactivation activity is required for estrogen stimulated breast tumor proliferation implies that the majority of breast cancer patients would benefit from a therapeutic strategy that disengages ER and 4ICD cooperative signaling. Indeed, we have shown that tamoxifen mediated disruption of the ER and 4ICD transcriptional complex results in breast tumor cell apoptosis [18]. Therefore, we predict that a HER4 inhibitor, such as lapatinib, would improve patient response to endocrine therapy.
Supplementary Material
Highlights
HER4/4ICD is an obligate coactivator for 37% of estrogen regulated genes
HER4/4ICD coactivated genes selectively regulate estrogen stimulated proliferation
Estrogen stimulated tumor cell migration occurs independent of HER4/4ICD
Disrupting HER4/4ICD and ER coactivated gene expression may suppress breast cancer
ACKNOWLEDGEMENTS
We thank Dr. Mary Sfondouris and Dr. Partha Das for excellent laboratory management and technical assistance. We thank Karen Wang and Felicia Huynh for assistance with experiments and other members of the Jones lab for helpful discussions. This work was supported by NIH/NCI grants RO1CA095783 (FEJ) and RO1CA096717 (FEJ).
ABBREVIATIONS
- 4ICD
HER4 intracellular domain
- BSA
bovine serum albumin
- cDNA
complementary DNA
- cRNA
complementary RNA
- CS-FBS
charcoal stripped fetal bovine serum
- Cy3
cyanine 3
- Cy5
cyanine 5
- EGFR
epidermal growth factor receptor
- ER
estrogen receptor alpha
- IPA
Ingenuity Pathway Analysis
- PgR
progesterone receptor
- RIP
regulated intramembrane proteolysis
- SE
standard error
- SRC
p160 steroid receptor coactivator
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Bardou VJ, Arpino G, Elledge RM, Osborne CK, Clark GM. Progesterone receptor status significantly improves outcome prediction over estrogen receptor status alone for adjuvant endocrine therapy in two large breast cancer databases. J Clin Oncol. 2003;21:1973–1979. doi: 10.1200/JCO.2003.09.099. [DOI] [PubMed] [Google Scholar]
- [2].Karmakar S, Foster EA, Smith CL. Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-alpha transcriptional activity. Endocrinology. 2009;150:1588–1596. doi: 10.1210/en.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].List HJ, Lauritsen KJ, Reiter R, Powers C, Wellstein A, Riegel AT. Ribozyme targeting demonstrates that the nuclear receptor coactivator AIB1 is a rate-limiting factor for estrogen-dependent growth of human MCF-7 breast cancer cells. J Biol Chem. 2001;276:23763–23768. doi: 10.1074/jbc.M102397200. [DOI] [PubMed] [Google Scholar]
- [4].Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- [5].Sutherland RL, Hall RE, Taylor IW. Cell proliferation kinetics of MCF-7 human mammary carcinoma cells in culture and effects of tamoxifen on exponentially growing and plateau-phase cells. Cancer Res. 1983;43:3998–4006. [PubMed] [Google Scholar]
- [6].Lonard DM, O'Malley BW. Nuclear receptor coregulators: modulators of pathology and therapeutic targets. Nat Rev Endocrinol. 2012;8:598–604. doi: 10.1038/nrendo.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vidal GA, Naresh A, Marrero L, Jones FE. Presenilin-dependent γ-secretase processing regulates multiple ERBB4/HER4 activities. J. Biol. Chem. 2005;280:19777–19783. doi: 10.1074/jbc.M412457200. [DOI] [PubMed] [Google Scholar]
- [8].Williams CC, Allison JG, Vidal GA, Burow ME, Beckman BS, Marrero L, Jones FE. The ERBB4/HER4 receptor tyrosine kinase regulates gene expression by functioning as a STAT5A nuclear chaperone. J Cell Biol. 2004;167:469–478. doi: 10.1083/jcb.200403155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rokicki J, Das PM, Giltnane JM, Wansbury O, Rimm DL, Howard BA, Jones FE. The ERα coactivator, HER4/4ICD, regulates progesterone receptor expression in normal and malignant breast epithelium. Mol Cancer. 2010;9:150. doi: 10.1186/1476-4598-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhu Y, Sullivan LL, Nair SS, Williams CC, Pandey A, Marrero L, Vadlamudi RK, Jones FE. Coregulation of estrogen receptor by estrogen-inducible ERBB4/HER4 establishes a growth promoting autocrine signal in breast cancer. Cancer Res. 2006;66:7991–7998. doi: 10.1158/0008-5472.CAN-05-4397. [DOI] [PubMed] [Google Scholar]
- [11].Tang CK, Goldstein DJ, Payne J, Czubayko F, Alimandi M, Wang L-M, Pierce JH, Lippman ME. ErbB-4 ribozymes abolish neuregulin-induced mitogenesis. Cancer Res. 1998;58:3415–3422. [PubMed] [Google Scholar]
- [12].Tang CK, Concepcion X-ZW, Milan M, Gong X, Montgomery E, Lippman ME. Ribozyme-mediated down-regulation of ErbB-4 in estrogen receptor-positive breast cancer cells inhibits proliferation both in vitro and in vivo. Cancer Res. 1999;59:5315–5322. [PubMed] [Google Scholar]
- [13].Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, Walenkamp AM. A review on CXCR4/CXCL12 axis in oncology: no place to hide. Eur J Cancer. 2013;49:219–230. doi: 10.1016/j.ejca.2012.05.005. [DOI] [PubMed] [Google Scholar]
- [14].Sundvall M, Iljin K, Kilpinen S, Sara H, Kallioniemi OP, Elenius K. Role of ErbB4 in breast cancer. J Mammary Gland Biol Neoplasia. 2008;13:259–268. doi: 10.1007/s10911-008-9079-3. [DOI] [PubMed] [Google Scholar]
- [15].Suo Z, Berner HS, Risberg B, Karlsson MG, Nesland JM. Estrogen receptor-alpha and C-ERBB-4 expression in breast carcinomas. Virchows Arch. 2001;439:62–69. doi: 10.1007/s004280000392. [DOI] [PubMed] [Google Scholar]
- [16].Mitra D, Das PM, Jones FE. JUMONJI/ARID1 B (JARID1B) promotes breast tumor cell cycle progression through epigenetic repression of micro RNA let-7e. J Biol Chem. 2010;286:40531–40535. doi: 10.1074/jbc.M111.304865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Labhart P, Karmakar S, Salicru EM, Egan BS, Alexiadis V, O'Malley BW, Smith CL. Identification of target genes in breast cancer cells directly regulated by the SRC-3/AIB1 coactivator. Proc Natl Acad Sci U S A. 2005;102:1339–1344. doi: 10.1073/pnas.0409578102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Naresh A, Thor AD, Edgerton SM, Torkko KC, Kumar R, Jones FE. The HER4/4ICD estrogen receptor coactivator and BH3-only protein is an effector of tamoxifen-induced apoptosis. Cancer Res. 2008;68:6387–6395. doi: 10.1158/0008-5472.CAN-08-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.