

Abstract
Energy coupling between distal DNA domains, may have profound regulatory consequences for biological processes, allowing for allosteric control of nucleic acid function. Repair of oxidative lesions at or near triplet repeat domains can enhance DNA expansion events that result in debilitating diseased states. We report here position, distance, and lesion-dependent energy crosstalk between pairs of lesions in a triplet repeat bulge loop and an adjacent duplex domain. We discuss the implications of such coupled communication between lesions in distal loop and duplex domains for lesion repair and DNA expansion associated with diseases.
Communication between proximal and distal sites within macromolecules is a hallmark of biological regulation. Generally, such coupling is empirically observed, with limited understanding of the underlying forces that modulate the biology. For example, the activation of base excision repair (BER) of damaged DNA bases induces expansion of proximal triplet repeat domains,1 yielding the genotype characteristic of so-called “triplet repeat” diseases.2-4 To understand the origins of this coupling, we have mapped, in the presence and absence of lesions, the complex energy landscapes of DNA structures containing triplet repeat bulge loops implicated in DNA expansion; so called Ω-DNA (scheme 1).5-7 We discovered a position-dependent energy coupling between distal DNA lesions in the loop and in adjacent duplex domains. Such crosstalk may have profound regulatory consequences, particularly in the context of the observed and yet unexplained coupling between DNA repair, a desirable process, and DNA expansion, a disease-inducing process.
SCHEME 1.
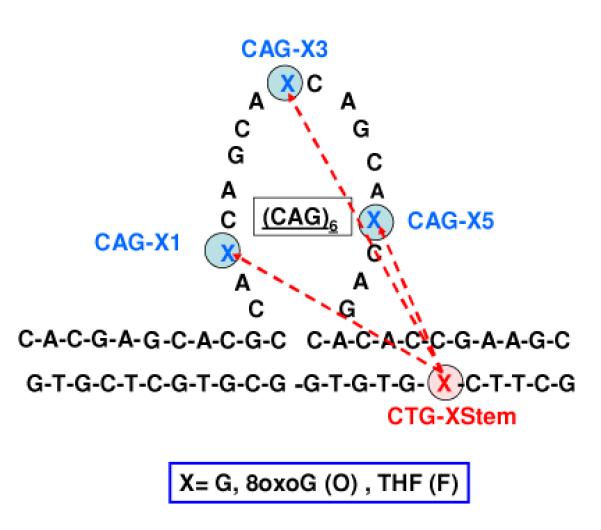
We refer to the bulge loop construct shown in Scheme 1 as an Ω-DNA based on its similarity to the Greek letter Ω when represented in two dimensions. We will adhere to the following nomenclature based on lesion type and position listed in the following order: identity of lesion in bulge loop; followed by a number in parenthesis of the CAG repeat containing the lesion; followed by the identity of the fixed position lesion in the downstream domain. Thus, O(3)-F refers to the member of the O(n)-F family of Ω-DNA constructs that contains the 8oxoG lesion in the 3rd CAG repeat and the F lesion in the downstream duplex domain. The designation (n) in place of a specific number within the parenthesis denotes the entire family of DNA lesion pairs; thus, O(n)-F refers to the three Ω-DNA constructs in which n=1, n=2, and n=5.
To probe this intriguing allostery/crosstalk, we devised an approach we call “DNA lesion scanning.” The two lesions studied here are 8oxodG (O) and a tetrahydrofuran abasic site analogue (F), both of which are mutagenic and destabilizing to the global stability of DNA.8,9 In our scanning experiment, we incorporate either the O or F lesion at a single fixed position in the opposing “non-bulged” strand, downstream from the loop domain, and then singularly and selectively replace guanines in the bulge loop domain with either O or F. This process creates a position-dependent family of Ω-DNA constructs containing pairs of lesions, as illustrated in scheme 1. We then use calorimetric melting curves to map the energy impact of these lesion pairs on DNA properties.10 Similar mutation scanning experiments, such as alanine scanning in proteins, and scanning for compensating double mutations in RNA, have found widespread use for identifying critical residues and structural elements.11-15 Our DNA lesion scanning experiments achieve a similar goal from an energetics perspective, and have conceptual similarities to the placement of fluorescent labels in FRET methods to map distance constraints between biopolymer domains.16
The choice of 8oxodG (O) and abasic site analogue (F) as suitable lesions is dictated by observations that faulty BER repair of oxidative lesions at or near CAG repeat domains can enhance rates of DNA expansion in mouse models of Huntington’s disease.1 The O lesion is a common form of oxidative DNA damage repaired by the BER pathway, and the abasic site is the universal intermediate in BER repair. 17-20 Formation of two or more closely spaced oxidative lesions, which forms the basis of our lesion scanning experiment, is a frequent consequence of DNA damage by ionizing radiation and/or chemical reagents.17,21
The resulting clustered lesions provide unique challenges for the DNA repair machinery.22-24 Our scanning method detects and quantitatively defines the collective consequences of such clustered oxidative lesions within triplet repeat bulge loop structures. Such characterizations are essential for assessing the differential recognition and processing of these unique substrates for DNA repair, and to evaluate the basis for the coupling of repair with DNA expansion.
Ω-DNA Constructs with Two Lesions Exhibit Biphasic Melting Profiles
Figure 1 shows the experimentally measured excess heat capacity curves for the different families of Ω-DNAs with one lesion in the bulge loop and a second lesion in the downstream duplex domain. The corresponding global thermodynamic data derived by integration of these excess heat capacity curves are listed in Table S1 of the supplementary material. Significantly, in contrast to the lesion-free parent and most single lesion Ω-DNAs we previously studied,6,7 all dual lesion containing constructs display multiphasic melting behavior, with the shape of the melting profile being dependent on lesion type and position.
Figure 1.
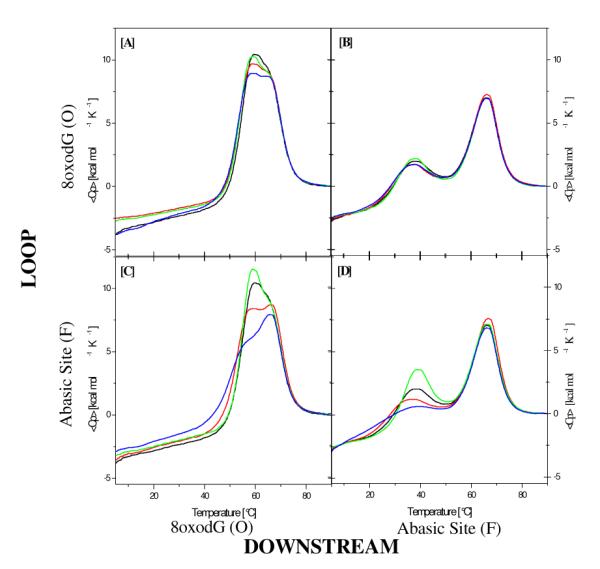
Experimentally measured excess heat capacity curves for lesion pairs O(n)-O (Fig 1A), O(n)-F (Fig 1B), F(n)-O (Fig 1C), and F(n)-F (Fig 1D) (n=1,3, or 5). X(1)-X lesion pairs are shown in red; X(3)-X lesion pairs are shown in green; and X(5)-X lesion pairs are shown in blue; For comparison, shown in black are the excess heat capacity curves for the Ω-DNA constructs with the corresponding single lesion in the downstream domain only.
To evaluate the thermodynamic impact of a particular lesion pair, we deconvoluted the multiphasic denaturation processes shown in Figure 1 into their component parts. To this end, we used a statistical mechanical model first developed by Wyman and Gill to analyze the complex multi-component excess heat capacity curves of tRNAs,25,26 modified here to take into account non-zero heat capacity changes. For two independent, two-state transitions, we find this model provides good fits to all Ω-DNA melting curves analyzed to date, including those that visually appear to be only a single cooperative transition.7,27 The fitting results (Table S2) and examples of fits to the dual lesion constructs for lesions located in loop position 3 (Figure S2) are shown in the supplementary material. While caution should be exercised when interpreting these results in microscopic terms, such deconvolutions resolve and define the macroscopic effects of the lesions on the overall melting behavior of the Ω-DNAs.
The High-Temperature Transition Exhibits a Constant Melting Temperature, Independent of the Lesion Pair, Consistent with Melting of the Lesion-Free Domain
Our data reveal the intriguing result that, regardless of the lesion pair, the high temperature fitted peak for all the dual lesion-containing Ω-DNA constructs is characterized by the same melting temperature, Tm(fit,2) = 65.6±0.2°C. Differential behavior is reflected in the enthalpy term rather than in the Tm data, with the fitted transition enthalpies falling into two groups (ΔH(fit,2) = 78.5±1.9 kcal mol−1 and ΔH(fit,2) = 60.5±2.1 kcal mol−1), depending on whether the two melting domains are overlapping (the O(n)-O and F(n)-O families) or well resolved (the O(n)-F and F(n)-F families). Significantly, when fitting the melting curves of single lesion or lesion-free Ω–DNA’s, we find the same sets of Tm and enthalpy values (also clustering within these two ΔH groups) for the high temperature component transition, so long as the lesion is located in either the loop or in the downstream domain. By contrast, when the lesion is located in the upstream duplex domain, we find shifts to lower values in the fitted Tm and the enthalpy for the upper transition.7,27 The ΔH of 78 kcal mol−1 we determine roughly corresponds to that of an 11mer duplex at this temperature, such as makes up the duplex arms of the Ω-DNA construct.28 Collectively, these observations suggest that the high temperature melting transition primarily reflects contributions from region(s) which lack lesions; namely, the upstream duplex domain. The two groups of enthalpies could arise from differential contributions due to cooperative coupling of the melting of this lesion-free domain with the melting of the loop self-structure and the duplex-loop-duplex three way junction.
The Low-Temperature Transition Exhibits Lesion-Dependent Behavior and Corresponds to Melting of the Domain Containing the Lesion
By contrast with the invariable, lesion-independent high temperature melting transition, we find that lesions in the loop and downstream domains cause considerable variation in the Tm and enthalpy values for the resolved lower temperature peak. We further observe that an abasic site (Fig 1 C&D) generally has a more significant impact on the domain melting enthalpy than does an 8oxodG lesion (Fig 1A&B), an observation consistent with the relative thermodynamic impact of abasic sites and 8oxodG lesions in duplex DNA. 8,9
Lesion Impact Depends on the Position Within the Loop
Independent of the nature of the second lesion, our data reveal that a given lesion exerts a greater thermodynamic impact when it is located in the downstream duplex domain than when it is located in the loop domain. This result is consistent with our observations on single lesions Ω-DNA constructs,7 and suggests that lesions favor formation of bulge loop structures by destabilizing the duplex state more than the loop state.
Within each family of dual lesion constructs, we observe the same characteristic pattern of Tm and enthalpy changes with lesion position in the loop domain. (Figure 2). Specifically, a lesion in the center of the loop (position 3) is only modestly perturbing, while a lesion within either the 5′ side of the loop (position 1) or the 3′ side of the loop (position 5) is significantly more perturbing, with differences between the 5′ and 3′ lesions being marginal. In summary, while the absolute magnitude of the lesion-induced perturbations in Tm (fit,1) and ΔH (fit,1) depend on the identities of the paired lesions, the position-dependent differential effects are preserved across each family (Figure 2), as reflected in the following order of decreasing impact on ΔΔH and ΔTm relative to the lesion-free Ω-construct: [n=5] ≈ [n=1] >[n=3].
Figure 2.
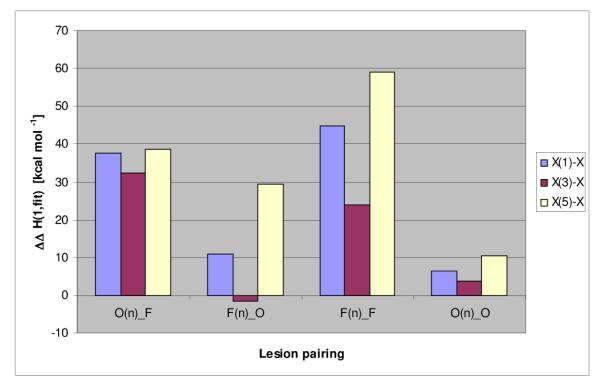
Comparison of the different Ω_DNA families. The difference in low temperature fitted enthalpy (ΔH(fit,1)) relative to ΔH(fit,1) of the unmodified parent Ω-DNA is shown.
Lesion Crosstalk is Reflected in the Nonadditivity of Lesion Impacts
Significantly, the enthalpy impact of a given lesion pair cannot be estimated from the sum of the enthalpies of the corresponding single lesion constructs (not shown). Except for the O(n)-O family of Ω-DNAs, the combined enthalpy impact of both lesions either significantly exceeds the summed enthalpies of the corresponding single lesion constructs (the O(n)-F and F(n)-F families) or is substantially less than the sum (the F(n)-O family). In other words, our dual lesion constructs exhibit energy coupling between the pairs of lesions that results in a more pronounced enthalpy change than would be expected based on contributions from the corresponding individual lesions alone. Such energy coupling between distal domains is a prerequisite for allostery,29-34 a phenomenon known to influence biological regulation, and is a hallmark of DNA telestability.35,36 This phenomenon may explain why lesion repair at/near repeat domains leads to enhanced rates of DNA expansion, as elaborated on below.
Energy Coupling Depends on Distance/Orientation Between the Paired Lesions
Based on the results shown in Figure 2, the energy coupling we observe does not depend simply on the identity and linear separation of the lesions in two dimensional sequence space. Instead, the coupling also depends on the three dimensional distance between and orientation of the two lesions imparted by the local and global helical twist of the bulge loop constructs. This feature is reflected in the complexity of the coupling data reported here. Future studies will assess the sequence dependence of this lesion crosstalk, as well as potential mechanisms of transmission of such allosteric energetic effects. When structural data become available, it may prove possible to correlate thermodynamic parameters with distances between lesion sites.
Macroscopic Energy Coupling and Microscopic Lesion-Induced Ensemble Redistribution
We previously have shown that the Ω–DNA is a metastable macrostate composed of an ensemble of energetically similar but not identical microstates.5 We subsequently showed that the metastable Ω macrostate can adapt to the energy perturbation induced by single lesions via redistribution of microstates within this ensemble.7 Building on our previous observations, we now postulate that the energy coupling observed here reflects lesion-induced redistribution of microstates within the ensemble that collectively makes up the repeat loop macrostate, including accommodations within the loop self-structure and the duplex-loop-duplex junction.
Energy Crosstalk and Coupling between BER and Expansion DNA Processing Pathways
In the BER pathway, isolated and clustered oxidative lesions are recognized, excised, and repaired in a highly orchestrated process which involves glycosylases, endonucleases, repair polymerases, and a number of auxiliary proteins that collectively coordinate the repair machinery.18,20 We propose that the effects of energy coupling between lesions in loop and adjacent duplex domains modulates recognition, binding, and processing of clustered oxidative lesions by select elements of the BER machinery. Our results suggest that BER proteins bound at one lesion site may act as allosteric effectors for BER processing of the 2nd site. Such modulation of the BER machinery could have profound effects on repair efficiency and outcome.
The observations of energy coupling/telestability between loop and adjacent duplex domains described here, and in our earlier publications, 7,27 provide a physico-chemical rationale for the observations of the Wells laboratory that DNA sequences flanking repeat DNA domains modulate/influence the propensity of repeat DNA sequences to expand or contract within in vivo model systems.37-40 Our results are consistent with allosteric control of biological processes in higher order nucleic acid structures, similar to what is observed for proteins.41-44 Such energy coupling/telestability could have significant implications for RNA function and biology as well,45 particularly given the critical role in RNA structure of loop domains connected via duplex regions.
Supplementary Material
ACKNOWLEDGMENT
The authors would like to thank Drs. Roger Jones and Barbara Gaffney (Rutgers University) for advice and helpful discussions. Supported by grants from the NIH GM23509, GM34469, and CA47995 (all to K.J.B.) and NRF (Pretoria, RSA) grant GUN 61103 to H.H.K.
Footnotes
SUPPORTING INFORMATION AVAILABLE: Supplementary Figures, Tables, Experimental Procedures, and Data Analysis Routines can be found in the supplementary material. This material is available free of charge via the internet at http://pubs.acs.org
REFERENCES
- (1).Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sutherland G, Richards R. Proc Natl Acad Sci U S A. 1995;92:3636–3641. doi: 10.1073/pnas.92.9.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ashley CT, Jr., Warren ST. Annu Rev Genet. 1995;29:703–728. doi: 10.1146/annurev.ge.29.120195.003415. [DOI] [PubMed] [Google Scholar]
- (4).Cummings CJ, Zoghbi HY. Annu Rev Genomics Hum Genet. 2000;1:281–328. doi: 10.1146/annurev.genom.1.1.281. [DOI] [PubMed] [Google Scholar]
- (5).Völker J, Klump HH, Breslauer KJ. Proc Natl Acad Sci U S A. 2008;105:18326–18330. doi: 10.1073/pnas.0810376105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Völker J, Klump HH, Breslauer KJ. J Am Chem Soc. 2007;129:5272–5280. doi: 10.1021/ja070258q. [DOI] [PubMed] [Google Scholar]
- (7).Völker J, Plum GE, Klump HH, Breslauer KJ. J Am Chem Soc. 2009;131:9354–9360. doi: 10.1021/ja902161e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gelfand CA, Plum GE, Grollman AP, Johnson F, Breslauer KJ. Biochemistry. 1998;37:7321–7327. doi: 10.1021/bi9803372. [DOI] [PubMed] [Google Scholar]
- (9).Plum GE, Grollman AP, Johnson F, Breslauer KJ. Biochemistry. 1995;34:16148–16160. doi: 10.1021/bi00049a030. [DOI] [PubMed] [Google Scholar]
- (10).Völker J, Blake RD, Delcourt SG, Breslauer KJ. Biopolymers. 1999;50:303–318. doi: 10.1002/(SICI)1097-0282(199909)50:3<303::AID-BIP6>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (11).Cunningham B, Wells J. Science. 1989;244:1081–1085. doi: 10.1126/science.2471267. [DOI] [PubMed] [Google Scholar]
- (12).Morrison KL, Weiss GA. Current Opinion in Chemical Biology. 2001;5:302–307. doi: 10.1016/s1367-5931(00)00206-4. [DOI] [PubMed] [Google Scholar]
- (13).Gutell RR, Weiser B, Woese CR, Noller HF. Prog. Nucleic Acid Res. Mol. Biol. 1985;32:155–216. doi: 10.1016/s0079-6603(08)60348-7. [DOI] [PubMed] [Google Scholar]
- (14).Engelen S, Tahi F. BMC Bioinformatics. 2007;8:464. doi: 10.1186/1471-2105-8-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Espinoza-Fonseca LM. Biochemistry. 2009;48:11332–11334. doi: 10.1021/bi901705z. [DOI] [PubMed] [Google Scholar]
- (16).Lakowicz JR. Principles of fluorescence spectroscopy. 2nd ed Kluwer Academic/Plenum; New York: 1999. [Google Scholar]
- (17).Grollman AP, Moriya M. Trends in Genetics. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- (18).Fromme JC, Verdine GL. In: Advances in Protein Chemistry. Yang W, editor. Volume 69. Academic Press; 2004. pp. 1–41. [DOI] [PubMed] [Google Scholar]
- (19).David SS, O’Shea VL, Kundu S. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA repair and mutagenesis. 2nd Ed ASM Press; Washington, D.C.: 2006. [Google Scholar]
- (21).Sutherland BM, Bennett PV, Sidorkina O, Laval J. Proc Natl Acad Sci U S A. 2000;97:103–108. doi: 10.1073/pnas.97.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Georgakilas AG. Mol Biosyst. 2008;4:30–35. doi: 10.1039/b713178j. [DOI] [PubMed] [Google Scholar]
- (23).Shikazono N, Noguchi M, Fujii K, Urushibara A, Yokoya A. J Radiat Res (Tokyo) 2009;50:27–36. doi: 10.1269/jrr.08086. [DOI] [PubMed] [Google Scholar]
- (24).Weinfeld M, Rasouli-Nia A, Chaudhry MA, Britten RA. Radiation Research. 2001;156:584–589. doi: 10.1667/0033-7587(2001)156[0584:robere]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- (25).Gill SJ, Richey B, Bishop G, Wyman J. Biophys Chem. 1985;21:1–14. doi: 10.1016/0301-4622(85)85001-8. [DOI] [PubMed] [Google Scholar]
- (26).Wyman J, Gill SJ. Binding and Linkage. Functional Chemistry of Biological Macromolecules. University Science Books; Mill Valley, CA: 1990. [Google Scholar]
- (27).Völker J, Plum GE, Klump HH, Breslauer KJ. Biopolymers. 2010;93:355–369. doi: 10.1002/bip.21343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Breslauer KJ, Frank R, Blocker H, Marky LA. Proc Natl Acad Sci U S A. 1986;83:3746–3750. doi: 10.1073/pnas.83.11.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Monod J, Wyman J, Changeux J-P. Journal of Molecular Biology. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- (30).Tsai CJ, Del Sol A, Nussinov R. Mol Biosyst. 2009;5:207–216. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tsai CJ, del Sol A, Nussinov R. J Mol Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).del Sol A, Tsai CJ, Ma B, Nussinov R. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cui Q, Karplus M. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Changeux JP, Edelstein SJ. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- (35).Burd JF, Larson JE, Wells RD. Journal of Biological Chemistry. 1975;250:6002–6007. [PubMed] [Google Scholar]
- (36).Burd JF, Wartell RM, Dodgson JB, Wells RD. Journal of Biological Chemistry. 1975;250:5109–5113. [PubMed] [Google Scholar]
- (37).Bacolla A, Jaworski A, Larson JE, Jakupciak JP, Chuzhanova N, Abeysinghe SS, O’Connell CD, Cooper DN, Wells RD. Proc Natl Acad Sci U S A. 2004;101:14162–14167. doi: 10.1073/pnas.0405974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bacolla A, Wells RD. J Biol Chem. 2004;279:47411–47414. doi: 10.1074/jbc.R400028200. [DOI] [PubMed] [Google Scholar]
- (39).Bacolla A, Wells RD. Mol Carcinog. 2009;48:273–285. doi: 10.1002/mc.20507. [DOI] [PubMed] [Google Scholar]
- (40).Bacolla A, Wojciechowska M, Kosmider B, Larson JE, Wells RD. DNA Repair (Amst) 2006;5:1161–1170. doi: 10.1016/j.dnarep.2006.05.032. [DOI] [PubMed] [Google Scholar]
- (41).Chaires JB. ACS Chem Biol. 2008;3:207–209. doi: 10.1021/cb800070s. [DOI] [PubMed] [Google Scholar]
- (42).Schurr JM, Delrow JJ, Fujimoto BS, Benight AS. Biopolymers. 1997;44:283–308. doi: 10.1002/(SICI)1097-0282(1997)44:3<283::AID-BIP7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- (43).Chaires JB. Biochemistry. 1985;24:7479–7486. doi: 10.1021/bi00346a067. [DOI] [PubMed] [Google Scholar]
- (44).Gray RD, Petraccone L, Trent JO, Chaires JB. Biochemistry. 2009;49:179–194. doi: 10.1021/bi901357r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rueda D, Bokinsky G, Rhodes MM, Rust MJ, Zhuang X, Walter NG. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10066–10071. doi: 10.1073/pnas.0403575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.