

Abstract
The β2-adrenergic receptor (β2-AR) agonist [3H]-(R,R′)-methoxyfenoterol was employed as the marker ligand in displacement studies measuring the binding affinities (Ki values) of the stereoisomers of a series of 4′-methoxyfenoterol analogs in which the length of the alkyl substituent at α′ position was varied from 0 to 3 carbon atoms. The binding affinities of the compounds were additionally determined using the inverse agonist [3H]-CGP-12177 as the marker ligand and the ability of the compounds to stimulate cAMP accumulation, measured as EC50 values, were determined in HEK293 cells expressing the β2-AR. The data indicate that the highest binding affinities and functional activities were produced by methyl and ethyl substituents at the α′ position. The results also indicate that the Ki values obtained using [3H]-(R,R′)-methoxyfenoterol as the marker ligand modeled the EC50 values obtained from cAMP stimulation better than the data obtained using [3H]-CGP-12177 as the marker ligand. The data from this study was combined with data from previous studies and processed using the Comparative Molecular Field Analysis approach to produce a CoMFA model reflecting the binding to the β2-AR conformation probed by [3H]-(R,R′)-4′-methoxyfenoterol. The CoMFA model of the agonist-stabilized β2-AR suggests that the binding of the fenoterol analogs to an agonist-stabilized conformation of the β2-AR is governed to a greater extend by steric effects than binding to the [3H]-CGP-12177-stabilized conformation(s) in which electrostatic interactions play a more predominate role.
Keywords: β2-adrenoceptor selective agonist; β2-adrenoceptor conformations; agonist-stabilized conformations; antagonist-stabilized conformations; [3H]-(R,R′)-4′-methoxyfenoterol
1. Introduction
(R,R′)-Fenoterol ((R,R′)-1, Table 1) is a potent and selective agonist of the β2-adrenergic receptor (β2-AR), which has entered clinical trials for the treatment of congestive heart failure. The potential therapeutic utility of (R,R′)-1 is based upon its 43-fold selectivity for the β2-AR relative to the β1-AR, determined as binding affinities (Ki values) that were calculated using the β2-AR antagonist [3H]-CGP-12177 as the marker ligand.1 (R,R′)-1 also selectively couples to Gs proteins2 and produces a 300% increase in contractile response in isolated rat ventricular myocytes.1,2
Table 1.
Chemical structures of discussed FEN derivatives.
| Compound | α′ carbon atom number | Structure |
|---|---|---|
| Old compounds
| ||
| (R,R′)-1 | 1 |
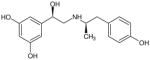
|
| (R,S′)-1 |
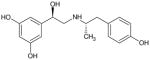
|
|
| (R,R′)-52 | 2 |
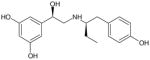
|
| (R,S′)-52 |
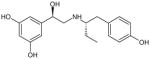
|
|
| (R,R′)-54 | 1 |
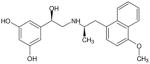
|
| (R,S′)-54 |
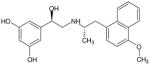
|
|
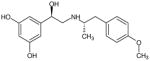
| (R,R′)-2 | 1 |

|
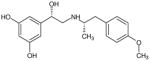
| (R,S′)-2 |
|
|
| (S,R′)-2 |

|
|
| (S,S′)-2 |
|
|
|
| ||
| New compounds
| ||
| (R,R′)-64 | 2 |
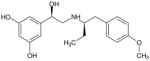
|
| (R,S′)-64 |
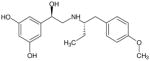
|
|
| (R,S′)-65 | 3 |

|
| (R,R′)-65 |
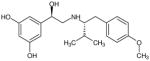
|
|
| (R,R′)-66 | 3 |
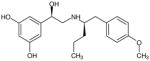
|
| (R,S′)-66 |
|
|
| (R)-67 | 0 |

|
| (S)-67 |
|
|
Using (R,R′)-1 as the scaffold, a project was initiated to study the effect of stereochemistry and the structure of the aminoalkyl moiety of the molecule on β2-AR binding affinity and selectivity as well as activity using induced stimulation of cAMP accumulation (EC50cAMP) and cadiomyocyte contractility (EC50cardio) as the functional markers.1,3 In the first stage of the project 26 compounds were synthesized and the Kiβ2-AR data used to develop a Comparative Molecular Field Analysis (CoMFA) model1, which was then used for virtual in silico prediction of binding affinities for a set of 21 new molecular structures.3 Six of the designed molecules were synthesized and their experimentally determined Kiβ2-AR values were in good agreement with the theoretical prediction and the data obtained using the additional compounds were used to refine the CoMFA model. While the expected Kiβ2-AR values were observed for the designed compounds, two of the derivatives, (R,R′)-52 and (R,R′)-54 had unexpectedly high β2-AR subtype selectivity with Kiβ1-AR/Kiβ2-AR of 334 and 573, respectively.3 The functional activities of a subset of the compounds were also determined and relationships between Kiβ2-AR and EC50cAMP and Kiβ2-AR and EC50cardio were observed for (R,R′)-1, (R,R′)-2 and (R,R′)-54 suggesting that the CoMFA model could be used to design compounds for use in the treatment of congestive heart failure. However, when (R,R′)-52 was studied, the compound had a >100-fold lower activity in the cardiomyocyte contractility model while the relationship between Kiβ2-AR and EC50cAMP was consistent with the other tested compounds. The results suggested that the substitution of an ethyl group on the aminoalkyl moiety of (R,R′)-1 had a profound effect on the interaction of (R,R′)-52 with a conformation(s) of β2-AR and that this effect was not reflected by the Kiβ2-AR values obtained using [3H]-CGP-12177 as the marker ligand or the CoMFA model constructed using these Kiβ2-AR values.
Based on the data obtained with (R,R′)-52, we have explored the effect of steric bulk at the α′-carbon of the aminoalkyl moiety on binding affinity and functional activity using 4′-methoxyfenoterol (2) as the scaffold. The alkyl length of the substituent on the α′-carbon of the aminoalkyl moiety was varied from 0 (compound 67) to 3 carbon atoms (compounds 65, 66), Table 1, and the Kiβ2-AR values were determined using [3H]-CGP-12177 and [3H]-(R,R′)-2 as the marker ligands. [3H]-(R,R′)-2 was used for this study as it has been previously demonstrated that this radioligand binds with high affinity to an agonist conformation of the β2-AR4, and that Kiβ2-AR values for the fenoterol analogs, including (R,R′)-52, determined using [3H]-(R,R′)-2 modeled their EC50cAMP values better than the data obtained using [3H]-CGP-12177.4 The EC50cAMP values of the newly synthesized compounds were also determined. The data were used to refine the CoMFA model developed using the Kiβ2-AR values obtained with [3H]-CGP-12177 as the marker ligand and to develop a new CoMFA model with the Kiβ2-AR values obtained when [3H]-(R,R′)-2 was the marker ligand. The new CoMFA model reflects binding to an agonist-stabilized conformation of the β2-AR. The results demonstrate that there are subtle but significant differences between the two models and suggest that the use of multiple models can be beneficial in the design of new chemical entities with specific pharmacological properties.
2. Results
2.1. Binding affinity of studied derivatives
Eight derivatives of compound 2 were synthesized by modifying the alkyl substituent at the α′ position from 0 to 3 carbon atom count (Table 1). The binding affinities of the synthesized compounds to β2-AR were determined using assays displacing either the antagonist radioligand [3H]-CGP-12177 or the selective agonist, [3H]-(R,R′)-2.1,4,5 When [3H]-CGP-12177 was the marker ligand, (R,R′)-2 and (R,R′)-64 had equal potency, 474 nM and 420 nM, while significantly lower affinities were observed for the other derivatives with an (R,R′) or equivalent configuration, 2,675 nM, 7,604 nM and not quantifiable for (R)-67 (R,R′)-66 and (R,S′)-65, respectively. It should be noted that the inclusion of (R,S′)-65 in this cohort is based on the change in the R and S designation at the α′ position based on the Cahn–Ingold–Prelog stereochemistry convention and not a change in actual configuration, (see Table 1). The measured affinities of (R,R′)-2 and (R,R′)-64 determined using [3H]-(R,R′)-2 as the marker ligand were 4.09 nM and 35.7 nM, respectively, indicating that while the affinities of the two compounds were similar, the magnitude of the binding affinity of (R,R′)-2 was more affected by the change in marker ligand than (R,R′)-64. However, this may only be a reflection of the fact that the calculated affinity of (R,R′)-2 is a Kd value. The Ki values for (R,S′)-65, (R,R′)-66, and (R)-67 could be determined using [3H]-(R,R′)-2 as the marker ligand and were 3,420 nM, 250 nM and 101 nM, respectively.
The data indicate that the optimal length of the alkyl moiety attached to the α′ position of the molecular scaffold is one or two carbon atoms. i.e. a methyl or ethyl group.
The observed trend in the effect of stereochemistry on receptor affinities was confirmed in studies of the potency and maximal activity for stimulation of cAMP accumulation HEK-293 cells stably transfected with human β2-AR gene. The EC50 values and % of stimulation in relation to isoproterenol, the reference full agonist, are presented in Table 2. All of the tested compounds were full β2-AR agonists and the most active analogs were (R,R′)-2, (R,S′)-2 and (R,R′)-64 with calculated EC50 values of 0.3 nM, (2 nM and 1.63 nM, respectively. The results are consistent with the observation that the use of [3H]-(R,R′)-2 as the marker ligand in displacement studies probes binding to an agonist conformation of the β2-AR and models EC50cAMP values better than the data obtained using [3H]-CGP-12177.4
Table 2.
Binding affinities of studied compounds.
| Compound | β2-AR binding affinity determined in displacement of
|
Induced cAMP accumulation
|
||||
|---|---|---|---|---|---|---|
| [3H]-CGP-12177 | [3H]-(R,R′)-2 | |||||
| Ki [nM] | HS | Ki [nM] | HS | EC50 [nM] | % stimulation | |
| Propranolol | 0.46 ± 0.06 | 1.24 ± 0.1 | 3.69 ± 1.36 | 1.88 ± 0.34 | ||
| Isoproterenol | 192 ± 24 | 0.85 ± 0.06 | 2.44 ± 0.28 | 0.78 ± 0.07 | 0.20 ± 0.09 | 100.00 |
| (R,R′)-1 | 345 ± 33.8a | 0.92 ± 0.1 | 4.00 ± 0.75b | 0.76 ± 0.1 | 0.30 ± 0.09a | 123 ± 9 |
| (R,S′)-1 | 3,695 ± 245a | 0.81 ± 0.1 | 183 ± 30.0b | 0.97 ± 0.1 | 4.70 ± 0.50a | 131 ± 30 |
| (R,R′)-52 | 1,273 ± 81a | 1.01 ± 0.01 | 39.1 ± 5.38b | 0.93 ± 0.1 | 2.80 ± 0.90a | 125 ± 6 |
| (R,S′)-52 | 5,758 ±833a | 2.07 ± 0.4 | 294 ± 45.1b | 0.87 ± 0.2 | 16.60 ± 4.90a | 98 ± 4 |
| (R,R′)-54 | 277 ± 11a | 1.07 ± 0.09 | 13.3 ± 2.72b | 0.86 ± 0.1 | 3.90 ± 1.80a | 106 ± 11 |
| (R,S′)-54 | 317 ± 6a | 1.06 ± 0.02 | 12.7 ± 1.83b | 0.90 ± 0.2 | 4.00 ± 1.29a | 118 ± 30 |
| (R,R′)-2 | 474 ± 40a | 0.98 ± 0.05 | 4.09 ± 0.55b | 0.80 ± 0.9 | 0.30 ± 0.23b | 135.70 ±11.03 |
| (R,S′)-2 | 1,930 ± 140a | 1.01 ± 0.14 | 26.1 ± 2.4b | 1.00 ± 0.05 | 2.00 ± 0.45b | 120.10 ± 18.97 |
| (S,R′)-2 | 5,270 ± 510a | 1.28 ± 0.09 | 91.3 ± 32b | 0.86 ± 0.13 | 7.20 ±1.01b | 129.00 ± 12.67 |
| (S,S′)-2 | 15,900 ± 2700a | 2.30 ± 0.32 | 2870 ± 234b | 1.68 ± 0.54 | 33.20 ± 11.48b | 156.40 ±35.93 |
| (R,R′)-64 | 420 ± 120 | 1.00 ± 0.11 | 35.7 ± 4.3 | 0.82 ± 0.04 | 1.63 ± 0.51 | 156 ± 33 |
| (R,S′)-64 | 20,100 ± 4,950 | ND | 4,300 ± 1,000 | 0.86 ± 0.05 | 1,010 ± 240 | 145.7 ± 8.8 |
| (R,R′)-65 | >100,000 | ND | 29,200 ± 4,400 | 1.41 ± 0.47 | 26,200 ± 6,500 | 103.5 ± 7.8 |
| (R,S′)-65 | >100,000 | ND | 3,420 ± 320 | 1.45 ± 0.56 | 6,100 ± 1,500 | 207 ± 56 |
| (R,R′)-66 | 7,600 ± 230 | 0.90 ± 0.03 | 250 ± 41 | 1.38 ± 0.32 | 462 ± 161 | 147.2 ± 2.7 |
| (R,S′)-66 | >100,000 | ND | 4,740 ± 700 | 0.89 ± 0.14 | 6,100 ± 1,300 | 197 ± 32 |
| (R)-67 | 2,670 ± 240 | 1.80 ± 0.70 | 101 ± 33 | 1.59 ± 0.50 | 10.19 ± 1.79 | 114.2 ± 8.4 |
| (S)-67 | >100,000 | ND | 1,150 ± 410 | 0.89 ± 0.03 | 210 ± 96 | 131 ± 43 |
2.2. Docking of ligands into β2-AR
Molecular models of studied derivatives were docked into the crystal model of the β2-AR binding site obtained in studies of the receptor cocrystallized with the agonist BI-167107 and the Gs protein. This model, PDB id: 3SN6, is commonly considered representative of an active state of the receptor6. Docking simulations show that the orientation of docked FEN (fenoterol) molecules corresponds well with the orientation of the BI-167107 molecule, Supplemental Figure S1. Comparison of the docking of (R,R′)-2 with BI-167107 indicates that both molecules assume very similar orientations within the binding site and thus have very similar ligand – receptor interactions. The most important interactions are those linking the protonated amino group and β-hydroxyl moiety of both ligands via a very strong network of hydrogen bonds with D113 and N312. In addition, the 3,5-hydroxyphenyl moiety of (R,R′)-2 interacts via hydrogen bonds with S203 and S207 residues located on TM5 while the 4′-methoxy moiety on the other end of the molecule forms a hydrogen bond with K305 residue on helix 7 (Fig. 1.a.).
Figure 1.

The lowest energy conformations of (A) (R,R′)-2, (B) (R,R′)-64 and (C) (R,S′)-64 obtained in docking simulations to molecular model of β2-AR binding site. For clarity of both figures, TM1, TM2, and extracellular loop 2 were hidden, and the remaining transmembrane segments are color coded as follows: TM3, red; TM4, green; TM5, magenta; TM6, yellow; and TM7, blue. Only the residues forming hydrogen bonds (shown as green arrows) with a ligand molecule are shown explicitly. All aliphatic hydrogen atoms are hidden.
Docking simulations attempted to identify optimal positions of the derivatives with increased alkyl chain length. Figure 1.b. and 1.c. show the lowest energy conformations obtained in docking of (R,R′)-64 and (R,S′)-64, respectively. It can be seen that in comparison to the docking position assumed by (R,R′)-2, increased bulkiness of the alkyl moiety significantly affects the location within the binding site and possible ligand – receptor interactions. In this agonist conformation, both (R,R′)-64 and (R,S′)-64 cannot assume positions allowing all interactions analogous to (R,R′)-2. In particular, the 3,5-dihydroxyphenyl ring of (R,R′)-64 is not positioned to form strong hydrogen bonds with S203 and S207 on TM5, while the 4′-methoxyphenyl ring of the molecule is bent preventing therefore the formation of a strong hydrogen bond with K305 (in all three cases distances between donor and acceptor atoms exceed 3.5 Ǻ). The amino group of (R,R′)-64 does form coulombic interactions with D113 (marked with a yellow arrow on Fig. 1.b.) but does not form a hydrogen bond with another possible partner, N312. This reduced hydrogen bond formation can explain the 10 fold decrease in binding affinity of (R,R′)-64 when tested using [3H]-(R,R′)-2 to measure binding in an agonist conformation. In docking of (R,S′)-64, the amino group of the ligand cannot interact with D113 but forms a hydrogen bond with N312; the 3,5-dihydroxyphenyl ring is closer to TM5 allowing stronger hydrogen bonds with serine residues (distances between donor and acceptor atoms are between 2.6 – 2.9 Ǻ); 4′-methoxyphenyl ring is bent deeper into the cavity, entirely out of range for K305 (distance reaches 5 Ǻ). Instead, the 4′-methoxy group forms a hydrogen bond with W313 residue of TM7 (the interaction is not shown on Fig. 1.c.).
Analogous observations as above can be made in docking the other derivatives, 65, 66 and 67. Either increasing the chain to propyl for (R,S′)-65 and (R,R′)-66, or reducing it to hydrogen as in case of (R)-67 prevents the molecules from occupying positions that exercise all interactions as observed in docking of (R,R′)-2, data not shown. This negative effect is increasingly profound in docking simulation of these derivatives with other stereochemistries. This is in agreement with experimental data where (R,R′)-64, (R,S′)-65, (R,R′)-66 or (R)-67 are more potent within the pair of stereoisomers.
Since the binding affinity of (R,R′)-64 is very similar to those of (R,R′)-2 or (R,R′)-1 the results suggest that the receptor most likely assumes slightly different conformation to adopt the molecule with α ′ethyl moiety.
2.3. 3D-QSAR
The affinity data determined for new derivatives were subjected to 3D-QSAR modeling. Previous CoMFA models generated based on affinity data determined using the [3H]-CGP-12177 displacement assay underlined the importance of the stereochemical configuration of a derivative and the type of modification at the aminoalkyl part of the molecule. If currently studied compounds explore additional dimensions of chemical modification this fact should be reflected in new CoMFA models. Additionally, the binding data for a significant number of FEN derivatives were determined using the radiolabeled agonist [3H]-(R,R′)-2. This presents the unique opportunity to compare our previous data with a second 3D-QSAR model based on pKi values determined to an agonist receptor conformation.
The number of data points in the current model (pKi values determined using [3H]-(R,R′)-2 derived data) is n = 32 (18 in current report, Table 1 and 14 previously presented3). The newly generated model maintains high statistical significance as reflected by R2 = 0.699, F = 191.7, SEP = 0.458, Q2 = 0.86.
The field distribution of the CoMFA [3H]-(R,R′)-2 model is presented in Figure 2. The model confirms previous observations that the stereochemistry of a molecule plays a very important role in the observed affinity. Both chiral centers of studied molecules are asymmetrically surrounded by molecular fields: β carbon atom by a combination of electrostatic fields and α′ carbon atom by a combination of steric fields. In the first case, the red field representing interactions with negative charge (or hydrogen bond acceptor) is located in front of the molecule, while a blue, electropositive field is behind the molecule in Fig. 2. The model, thus, illustrates that the β-hydroxyl moiety of FEN and analogs can donate a proton to make a hydrogen bond while in the (R) stereoconfiguration, and such an interaction is disfavored in the (S) configuration of the chiral center. With regards to the other chiral center, both sides of the α′ carbon atom, are filled by steric fields: the large yellow unfavorable field behind the molecule on Fig. 2, underlines the fact that pointing the alkyl moiety in that direction (i.e., the (S′) stereoconfiguration of the center for methyl, ethyl and n-propyl moieties; the (R′) configuration for i-propyl) negatively affects the binding affinity. On the other hand, green sterically favorable field in front of the chiral center suggests that the (R′) configuration of these analogs derivatives has a positive effect on observed pKi values. Interestingly, the green field is associated with another smaller yellow field at a greater distance from the chiral center what clearly supports the observation that increased size of alkyl moiety (i.e., propyl chains) significantly reduces the affinity these derivatives even if it is in preferred stereoconfiguration.
Figure 2.

CoMFA model constructed for affinity data determined using [3H]-(R,R′)-2 displacement assay for 32 FEN derivatives assayed in the current and in previous reports.1,3 The fields are color coded in the following manner: green and yellow represent regions of steric (bulk) interactions (favorable and unfavorable, respectively), electrostatic interactions with a positive charge (or H-bond donor) are blue, and electrostatic interactions with a negative charge (or H-bond acceptor) are red. Molecular model of (R,R′)-64 is rendered in stick mode for orientation purpose.
Another region of interest in this CoMFA model is the aminoalkyl part of these molecules where structural modifications are concentrated. This region of the model is represented by several steric fields, among them one significant favorable (green) field within the plane of the phenyl ring of the molecule (Fig. 2). This field likely corresponds with positive effects associated with occurrence of a naphthyl ring present in several analogs (previously described in detail3). There is also one electropositive field in this region of the model (blue, on the bottom right of Fig. 2.), which represents the positive effect of forming a hydrogen bond with an acceptor atom (either oxygen or nitrogen) in 4′-position of a derivative.
The model has high statistical significance and may be considered as a 3D representation of derivatives’ affinity to an active form of the receptor. Supporting material includes a Table S2 collecting pKi values predicted by the model for all derivatives including those not yet tested in the [3H]-(R,R′)-2 displacement assay.
3. Discussion
Previous studies of β2-AR agonists built upon a catecholamine scaffold established that compounds with an R configuration at the β-OH carbon are more active than the corresponding isomers with an S configuration at this position.1,3 Data obtained in the initial studies of the stereoisomers of 2 were consistent with this observation as the compounds with an (R-) configuration at the β-OH carbon, (R,R′)-2 and (R,S′)-2, had higher binding affinities than (S,R′)-2 and (S,S′)-2 and were more pharmacologically active in both stimulation of cAMP accumulation and in cardiomyocyte contractility.1–3 The results also indicated that the stereochemical orientation the α′-alkyl moieties had an effect on β2-AR interactions as the relative binding affinity and pharmacological activity of (R,R′)-2 was greater than (R,S′)-2 and only (R,R′)-2 selectively signaled through Gs proteins.1–3 Based on these results, only the (R,R′)- and (R,S′)-isomers of the test compounds were synthesized and studied. The compounds were produced by changing the alkyl chain at the α ′ position from 0 to 3 carbon atoms using compound 2 as the scaffold. In these studies, the binding affinities of the test compounds to the β2-AR were determined using [3H]-CGP-12177 and [3H]-(R,R′)-2 as the marker ligands.
When [3H]-CGP-12177 was used as the marker ligand, the Ki value of (R,R′)-64 was equivalent to (R,R′)-2 and 3-fold lower than (R,R′)-52. In addition, there was a 50-fold difference between the binding affinities of (R,S′)-64 and (R,R′)-64, calculated as Ki(R,S′)/Ki(R,R′), which differed from the ~5-fold differences calculated for the (R,R′)- and (R,S′)-isomers of 2 and 52. The Ki values of (R,R′)-66 and (R)-67 were also lower than their respective stereoisomers, indeed no measurable binding affinities were observed for (R,S′)-66 and (S)-67. The data suggest that both the 4′-methoxy moiety and the stereochemical configuration of the alkyl moiety at the α′ carbon affect the binding interactions with the conformations probed by [3H]-CGP-12177.
When the methyl moiety on (R,R′)-2 was replaced by a hydrogen atom, the Ki value of (R)-67 was increased by 50-fold relative to (R,R′)-2 and decreased by ~3-fold relative to the Ki value of (R)-7, the N-desmethyl analog of (R,R′)-1.1 These results suggest that the presence of some steric bulk at the α′ carbon has a positive effect on the binding affinity of the area(s) probed using [3H]-CGP-12177 as the marker ligand. However, the data obtained with the other analogs indicate that this effect is limited as the substitution of an n-propyl moiety, (R,R′)-66, significantly reduced the Ki value to >7,000 nM and no measurable binding affinities were observed with the derivatives containing an i-propyl substituent, (R,R′)-65 and (R,S′)-65 or for (R,S′)-66 and (S)-67.
Previous studies of the binding affinities of FEN analogs to the β2-AR utilizing [3H]-(R,R′)-2 as the marker ligand have demonstrated that the calculated Ki values are significantly lower than the values calculated using [3H]-CGP-12177 as the marker ligand.4 The same results were obtained in this study including the ability to determine Ki values for (R,R′)-65, (R,S′)-65, (R,S′)-66 and (S)-67, Table 2. The same effect of the stereochemical configuration at the α′ carbon was also observed as lower Ki values were observed for (R,R′)-2, (R,R′)-64, (R,S′)-65 and (R,R′)-66 relative to their respective stereoisomers. In this regards, it is important to note that the configuration at the α ′ carbon of (R,S′)-65 is the same as the configurations in (R,R′)-64 and (R,R′)-66 as the only change is in the Cahn-Ingold-Prelog nomenclature produced by the α′-i-propyl moiety which changes the priority at this chiral center from (R) to (S). The Ki value for (R)-67 was also 10-fold lower than (S)-67, consistent with the importance of the configuration at the β-OH carbon.
The Ki values for compounds 64, 65, 66 and 67 determined using [3H]-(R,R′)-2 were combined with the Ki values of 22 previously studied FEN analogs4 to create a new CoMFA model correlating molecular fields with the affinity data determined using [3H]-(R,R′)-2. The resulting model, Fig. 2, confirms the importance of a proper stereochemistry at both chiral centers of the FEN scaffold: 1) the stereoconfiguration of the β-OH carbon corresponding to the (R)-configuration is preferred due to the possibility of donating a hydrogen bonding partner from the β-OH group; 2) the stereoconfiguration of the α′ carbon atom corresponding to the (R′) configuration is preferred due to a steric reason as the model clearly indicates that the opposite configuration of this chiral center is sterically unfavored in terms of binding affinity. In addition, the binding data and the modeling results suggest that for FEN analogs with a 4′-methoxy moiety the optimal binding affinity is obtained with either a methyl (2) or ethyl (64) moiety at α′ position and an (R,R′)-stereoconfiguration, while the molecules with no alkyl chain (67) or propyl chains (65 or 66) show drastically reduced affinities.
The extensive 3D-QSAR modeling performed on a larger cohort of FEN derivatives offers the opportunity to compare the new CoMFA model generated using the agonist [3H]-(R,R′)-2 and the previously reported model generated from affinity data determined using the antagonist [3H]-CGP-12177.1,3 The two models are very similar in regions responsible for stereoselectivities with similar fields distribution can be identified in vicinities of the two chiral centers. There are, however, significant differences in the third region of structural variation, the aminoalkyl tail of the derivatives. While CoMFA{[3H]-CGP-12177} models show mainly electrostatic fields in this region, the CoMFA{[3H]-(R,R′)-2} model indicates relative prevalence of steric fields in the vicinity of the aminoalkyl part of derivatives. The results from the 3D-QSAR studies suggest that steric non-polar effects dominate in interactions of studied ligands with the COMFA{[3H]-(R,R′)-2} model, while prevalence of electrostatic interactions can be assigned to the COMFA{[3H]-CGP-12177} model.
In a recent publication, Kim et al.7 discussed three NMR-probed states of the β2-AR, which were designated as S1, S2 and S3. S1 and S2 were associated with the inverse agonist carazolol and the partial agonist salmeterol, respectively, and were described as two distinct inactive-state conformers, while S3 was identified as an agonist-activated intermediate. The results from our previous studies of the binding of FEN analogs to the β2-AR1,3 and the data from this study are consistent with the hypothesis that Ki values determined using [3H]-CGP-12177 and [3H]-(R,R′)-2 represent binding affinities to different states of the β2-AR with [3H]-(R,R′)-2 reflecting binding to an agonist-stabilized conformation (S3) and [3H]-CGP-12177 probing antagonist-stabilized populations (S1,S2) as well as S3.4 As part of these studies we characterized the thermodynamics of the binding process of (R,R′)-1, (R,S′)-1, (S,R′)-1 and (S,S′)-1 to the β2-AR. Van’t Hoff analysis of the data obtained using [3H]-CGP-12177 as the marker determined that binding of (R,R′)-1 and (R,S′)-1 were exclusively entropy driven while the binding of (S,R′)-1 and (S,S′)-1 were predominantly enthalpy driven.3 When [3H]-(R,R′)-2 was used as the radioligand, the binding for all four stereoisomers of 1 were entropy-controlled.4 The data from the thermodynamic studies of the stereoisomer of 1 are consistent with the results presented by Kim et al.7 which indicated that the activation to S3 is enthalpically unfavorable and entropically favored. Therefore, results from the 3D-QSAR studies suggest that the COMFA{[3H]-(R,R′)-2} model reflects an agonist-stabilized state of the β2-AR (S3) and the COMFA{[3H]-CGP-12177} model is associated with conformation states reflecting antagonist-stabilized forms of the β2-AR. The difference between the models is also reflected by the pharmacological activities of the compounds tested in this study. All of the analogs were full β2-AR receptor agonists with respect to the stimulation of cAMP accumulation, relative to isoproterenol, Table 2. However, when [3H]-CGP-12177 was used as the marker ligand no significant binding was observed for (R,R′)-65, (R,S′)-65, (R,S′)-66 and (S)-67, Table 2.
One of the objectives of the current study was the determination of the pharmacological profile of (R,R′)-64. This objective was based upon the previous observations that (R,R′)-52 was essentially inactive in the stimulation of rat cardiomyocyte contractility3 while potently inhibiting proliferation of 1321N1 astrocytoma cells (IC50 1.4 nM).5 In addition, in the rat, the oral bioavailability of (R,R′)-2 is 3-fold higher than that of (R,R′)-1, indicating that the substitution of a 4′-methoxy moiety reduced presystemic glucuronidation.8 Initial studies using the previously described procedures1,3,5 demonstrated that (R,R′)-64 has no activity in the cardiomyocyte contractility model and effectively inhibits the proliferation of 1321N1 astrocytoma cells with an IC50 of 1.5 nM (unpublished data). The data suggest that (R,R′)-64 and the COMFA{[3H]-(R,R′)-2} model can be used for the design and optimization of orally available anti-cancer agents which have no cardiovascular effects.
4. Experimental Section
4.1. β2-AR binding assays using [3H]-CGP-12177 as a marker
Compounds synthesized in this study were tested at least three times in triplicate to determine their binding affinities at the β2-AR following a previously described approach.1 In brief, β2-AR binding was conducted on membranes derived from HEK cells containing human β2-AR (provided by Dr. Brian Kobilka, Stanford Medical Center, Palo Alto, CA). Cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and 0.05% penicillin–streptomycin with 400 lg/mL G418. The binding assays contained 0.3 nM [3H]-CGP-12177 and 60 μg of cell membranes in a volume of 1.0 mL. Nonspecific binding was determined using 10 μM propranolol. Bound radioactivity was counted on a Wallac plate liquid scintillation counter (PerkinElmer Life and AnalyticalSciences, Waltham, MA) and expressed in counts per minute. Competition curves with standard and unknown compounds included at least six concentrations, in triplicate. IC50 values and Hill coefficients were calculated using Prism software. Ki values were calculated using the Chang-Prusoff transformation.9
4.2. β2-AR binding assays using [3H]-(R,R′)-2 as a marker and determination of the stimulation of cAMP accumulation
Binding affinities and cAMP accumulation measurement experiments were conducted using exactly the same protocol as described in Toll et al.4
4.3. CoMFA analysis
The CoMFA [3H]-(R,R′)-2 model was generated using methodology implemented in Sybyl-X 2.0 (TRIPOS Inc., St. Louis, MO). The molecular models of structures were prepared in HyperChem v. 6.03 (HyperCube Inc., Gainesville, FL) using Model Build procedure to ensure the same conformation of the common scaffold.10 The models were extracted to SYBYL and the Gasteiger-Huckel atomic charges were calculated. The models were aligned with molecules of original training set using the two asymmetric carbon atoms in the core of the fenoterol molecule (–Cβ–CH2–NH–Cα′–CH2–) as a common substructure. A PREDICT procedure implemented in CoMFA package was used to compute the estimated pKi values. Two types of molecular fields (steric and electrostatic) were sampled on the grid (2 Å spacing) lattice surrounding each structure.
4.4. Docking simulations
In the present study we used the automated docking simulations of the flexible ligands (stereoisomers of fenoterol and its derivatives) to the crystal model of β2-AR. The crystallographic structure of β2-AR with bounded agonist, BI-167107 was obtained from Protein Data Bank (PDB id: 3SN6).6 In order to stabilize the active state of the receptor, β2-AR was crystallized in the complex with Gs protein. In the present study the stereoisomers of fenoterol and its derivatives were used as a molecular probe to identify differences in stereo-recognition of structurally similar agonists. Models of ligands were built using the HyperChem 6.03 (HyperCube Inc., Gainesville, FL) software and the in-built Model Build procedure. Molecules were optimizing by using the AM1 semi-empirical potential11 implemented in the HyperChem v. 6.03. The Molegro Virtual Docker (MVD v. 2010.4.0.0) software was employed for docking ligands (n = 74) to the rigid model of β2-AR. The MolDock SE as a search algorithm was used, and the number of runs was set to 100. The parameters of docking procedure are as follows: population size of 50, maximum iteration of 1500, energy threshold of 100.00, and maximum number of steps of 300. The maximum number of poses to generate was increased to 10 from a default value of 5. Docking procedure was performed for the ligands with protonated amine group. The appropriate charge was added in MVD. The estimation of ligand-receptor interactions was described by the MVD-related scoring functions: MolDock Score, Rerank Score, Hbond Score, Similarity Score, Docking Score. The scoring functions include the influence of several factors such as van der Waals forces, electrostatic interactions and solvent terms.12 The values of these functions are used to discriminate among orientations and molecules and to interpret of ligand poses in the binding cavity. The predicted positions of ligands in the cavity of β2-AR are characterized by a simultaneous lowering of the scoring function values; this corresponds to the high values of the ligand binding energy. The analysis of docking results was focused on the scoring function values obtained for complexes fulfilling all the three following conditions: (1) the lowest value of MolDock Score; (2) the 3,5-dihydroxyphenyl moiety of the ligand interacts with serine residues on the fifth trasmembrane domain (TM5); (3) the protonated nitrogen atom of ligand creates the salt bridge with D113 (TM3). According to the docking procedure, all studied ligands (fenoterol derivatives) and the BI-167107 molecule (for the validation of docking procedure), were docked to the binding cavity limited by the sphere, the centre of which corresponds to the position of agonist molecule in the BI-167107-β2-AR complex (PDB id: 3SN6). This sphere contains the amino acid residues which play an important role in creating the ligand-β2-AR attractive interaction e.g.: D113, N293, S203, S204, S207, Y308, K305 and the second extracellular loop (ECL2). The significance of these residues was confirmed in functional and biophysical studies.
4.5. Chemistry of new derivatives
All reactions were carried out using commercial grade reagents and anhydrous solvents. Ultraviolet spectra were recorded on a Cary 50 Concentration spectrophotometer. Optical rotations were done at 25 °C on a Rudolph Research Autopol IV. NMR Spectra were recorded on either a Varian Mercury VMX 300-MHz or a Varian XL-400 MHz spectrophotometer. In reporting the NMR multiplicities, we used the following abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet; apt, apparent; and br, broad. Low-resolution mass spectra (MS) were obtained on a Thermo LCQ Fleet MS system equipped with an electrospray ionization (ESI) probe. High-resolution mass spectra (HRMS) were obtained by the Old Dominion University Mass Spectrometry Service (Norfolk, VA), also with an ESI probe. Analytical HPLC data were obtained using a Waters 2690 Separations Module with PDA detection. Merck silica gel (230–400 mesh) was used for open column chromatography.
Chiral amines (72a–c) were accessed using a modified preparation for a similar substrate13, the major modifications being the stoichiometry of the initial aryl-alkylation reaction was changed to 1:1 with respect to the chiral oxirane and using excess borane-etherate. In early attempts, specifically for 1-(4-methoxyphenyl)butane-2-ol (68a), 4-iodoanisole was used in place of 4-bromoanisole, but yield variability on scaling-up resulted in our using 4-bromoanisole. We also found that preparing the mesylate in dichoromethane in the presence of triethylamine to be preferable to methylsulfonation in pyridine. Note: All traces of dichloromethane should be carefully removed prior to treatment with sodium azide to prevent the formation of potentially explosive diazidomethane.14
In the case of the ethyl analog, the chiral oxirane is commercially available. For the i-propyl and n-propyl analogs, the oxiranes were prepared from valine and norvaline isomers, respectively, via a Sandmeyer type chlorination with retention of absolute configuration15,16 followed by reduction of the acid to the chlorohydrin, then epoxidation with base resulting in an inversion of absolute configuration. Benzylamine 72d was prepared using established benzylation chemistry on the corresponding primary amine precursor.
With the new amines in hand, fenoterol analogs 64–67 were prepared using our previously reported synthesis1,3 with minimal modifications by coupling the chiral amines to (R)- or (S)-2-(3,5-bis(benzyloxy)phenyl)oxirane (R)-73 or (S)-73.
General Preparation of 2-Alkyloxiranes, (R)-2-Isopropyloxirane Represented [(R)-78]
(S)-2-Chloro-3-methylbutanoic acid [(S)-76]
To 12.5 g (107 mmol) of D-(−)-valine dissolved in 134 mL of 6N HCl with heat then cooled to 0 °C was added 11.8 g (171 mmol, 1.6 eq) of sodium nitrite in small portions over 2–3 h (hours). The solution was stirred at 0 °C for 6 h then warmed to RT (room temperature) and extracted into DCM (dichloromethane), the extracts washed with water, dried (MgSO4), filtered, and evaporated to give a colorless oil, 12.6 g (86%). 1H NMR: (300 MHz, CDCl3) δ 1.07 (d, J = 6.6 Hz, 3H), 1.09 (d, J = 6.6 Hz, 3H), 2.36 (m, J = 6.0 Hz, 1H), 4.20 (d, J = 6.0 Hz, 1H) ppm.
(S)-2-Chloro-3-methylbutan-1-ol [(S)-77]
To 12.55 g (92 mmol) of (S)-76 in 80 mL of hexanes was added at RT, 20.2 mL (102 mmol, 1.1 eq) of BH3SMe2 (borane dimethylsulfide) (5 M in Et2O). The reaction mixture refluxed for 1 h, cooled, and poured into 100 mL of ice-cold methanol and stirred overnight. The solvent was removed to give 11.26 g (100%) of a colorless oil, which was used without further purification. 1H NMR: (300 MHz, CDCl3) δ 1.02 (d, J = 6.0 Hz, 3H), 1.04 (d, J = 6.0 Hz, 3H), 1.76 (br s, 1H), 2.05 (m, 1H), 3.49 (s, 1H), 3.77 (m, 2H), 3.91 (m, 1H) ppm.
(R)-2-Isopropyloxirane [(R)-78]
To 11.2 g (91.3 mmol) of (S)-77 chilled to −70 °C was added 10.25 g (183 mmol) of finely ground potassium hydroxide. The slurry was allowed to warm to RT, then distilled (bp 71–73 °C) giving 4.79 g (61%) of (R)-78. 1H NMR: (300 MHz, CDCl3) δ 0.96 (d, J = 6.6 Hz, 3H), 1.03 (d, J = 6.6 Hz, 3H), 1.47 (m, 1H), 2.51 (m, 1H), 2.71 (m, 2H) ppm.
(R)-2-Chloro-3-methylbutanoic acid [(R)-76]
Prepared from 46.8 g (0.40 mol) of L-(+)-valine to give 44.0 g (80%). 1H NMR: (300 MHz, CDCl3) δ 1.07 (d, J = 6.6 Hz, 3H), 1.09 (d, J = 6.6 Hz, 3H), 2.36 (m, J = 6.0 Hz, 1H), 4.20 (d, J = 6.0 Hz, 1H) ppm.
(R)-2-chloro-3-methylbutan-1-ol [(R)-77]
Prepared from 12.3 g (90.0 mmol) of (R)-76 to give 10.7 g (97%). Bp 85–88 °C/18 mm; 1H NMR: (300 MHz, CDCl3) δ 1.02 (d, J = 6.0 Hz, 3H), 1.04 (d, J = 6.0 Hz, 3H), 1.76 (br s, 1H), 2.05 (m, 1H), 3.49 (s, 1H), 3.77 (m, 2H), 3.91 (m, 1H) ppm.
(S)-2-Isopropyloxirane [(S)-78]
Prepared from 10.7 g (87.6 mmol) of (R)-77 to give 5.78 g (77%). 1H NMR: (300 MHz, CDCl3) δ 0.96 (d, J = 6.6 Hz, 3H), 1.03 (d, J = 6.6 Hz, 3H), 1.47 (m, 1H), 2.51 (m, 1H), 2.71 (m, 2H) ppm.
(S)-2-Chloropentanoic acid [(S)-79]
Prepared from 10.0 g (85.4 mmol) of L-(+)-norvaline to give 9.58 g (82%). 1H NMR: (300 MHz, CDCl3) δ 0.97 (t, J = 7.2 Hz, 3H), 1.53 (m, 2H), 2.00 (m, 2H), 4.33 (dd, J = 6.0, 7.6 Hz, 1H) ppm.
(S)-2-Chloropentan-1-ol [(S)-80]
Prepared from 9.58 g (70.1 mmol) of (S)-79 to give 7.14 g (83%). Bp 79–80 °C/20 mm; 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.2 Hz, 3H), 1.52 (m, 2H), 1.72 (m, 2H), 1.81 (br s, 1H), 3.66 (m, 1H), 3.79 (m, 1H), 4.04 (m, 1H) ppm.
(R)-2-Propyloxirane [(R)-81]
Prepared from 7.14 g (58.3 mmol) of (S)-80 to give 3.88 g (77%). Bp 78–79 °C; 1H NMR: (300 MHz, CDCl3) δ 0.91 (t, J = 7.2 Hz, 3H), 1.40 (m, 2H), 1.52 (m, 2H), 2.46 (m, 1H), 2.74 (m, 1H), 2.89 (m, 1H) ppm.
(R)-2-Chloropentanoic acid [(R)-79]
Prepared from 10.0 g (85.4 mmol) D-(−)-norvaline to give 8.05 g (69%). 1H NMR: (300 MHz, CDCl3) δ 0.97 (t, J = 7.2 Hz, 3H), 1.53 (m, 2H), 2.00 (m, 2H), 4.33 (dd, J = 6.0, 7.6 Hz, 1H) ppm.
(R)-2-Chloropentan-1-ol [(R)-80]
Prepared from 8.05 g (58.9 mmol) of (R)-79 to give 7.02 g (95%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.2 Hz, 3H), 1.52 (m, 2H), 1.72 (m, 2H), 1.81 (br s, 1H), 3.66 (m, 1H), 3.79 (m, 1H), 4.04 (m, 1H) ppm.
(S)-2-Propyloxirane [(S)-81]
Prepared from 7.02 g (57.3 mmol) of (R)-80 to give 3.11 g (63%). Bp 78–79 °C; 1H NMR: (300 MHz, CDCl3) δ 0.91 (t, J = 7.2 Hz, 3H), 1.40 (m, 2H), 1.52 (m, 2H), 2.46 (m, 1H), 2.74 (m, 1H), 2.89 (m, 1H) ppm.
Preparation of Chiral Alcohols
(S)-1-(4-Methoxyphenyl)butan-2-ol [(S)-68a]
Under an argon atm, a solution of 5.85 g (25 mmol) 4-iodoanisole in 120 mL of dry THF was cooled to −70 °C internal (CO2-iPrOH bath). A solution of 10.0 mL (25 mmol) of n-butyllithium (n-BuLi) (2.5 M in hexanes) was added slowly over 20 min. The mixture was stirred for 10 min, then 1.1 mL (12.5 mmol, 0.5 eq) of (S)-(−)-1,2-epoxybutane was added followed by the addition of 2.3 mL (19 mmol, 0.75 eq) of BF3•Et2O. The solution was stirred for 20 min then removed from the cooling bath and quenched by the slow addition of saturated NH4Cl solution (~40 mL). Partitioned between Et2O and 100 mL of added water, washed with brine, dried (Na2SO4), filtered, and evaporated. Column chromatography on silica with 1:3 hexanes:DCM gave 1.5 g (72%). 1H NMR: (300 MHz, CDCl3) δ 0.99 (t, J = 7.2 Hz, 3H), 1.50 (m, 2H), 1.59 (br s, 1H), 2.59 (m, 1H), 2.76 (m, 1H), 3.68 (m, 1H), 3.79 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)butan-2-ol [(R)-68a]
Prepared as for (S)-68a from 3.25 g (13.9 mmol) 4-iodoanisole, 6.0 mL (13.9 mmol) of n-BuLi (2.3 M in hexanes), 1.2 mL (13.9 mmol, 1 eq) of (R)-(+)-1,2-epoxybutane and 2.6 mL (20.9 mmol, 1.5 eq) of BF3•Et2O. Column chromatography on silica with 1:3 hexanes:DCM gave 1.6 g (64%). 1H NMR: (300 MHz, CDCl3) δ 0.99 (t, J = 7.2 Hz, 3H), 1.50 (m, 2H), 1.59 (br s, 1H), 2.59 (m, 1H), 2.76 (m, 1H), 3.68 (m, 1H), 3.79 (s, 3H), 6.86, (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
General Procedure for the Preparation of Alcohols (68b–c)
Under an argon atm, 4-bromoanisole was dissolved in dry THF (conc 0.6 M) and cooled to an internal temperature of −70 °C. A solution of 2.5 M n-BuLi in hexanes (1 eq) was added slowly taking care to maintain the low internal temperature. The chiral oxirane (0.83–0.9 eq) was added over 5 min followed by the slow addition of BF3•Et2O (1.25 eq). Stirring was continued for 30 min, at which point the reaction was removed from the cooling bath and quenched by the slow addition of NH4Cl. The reaction mixture was partitioned with ether, washed with brine, dried (MgSO4), filtered, and evaporated. The residues were purified on silica gel with a gradient of 60–100% DCM in hexanes.
(S)-1-(4-Methoxyphenyl)-3-methylbutan-2-ol [(S)-68b]
Prepared from 5.42 g (29.0 mmol) of 4-bromoanisole, 11.6 mL (29.0 mmol) of n-BuLi (2.5 M in hexanes), 2.24 g (26.0 mmol) of (S)-78 and 4.8 mL (39 mmol) of BF3•Et2O. Column chromatography on silica with hexanes/DCM gave 3.93 g (77%). 1H NMR: (300 MHz, CDCl3) δ 0.99 (t, J = 6.9 Hz, 6H), 1.44 (br s, 1H), 1.74 (m, 1H), 2.53 (m, 1H), 2.80 (m, 1H), 3.55 (m, 1H), 3.80 (s, 3H), 6.86, (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)-3-methylbutan-2-ol [(R)-68b]
Prepared from 10.3 g (55.0 mmol) of 4-bromoanisole, 22.0 mL (55.0 mmol) of n-BuLi (2.5 M in hexanes), 4.31 g (50.0 mmol) of (R)-78 and 9.2 mL (75 mmol) of BF3•Et2O. Column chromatography on silica with hexanes/DCM gave 6.08 g (63%). 1H NMR: (300 MHz, CDCl3) δ 0.99 (t, J = 6.9 Hz, 6H), 1.44 (br s, 1H), 1.74 (m, 1H), 2.53 (m, 1H), 2.80 (m, 1H), 3.55 (m, 1H), 3.80 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(4-Methoxyphenyl)pentan-2-ol [(S)-68c]
Prepared from 8.10 g (43.3 mmol) 4-bromoanisole, 17.3 mL (43.3 mmol) of n-BuLi (2.5 M in hexanes), 3.11 g (36.1 mmol) of (S)-81 and 6.7 mL (54 mmol) of BF3•Et2O. Column chromatography on silica with hexanes/DCM gave 6.20 g (88%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 6.6 Hz, 3H), 1.47 (m, 5H), 2.57 (m, 1H), 2.79 (m, 1H), 3.75 (m, 1H), 3.79 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)pentan-2-ol [(R)-68c]
Prepared from 10.1 g (53.8 mmol) 4-bromoanisole, 21.5 mL (53.8 mmol) of n-BuLi (2.5 M in hexanes), 3.86 g (44.8 mmol) of (R)-81 and 8.3 mL (67 mmol) of BF3•Et2O. Column chromatography on silica with hexanes/DCM gave 7.68 g (74%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 6.6 Hz, 3H), 1.47 (m, 5H), 2.57 (m, 1H), 2.79 (m, 1H), 3.75 (m, 1H), 3.79 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
General Procedure for the Preparation of Mesylates (69a–c)
To the chiral alcohol (68a–c) in dry DCM (conc. 0.15 M) at −70 °C was added over 1 min triethylamine (1.2 eq) followed by the slow addition of methanesulfonyl chloride (1.2 eq). The mixture was allowed to warm to RT over 1–2 h and stirring was continued for an additional 1 h. The reaction mixture was poured into 100 mL of ice water, the organics separated, washed with brine, dried (MgSO4), filtered, and evaporated. The residues were used without further purification.
(S)-1-(4-Methoxyphenyl)butan-2-yl methanesulfonate [(S)-69a]
Prepared from 1.92 g (10.7 mmol) of (S)-68a, 0.9 mL (12 mmol) of triethylamine, and 1.68 mL (12 mmol) of methansulfonyl chloride gave 2.66 g (97%). 1H NMR: (300 MHz, CDCl3) δ 1.03 (t, J = 7.5 Hz, 3H), 1.78 (m, 2H), 2.53 (s, 3H), 2.91 (d, J = 6.6 Hz, 2H), 3.79 (s, 3H), 4.73 (p, J = 6.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)butan-2-yl methanesulfonate [(R)-69a]
Prepared from 1.89 g (10.5 mmol) of (R)-68a, 1.8 mL (13 mmol) of triethylamine, and 0.98 mL (13 mmol) of methanesulfonyl chloride gave 2.70 g (100%). 1H NMR: (300 MHz, CDCl3) δ 1.03 (t, J = 7.5 Hz, 3H), 1.78 (m, 2H), 2.53 (s, 3H), 2.91 (d, J = 6.6 Hz, 2H), 3.79 (s, 3H), 4.73 (p, J = 6.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(4-Methoxyphenyl)-3-methylbutan-2-yl methanesulfonate [(S)-69b]
Prepared from 3.93 g (20.2 mmol) of (S)-68b, 3.4 mL (24 mmol) of triethylamine, and 1.9 mL (24 mmol) of methanesulfonyl chloride gave 5.37 g (98%). 1H NMR: (300 MHz, CDCl3) δ 1.03 (m, 6H), 2.05 (m, 1H), 2.40 (s, 3H), 2.88 (m, 2H), 3.78 (s, 3H), 4.68 (m, 1H), 6.85 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)-3-methylbutan-2-yl methanesulfonate [(R)-69b]
Prepared from 6.07 g (31.2 mmol) of (R)-68b, 5.2 mL (37 mmol) of triethylamine, and 2.9 mL (37 mmol) of methanesulfonyl chloride gave 8.50 g (100%). 1H NMR: (300 MHz, CDCl3) δ 1.03 (m, 6H), 2.05 (m, 1H), 2.40 (s, 3H), 2.88 (m, 2H), 3.78 (s, 3H), 4.68 (m, 1H), 6.85 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(4-Methoxyphenyl)pentan-2-yl methanesulfonate [(S)-69c]
Prepared from 6.20 g (31.9 mmol) of (S)-68c, 5.3 mL (38 mmol) of triethylamine, and 3.0 mL (38 mmol) of methanesulfonyl chloride gave 7.66 g (88%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.2 Hz, 3H), 1.47 (m, 2H), 1.67 (m, 2H), 2.53 (s, 3H), 2.91 (d, J = 7.5 Hz, 2H), 3.79 (s, 3H), 4.79 (p, J = 6.0 Hz, 1H), 6.85 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)pentan-2-yl methanesulfonate [(R)-69c]
Prepared from 7.68 g (39.5 mmol) of (R)-68c, 6.6 mL (47 mmol) of triethylamine, and 3.7 mL (47 mmol) of methanesulfonyl chloride gave 10.76 g (100%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.2 Hz, 3H), 1.47 (m, 2H), 1.67 (m, 2H), 2.53 (s, 3H), 2.91 (d, J = 7.5 Hz, 2H), 3.79 (s, 3H), 4.79 (p, J = 6.0 Hz, 1H), 6.85 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
General Procedure for the Preparation of Azides (70a–c)
The mesylate was combined with an excess of sodium azide (NaN3) in dry DMF (conc = 0.1 M) and stirred at 35 °C for 36 h. Caution: All traces of dichloromethane should be carefully removed from the mesylate prior to treatment with sodium azide to prevent the formation of potentially explosive diazidomethane.14 The reaction was partitioned between Et2O and water, washed with brine, dried (MgSO4), filtered, and evaporated. The residues were purified on silica gel with a gradient of 60–100% DCM in hexanes.
(R)-1-(2-Azidobutyl)-4-methoxybenzene [(R)-70a]
Prepared from 2.66 g (10.3 mmol) of (S)-69a, 2.60 g (40 mmol) of NaN3. Chromatography on silica gel with hexanes/DCM gave 1.56 g (71%). 1H NMR: (300 MHz, CDCl3) δ 1.01 (t, J = 7.2 Hz, 3H), 1.55 (m, 2H), 2.75 (d, J = 7.5 Hz, 2H), 3.39 (p, J = 6.0 Hz, 1H), 3.80 (s, 3H), 6.85 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(2-Azidobutyl)-4-methoxybenzene [(S)-70a]
Prepared from 2.70 g (10.5 mmol) of (R)-69a and 3.07 g (47.2 mmol) of NaN3. Chromatography on silica gel with hexanes/DCM gave 1.84 g (84%). 1H NMR: (300 MHz, CDCl3) δ 1.01 (t, J = 7.2 Hz, 3H), 1.55 (m, 2H), 2.75 (d, J = 7.5 Hz, 2H), 3.39 (p, J = 6.0 Hz, 1H), 3.80 (s, 3H), 6.85 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(2-Azido-3-methylbutyl)-4-methoxybenzene [(R)-70b]
Prepared from 5.37 g (19.7 mmol) of (S)-69b and 1.95 g (30 mmol) of NaN3. Chromatography on silica gel with hexanes/DCM gave 3.63 g (84%). 1H NMR: (300 MHz, CDCl3) δ 1.01 (t, J = 6.6 Hz, 6H), 1.83 (m, 1H), 2.68 (m, 1H), 2.81 (m, 1H), 3.34 (m, 1H), 3.80 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(2-Azido-3-methylbutyl)-4-methoxybenzene [(S)-70b]
Prepared from 8.50 g (31.2 mmol) of (R)-69b and 4.06 g (62.5 mmol) of NaN3. Chromatography on silica gel with hexanes/DCM gave 5.94 g (87%). 1H NMR: (300 MHz, CDCl3) δ 1.01 (t, J = 6.6 Hz, 6H), 1.83 (m, 1H), 2.68 (m, 1H), 2.81 (m, 1H), 3.34 (m, 1H), 3.80 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(2-Azidopentyl)-4-methoxybenzene [(R)-70c]
Prepared from 7.66 (28.1 mmol) of (S)-69c and 4.15 g (63.8 mmol) of NaN3. Chromatography on silica gel with hexanes/DCM gave 5.46 g (81%). 1H NMR: (300 MHz, CDCl3) δ 0.93 (t, J = 6.6 Hz, 3H), 1.50 (m, 4H), 2.76, (d, J = 6.9 Hz, 2H), 3.47 (p, J = 6.6 Hz, 1H), 3.80 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(2-Azidopentyl)-4-methoxybenzene [(S)-70c]
Prepared from 10.79 (39.6 mmol) of (R)-69c and 5.14 g (79.0 mmol) of NaN3. Chromatography on silica gel with hexanes/DCM gave 7.07 g (81%). 1H NMR: (300 MHz, CDCl3) δ 0.93 (t, J = 6.6 Hz, 3H), 1.50 (m, 4H), 2.76, (d, J = 6.9 Hz, 2H), 3.47 (p, J = 6.6 Hz, 1H), 3.80 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H) ppm.
General Procedure for the Preparation of Primary Amines (71a–c)
The azide was dissolved in absolute EtOH (conc = 0.22M) and added was palladium on carbon. The mixture was hydrogenated with 1 atm of hydrogen gas for 12–48 h. The reaction was filtered through Celite and concentrated.
(R)-1-(4-Methoxyphenyl)butan-2-amine [(R)-71a]
Prepared from 1.56 g (7.60 mmol) of (R)-70a and 40 mg of 5% Pd/C (Sigma) giving 1.36 g (100%). 1H NMR: (300 MHz, CDCl3) δ 0.97 (t, J = 7.2 Hz, 3H), 1.36 (m, 1H), 1.51 (m, 1H), 1.65 (br s, 1H), 2.38 (m, 1H), 2.74 (m, 1H), 2.86 (m, 1H), 3.79 (s, 3H), 6.84 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(4-Methoxyphenyl)butan-2-amine [(S)-71a]
Prepared from 1.80 g (8.77 mmol) of (S)-70a and 360 mg of 10% Pd/C (Strem Co, 50% paste in water) giving 1.33 g (85%). 1H NMR: (300 MHz, CDCl3) δ 0.97 (t, J = 7.2 Hz, 3H), 1.36 (m, 1H), 1.51 (m, 1H), 1.65 (br s, 2H), 2.38 (m, 1H), 2.74 (m, 1H), 2.86 (m, 1H), 3.79 (s, 3H), 6.84 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)-3-methylbutan-2-amine [(R)-71b]
Prepared from 3.62 g (16.5 mmol) of (R)-70b and 50 mg of 5% Pd/C (Sigma) giving 3.13 g (98%). 1H NMR: (300 MHz, CDCl3) δ 0.97 (t, J = 6.6 Hz, 6H), 1.21 (br s, 2H), 1.65 (m, 1H), 2.34 (m, 1H), 2.76 (m, 2H), 3.79 (s, 3H), 6.84 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(4-Methoxyphenyl)-3-methylbutan-2-amine [(S)-71b]
Prepared from 5.94 g (27.1 mmol) of (S)-70b and 50 mg of 5% Pd/C (Sigma) giving 5.23 g (100%). 1H NMR: (300 MHz, CDCl3) δ 0.97 (t, J = 6.6 Hz, 6H), 1.21 (br s, 2H), 1.65 (m, 1H), 2.34 (m, 1H), 2.76 (m, 2H), 3.79 (s, 3H), 6.84 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H) ppm.
(R)-1-(4-Methoxyphenyl)pentan-2-amine [(R)-71c]
Prepared from 5.46 g (24.9 mmol) of (R)-70c and 50 mg of 5% Pd/C (Sigma) giving 4.81 g (100%). 1H NMR: (300 MHz, CDCl3) δ 0.93 (t, J = 6.6 Hz, 3H), 1.35 (m, 2H), 1.45 (m, 2H), 1.72 (br s, 2H), 2.42 (m, 1H), 2.75 (m, 1H), 2.95 (m, 1H), 3.79 (s, 3H), 6.84 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H) ppm.
(S)-1-(4-Methoxyphenyl)pentan-2-amine [(S)-71c]
Prepared from 7.07 g (32.2 mmol) of (S)-70c and 50 mg of 5% Pd/C (Sigma) giving 6.23 g (100 %). 1H NMR: (300 MHz, CDCl3) δ 0.93 (t, J = 6.6 Hz, 3H), 1.35 (m, 2H), 1.45 (m, 2H), 1.72 (br s, 2H), 2.42 (m, 1H), 2.75 (m, 1H), 2.95 (m, 1H), 3.79 (s, 3H), 6.84 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.4 Hz, 2H) ppm.
General Procedure for the Preparation of N-Benzylamines (72a–c)
A flask was charged with the primary amine (71a–c) in absolute EtOH (conc. 0.15M) and 1 equivalent of benzaldehyde. The solution was stirred at reflux under argon atm for 2 h monitoring by TLC. The reaction was cooled to 0 °C under argon and an excess of sodium triacetoxyborohydride (Na(OAc)3BH) was added in portions. After stirring under argon atm for 16 h the reaction mixture was concentrated, partitioned between DCM and 10% K2CO3 solution (5–6 eq), dried (K2CO3), filtered, and evaporated. The residue was either purified by column chromatography with 10–30% EtOAc in DCM or by recystallization from optically active mandelic acids. Chiral-HPLC Method A: Chirobiotic V column, 250 × 4.6 mm; 1.0 mL/min; Gradient program 20–40% MeOH over 40 min/20 mM KH2PO4; Det: 225 nm. Chiral-HPLC Method B: Chiralpak AD-H column, 250 × 4.6 mm; 0.25 mL/min; isocratic 7:93:0.5 iPrOH/hexane/DEA; Det: 279 nm.
(R)-N-Benzyl-1-(4-methoxyphenyl)butan-2-amine [(R)-72a]
Prepared from 1.36 g (7.60) mmol of (R)-71a, 806 mg (7.60 mmol) of benzaldehyde and 3.22 g (15.2 mmol) of Na(OAc)3BH. Chromatography on silica gel with DCM/EtOAc gave 1.69 g (82%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.5 Hz, 3H), 1.48 (m, 3H), 2.67 (m, 3H), 3.69 (d, J = 13.2 Hz, 1H), 3.79 (d, J = 13.2 Hz, 1H), 3.80 (s, 3H), 6.83 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.40–7.20 (m, 5H) ppm; MS m/z (rel): 270 (100, M+H); Chiral-HPLC Method A: Rt = 19.3 min (98.0% R).
(S)-N-Benzyl-1-(4-methoxyphenyl)butan-2-amine [(S)-72a]
Prepared from 1.04 g (5.80) mmol of (S)-71a, 613 mg (5.80 mmol) of benzaldehyde and 3.07 g (14.5 mmol) of Na(OAc)3BH. Chromatography on silica gel with DCM/EtOAc gave 400 mg (26%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.5 Hz, 3H), 1.48 (m, 3H), 2.67 (m, 3H), 3.69 (d, J = 13.2 Hz, 1H), 3.79 (d, J = 13.2 Hz, 1H), 3.80 (s, 3H), 6.83 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.40–7.20 (m, 5H) ppm; MS m/z (rel): 270 (100, M+H); Chiral-HPLC Method A: Rt = 17.5 min (99% S).
(R)-N-Benzyl-1-(4-methoxyphenyl)-3-methylbutan-2-amine [(R)-72b]
Prepared from 3.13 g (16.2) mmol of (R)-71b, 1.68 g (16.2 mmol) of benzaldehyde and 7.00 g (33.0 mmol) of Na(OAc)3BH. Chromatography on silica gel with DCM/EtOAc gave 3.24 g (72%). 1H NMR: (300 MHz, CDCl3) δ 1.00 (m, 6H), 1.20 (br s, 1H), 1.90 (m, 1H), 2.54 (m, 1H), 2.63 (m, 1H), 2.74 (m, 1H), 3.66 (d, J = 13.2 Hz, 1H), 3.76 (d, J = 13.2 Hz, 1H), 3.82 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 7.42–7.18 (m, 5H) ppm; MS m/z (rel): 284 (100, M+H); Chiral-HPLC Method A: Rt = 26.0 min (98.4% R).
(S)-N-Benzyl-1-(4-methoxyphenyl)-3-methylbutan-2-amine [(S)-72b]
Prepared from 5.23 g (27.1 mmol) of (S)-71b, 2.88 g (27.1 mmol) of benzaldehyde and 10.6 g (50.0 mmol) of Na(OAc)3BH. Chromatography on silica gel with DCM/EtOAc gave 6.47 g (84%). 1H NMR: (300 MHz, CDCl3) δ 1.00 (m, 6H), 1.20 (br s, 1H), 1.90 (m, 1H), 2.54 (m, 1H), 2.63 (m, 1H), 2.74 (m, 1H), 3.66 (d, J = 13.2 Hz, 1H), 3.76 (d, J = 13.2 Hz, 1H), 3.82 (s, 3H), 6.86 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 7.42–7.18 (m, 5H) ppm; MS m/z (rel): 284 (100, M+H); Chiral-HPLC Method A: Rt = 28.3 min (99% S).
(R)-N-Benzyl-1-(4-methoxyphenyl)pentan-2-amine [(R)-72c]
Prepared from 4.83 g (25.0 mmol) of (R)-71c, 2.66 g (25.0 mmol) of benzaldehyde and 10.4 g (49.0 mmol) of Na(OAc)3BH. Chromatography on silica gel with DCM/EtOAc gave 4.74 g (67%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.5 Hz, 3H), 1.42 (m, 4H), 1.52 (br s, 1H), 2.70 (m, 3H), 3.72 (d, J = 13.2 Hz, 1H), 3.78 (d, J = 13.2 Hz, 1H), 3.80 (s, 3H), 6.83 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.38–7.15 (m, 5H) ppm; MS m/z (rel): 284 (100, M+H); Chiral-HPLC Method B: Rt = 17.2 min (97.1% R).
(S)-N-Benzyl-1-(4-methoxyphenyl)pentan-2-amine [(S)-72c]
Prepared from 6.23 (32.2 mmol) of (S)-71c, 3.43 g (32.3 mmol) of benzaldehyde and 12.7 g (60.0 mmol) of Na(OAc)3BH. Chromatography on silica gel with DCM/EtOAc gave 6.84 g (79%). 1H NMR: (300 MHz, CDCl3) δ 0.94 (t, J = 7.5 Hz, 3H), 1.42 (m, 4H), 1.52 (br s, 1H), 2.70 (m, 3H), 3.72 (d, J = 13.2 Hz, 1H), 3.78 (d, J = 13.2 Hz, 1H), 3.80 (s, 3H), 6.83 (d, J = 8.4 Hz, 2H), 7.08 (d, J = 8.4 Hz, 2H), 7.38–7.15 (m, 5H) ppm; MS m/z (rel): 284 (100, M+H); Chiral-HPLC Method B: Rt = 16.4 min (95.8% S).
N-Benzylidene-2-(4-methoxyphenyl)ethanamine (82)
A 50 mL round bottom flask fitted with a Dean-Stark trap containing sodium sulfate in its side arm, was charged with 1.73 g (11.5 mmol) of 4-methoxyphenethylamine, 40 mL of toluene and 1.19 g (11.3 mmol) of benzaldehyde. The mixture was refluxed under argon atm for 48 h, cooled, and the toluene removed to give 2.63 g (97%) of an orange solid that was used in the next step without further purification. 1H NMR (300 MHz, CDCl3): δ 2.96 (t, 2H, J = 7.5 Hz), 3.81 (t, 2H, J = 7.5 Hz), 3.83 (s, 3H), 6.83 (d, 2H, J = 6.6 Hz), 7.16 (d, 2H, J = 6.6 Hz), 7.26–7.42 (m, 3H), 7.69–7.71 (m, 2H) ppm; 13CNMR (75 MHz, CDCl3): δ 36.6, 55.2, 63.4, 113.7, 128.0, 129.9, 130.5, 132.0, 136.2, 157.9, 161.4 ppm.
N-Benzyl-2-(4-methoxyphenyl)ethanamine (72d)
A 1.56 g (6.53 mmol) sample of 82 was dissolved in 25 mL of ethanol and degassed with argon. Added in portions was 3.45 g (16.3 mmol, 3 eq) mg Na(OAc)3BH, and the mixture was allowed to stir overnight. The solvent was removed, the residue partitioned between 30 mL of chloroform 180 mL of 5% K2CO3 solution, followed by 50 mL of 10% K2CO3 and 50 mL of water. The organic layer was dried (Na2SO4), evaporated and purified on silica gel (CHCl3-MeOH) giving 1.41 g (90%) of a pale yellow oil. 1H NMR (400 MHz, CDCl3): δ 2.75 (t, 2H, J = 7.2 Hz), 2.85 (t, 2H, J = 7.2 Hz), 3.76 (s, 3H), 3.78 (s, 2H) 6.81 (d, 2H, J = 8.4 Hz), 7.10 (d, 2H, J = 8.8 Hz), 7.21–7.32 (m, 5H) ppm; 13C NMR (75 MHz, CDCl3): δ 35.45, 50.77, 53.90, 52.25, 113.83, 126.87, 128.07, 128.37, 129.62, 132.08, 140.39, 158.03 ppm; MS m/z (ESI+): 242 (M+H).
General Procedure for the Preparation of N-Alkyl Modified Fenoterol Analogs (64–67)
(R)-2-(3,5-bis(benzyloxy)phenyl)oxirane (R)-73 was combined with (0.95–1 eq) of the appropriate chiral N-benzylamine 72a–d in toluene and then evaporated under high vacuum. The residue was heated at the temperature and time indicated in the specific procedure. The amber residue was applied to a column of silica gel (1:75) eluting with EtOAc/hexanes to give the pure 74a–d. Unless otherwise noted, the entire residue was deprotected by hydrogenating at 50 psi over 10% wt Pd/C (Strem Co 50% paste in water, 1:5 wt:wt). Filtered through Celite®, washed the cake well with EtOH and evaporated to a dry residue. The specific rotation of the free amine was taken in MeOH and then the base was immediately converted to the hemi-fumarate salt by heating with 0.5 eq of the diacid in MeOH (conc = 0.1M) to give the fenoterol salt (64–67).
(R,R′)-(−)-5-(1-Hydroxy-2-((1-(4-methoxyphenyl)butan-2-yl)amino)ethyl)benzene-1,3-diol [(R,R′)-64]
A mixture of 768 mg (2.31 mmol) of (R)-73 and 591 mg (2.19 mmol) of (R)-72a was heated at 120 °C for 30 h and further treated using the general procedure above to give 470 mg (55%) of hemi-fumarate salt. 1H NMR (400 MHz, CD3OD): δ 0.962 (t, 3H, J =7.6 Hz), 1.65 (p, 2H, J =5.6), 2.86–2.92 (m, 3H), 3.03–3.08 (m, 2H), 3.76 (s, 3H), 4.76 (dd, 1H J = 3.6, 8.8 Hz), 6.20 (t, 1H, J = 2.0 Hz), 6.33 (d, 2H, J= 2.0 Hz), 6.70 (s, 1H, fum), 6.87 (d, 2H, J = 8.8 Hz), 7.15 (d, 2H, J = 8.8 Hz) ppm. 13C CMR (75 MHz, CD3OD): δ 8.16, 22.48, 35.68, 51.40, 54.28, 60.69, 69.21, 101.86, 103.86, 113.92, 128.17, 129.91, 135.59, 143.53, 158.54, 158.88 ppm. UV (MeOH): λmax (ε) 277 nm (3,690), 223 (20,100), 210 (26,000), 202 (6,280). MS m/z (rel): 332 (100, M+H), 663 (17). RP-HPLC: Ace-5 C18 column, 100 × 4.6 mm; 1.0 mL/min; 10–50% (5 min) acetonitrile/0.1%TFA/water; 275 nm; Rt = 3.44 min (94.9%), 1.47 min (3.8% fumarate). Chiral-HPLC: Chirobiotic V column, 250 × 4.6 mm; 1.0 mL/min; 10–30% (40 min) MeOH/20 mM KH2PO4; 275 nm; Rt = 19.2 min (93.6%, R,R), Rt = 18.4 min (6.4%, R,S). [α]D = −31.1 (7.0%, free base MeOH). HRMS m/z calcd for [C19H25NO4]H+ 332.1856, found 332.1852.
(R,S′)-(+)-5-(1-Hydroxy-2-((1-(4-methoxyphenyl)butan-2-yl)amino)ethyl)benzene-1,3-diol [(R,S′)-64]
A mixture of 380 mg (1.15 mmol) of (R)-73 and 294 mg (1.1 mmol) of (S)-72a was heated at 120 °C for 24 h and further treated using the general procedure above to give 376 mg (88%) of hemi-fumarate salt. 1H NMR (400 MHz, CD3OD): δ 0.974 (t, 3H, J = 9.6 Hz), 1.67 (m, 2H), 2.87–2.96 (m, 3H), 3.12 (dd, 1H, J = 4.4, 16.4 Hz), 3.77 (s, 3H), 4.74 (dd, 1H J = 4.0, 12.8 Hz), 6.19 (t, 1H, J = 2.8 Hz), 6.31 (d, 2H, J = 2.8 Hz), 6.70 (s, 1H, fum), 6.89 (d, 2H, J = 11.6 Hz), 7.17 (d, 2H, J = 11.6 Hz) ppm. 13C NMR (75 MHz, CD3OD): δ 9.91, 24.68, 37.08, 52.85, 55.71, 62.20, 70.55, 103.24, 105.27, 115.41, 129.64, 131.34, 137.01, 144.93, 159.93, 160.34 ppm. UV (MeOH) λmax (ε): 277 nm (3,530), 223 (19,750), 204 (41,300). MS m/z (rel): 332 (100, M+H), 663 (17). RP-HPLC: Ace-5 C18 column, 100 × 4.6 mm; 1.0 mL/min; 10–50% (5 min) ACN/0.1% TFA/water; 275 nm; Rt = 3.47 min (94.3%), 1.46 min (4.04%, fumarate). Chiral-HPLC: Chirobiotic V column, 250 × 4.6 mm; 1.0 mL/min; 10–30% (40 min) MeOH/20 mM KH2PO4; 275 nm; Rt = 18.3 min (99%, R,S). [α]D = +1.43 (7.0%, free base in MeOH). HRMS m/z calcd for [C19H25NO4]H+ 332.1856, found 332.1853.
(R,R′)-5-(1-Hydroxy-2-((1-(4-methoxyphenyl)-3-methylbutan-2-yl)amino)ethyl)-benzene-1,3-diol [(R,R′)-65]
A mixture of 579 mg (1.74 mmol) of (R)-73 and 494 mg (1.74 mmol) of (R)-72b was heated at 145 °C for 48 h and further treated using the general procedure above to give 401 mg (57%) of hemi-fumarate salt. 1H NMR (400 MHz, CD3OD): δ 1.05 (d, 6H, J = 6.9 Hz), 2.08 (hept, 1H, J = 3.7 Hz), 2.76–2.84 (m, 2H), 2.91–3.00 (m, 2H), 3.25 (m, 1H), 3.77 (s, 3H), 4.64 (dd, 1H, J = 3.3, 10.0 Hz), 6.20 (d, 2H, J = 2.2 Hz), 6.67 (s, 1H, fumarate), 6.90 (d, 2H, J = 8.6 Hz), 7.19 (d, 2H, J = 8.6 Hz) ppm. 13C NMR (75 MHz, CD3OD): δ 17.68, 18.54, 30.68, 34.42, 54.00, 55.74, 66.03, 69.90, 103.17, 105.26, 115.54, 130.38, 131.20, 136.91, 144.86, 159.85, 160.34, 173.70 ppm. UV (MeOH) λmax (ε): 278 nm (3,490), 223 (18,800), 206 (22,800). MS M/z (rel): 346 (M+H base). RP-HPLC: Ace-5 C18 column, 100 × 4.6 mm; 10–60%/10 min ACN/20 mM KH2PO4 (pH 4.5); 1.0 mL/min; 275 nm; Rt = 1.15 min (5.82% fumarate), 6.44 min (92.8%), purity 98.6%. Chiral-HPLC: Chiral AGP column, 100 × 4.0 mm; 0.70 mL/min; 8% acetonitrile/50 mM ammonium acetate (pH 7); 276 nm; Rt = 9.1 min (98.4% R,R). [α]D = +3.10 (1.0% free base in MeOH); HRMS m/z calcd for [C20H27NO4]H+ 346.2013, found 346.2006.
(R,S′)-(−)-5-(1-hydroxy-2-((1-(4-methoxyphenyl)-3-methylbutan-2-yl)amino)-ethyl)benzene-1,3-diol [(R,S′)-65]
A mixture of 644 mg (1.94 mmol) of (R)-73 and 550 mg (1.94 mmol) of (S)-72b was heated at 145 °C for 48 h and further treated using the general procedure above to give 478 mg (61%) of hemi-fumarate salt. 1H NMR (400 MHz, CD3OD): δ 1.04 (t, 6H, J = 6.8 Hz), 2.07 (hept, 1H, J = 4.0 Hz), 2.77–2.82 (m, 2H), 2.88–2.94 (m, 2H), 3.24 (m, 1H), 3.77 (s, 3H), 4.68 (dd, 1H, J = 5.6, 7.6 Hz), 6.18 (t, 1H, J = 2.4 Hz), 6.26 (d, 2H, J = 2.0 Hz), 6.69 (s, 1H, fumarate), 6.87 (d, 2H, J = 8.8 Hz), 7.15 (d, 2H, J = 8.8 Hz) ppm. 13C NMR (75 MHz, CD3OD): δ 17.62, 18.59, 30.19, 34.45, 54.44, 55.70, 66.95, 70.89, 103.17, 105.22, 115.39, 130.28, 131.17, 136.85, 145.02, 159.91, 160.24 ppm. UV (MeOH) λmax (ε): 278 nm (3,240), 224 (17,000), 205 (33,600). MS M/z (rel): 346 (M+H base). RP-HPLC: Ace-5 C18 column, 100 × 4.6 mm; 10–60%/10 min acetonitrile/20 mM KH2PO4 (pH 4.5); 1.0 mL/min; 275 nm; Rt = 1.15 min (6.33% fumarate), 6.49 min (92.1%), purity = 98.4%. Chiral-HPLC: Chiral AGP column, 100 × 4.0 mm; 0.70 mL/min; 8% ACN/50 mM ammonium acetate (pH 7), 276 nm; Rt = 7.5 min (98.8% R,S). [α]D= −30.6 (1.0% free base in MeOH). HRMS m/z calcd for [C20H27NO4]H+ 346.2013, found 346.2008.
(R,R′)-5-(1-Hydroxy-2-((1-(4-methoxyphenyl)pentan-2-yl)amino)ethyl)benzene-1,3-diol [(R,R′)-66]
A mixture of 550 mg (1.65 mmol) of (R)-73 and 468 mg (1.65 mmol) of (R)-72c was heated at 135 °C for 29 h and further treated using the general procedure above to give 304 mg (46%) of hemi-fumarate salt. 1H NMR (400 MHz, CD3OD): δ 0.899 (t, 3H, J = 7.2 Hz), 1.26–1.46 (m, 2H), 1.57–1.65 (m, 2H), 2.80–2.98 (m, 2H), 3.02–3.15 (m, 2H), 3.35–3.36 (m, 1H), 4.73 (dd, 1H, J = 4.0, 8.8 Hz), 6.21 (t, 1H, J = 2.0 Hz), 6.32 (d, 2H, J = 2.4 Hz), 6.68 (s, 1H, fum), 6.89 (d, 2H, J = 8.8 Hz), 7.15 (d, 2H, J = 8.8 Hz) ppm. 13C NMR (75 MHz, CD3OD): δ 14.12, 19.42, 33.24, 37.46, 52.50, 55.68, 60.90, 70.36, 103.29, 105.21, 115.37, 129.20, 131.32, 136.46, 144.71, 160.00, 160.41, 172.21 ppm. UV (MeOH) λmax (ε): 277 nm (3,280), 224 (17,800), 203 (40,500). MS M/z (rel): 346 (100, M+H base). RP-HPLC: Ace-5 C18 column, 100 × 4.6 mm; 10–60%/10 min ACN/20 mM KH2PO4 (pH 4.5); 1.0 mL/min; 275 nm; Rt = 6.79 min (98.8%). Chiral-HPLC: Chirobiotic V column, 250 × 4.6 mm; 1.0 mL/min; 20–40% (40 min) MeOH/20 mM KH2PO4; 275 nm; Rt = 20.0 min (96.7% R,R). [α]D= −32.1 (1.0% MeOH, free base). HRMS m/z calcd for [C20H27NO4]H+ 346.2013, found 346.2009.
(R,S′)-5-(1-Hydroxy-2-((1-(4-methoxyphenyl)pentan-2-yl)amino)ethyl)benzene-1,3-diol [(R,S′)-66]
A mixture of 587 mg (1.77 mmol) of (R)-73 and 500 mg (1.77 mmol) of (S)-72c was heated at 135 °C for 29 h and treated using the general procedure above to give 352 mg (49%) of hemi-fumarate salt. 1H NMR (400 MHz, CD3OD): δ 0.904 (t, 3H, J = 7.2 Hz), 1.35–1.45 (m, 2H), 1.54–1.58 (m, 2H), 2.83–2.92 (m, 2+1H), 3.03–3.07 (m, 1H), 3.22 (m, 1H), 3.77 (s, 3H), 6.67 (dd, 1H, J = 3.2, 9.2 Hz), 6.19 (t, 1H, J = 2.4), 6.29 (d, 2H, J = 2.0 Hz), 6.68 (s, 1H, fum), 6.88 (d, 2H, J = 8.8 Hz), 7.15 (d, 2H, J = 8.8 Hz) ppm. 13C NMR (75 MHz, CD3OD): δ 14.22, 19.71, 34.36, 37.78, 52.98, 55.71, 60.77, 70.72, 103.21, 105.27, 115.40, 129.75, 131.34, 136.91, 145.02, 159.95, 160.34 ppm. UV (MeOH) λmax (ε): 277 nm (3,510), 224 (18,900), 203 (38,900). MS m/z (rel): 346 (100, M+H base). RP-HPLC: Ace-5 C18 column, 100 × 4.6 mm; 10–60%/10 min ACN/20 mM KH2PO4 (pH 4.5); 1.0 mL/min; 275 nm; Rt = 6.82 min (97.5%). Chiral-HPLC: Chirobiotic V column, 250 × 4.6 mm; 1.0 mL/min; 20–40% (40 min) MeOH/20 mM KH2PO4; 275 nm; Rt = 18.0 min (98.0% R,S). [α]D= −1.20 (1.0% MeOH, free base). HRMS m/z calcd for [C20H27NO4]H+ 346.2013, found 346.2009.
(R)-(−)-5-(1-Hydroxy-2-((4-methoxyphenethyl)amino)ethyl)benzene-1,3-diol [(R)-67]
A mixture of 642 mg (1.93 mmol) of (R)-73 and 443 mg (1.83 mmol) of 72d was heated at 135 °C for 30 h and further treated using the general procedure above to give 350 mg (53% overall) of the hemi-fumarate salt (R)-67. 1H NMR (400 MHz, CD3OD): δ 2.88–2.95 (m, 2H), 3.02 (dd, 1H, J = 9.6, 12.4 Hz), 3.09–3.18 (m, 3H), 3.76 (s, 3H), 4.76 (dd, 1H, J = 3.2, 9.2 Hz), 6.20 (t, 1H J = 2.0 Hz), 6.33 (d, 2H, J = 2.0 Hz), 6.68 (s, 1H, fum), 6.87 (d, 2H, J = 8.4 Hz), 7.16 (d, 2H, J = 8.4 Hz) ppm. 13C NMR (75 MHz CD3OD): δ 32.43, 50.27, 55.29, 55.68, 70.26, 103.27, 105.27,115.30, 129.85, 130.79, 137.04, 144.81, 159.94, 160.26, 174.13 ppm. UV (MeOH) λmax (ε): 277 nm (3,040), 222 (17,180), 208 (24,800). MS M/z (rel): 304 (100, M+H base). RP-HPLC: Eclipse XDB-C18, 150×4.6 mm; 10–50%/5min ACN/0.1%TFA-water, 1.0 mL/min, 275 nm, Rt = 5.11 min (95.66%), 2.16 min (2.80%, fumarate). Chiral-HPLC: 100×4.0 mm; 95/5 50 mM NH4OAc (pH 7); 0.70 mL/min; 275 nm; Rt = 15.14 min (96.8% R), 13.91 min (3.2% S); [α]D= −14.42 (0.95% free base in MeOH). HRMS: m/z calcd for [C17H21NO4]H+ 304.1543, found 304.1540.
(S)-5-(1-Hydroxy-2-((4-methoxyphenethyl)amino)ethyl)benzene-1,3-diol [(S)-67]
A mixture of 768 mg (2.38 mmol) of (S)-73 and 546 mg (2.26 mmol) of 72d was heated at 135 °C for 30 h and further treated using the general procedure above to give 421 mg (52% overall) of the hemi-fumarate salt (S)-67. 1H NMR (400 MHz, CD3OD): δ 2.89–2.95 (m, 2H), 3.02 (dd, 1H, J = 10.0, 12.8 Hz), 3.76 (s, 3H), 3.14–3.19 (m, 3H), 4.77 (dd, 1H, J = 3.6, 9.6 Hz), 6.19 (t, 1H J = 2.4 Hz), 6.34 (d, 2H, J = 2.4 Hz), 6.83 (s, 1H, fum), 6.87 (d, 2H, J = 8.4 Hz), δ 7.16 (d, 2H, J = 8.8 Hz) ppm. 13C NMR (75 MHz, CD3OD): δ 32.63, 50.38, 55.44, 55.71, 70.45, 103.26, 105.28, 115.33, 130.00, 130.77, 136.95, 144.91, 159.97, 160.30, 173.87 ppm. UV (MeOH) λmax (ε): 277 nm (2,850), 224 (14,500), 207 (21,900). MS m/z (rel): 304 (100, M+H base). RP-HPLC: Eclipse XDB-C18, 150×4.6 mm; 10–50%/5min ACN/0.1%TFA-water, 1.0 mL/min, 275 nm, Rt = 5.11 min (95.38%), 2.16 min (2.79%, fumarate). Chiral-HPLC: 100×4.0 mm; 95/5 50 mM NH4OAc (pH 7); 0.70 mLmin; 275 nm; Rt = 13.2 (96.1% S), 16.2 min (3.9% R). [α]D= +13.8 (1.0% free base in MeOH). HRMS: m/z calcd for [C17H21NO4]H+ 304.1543, found 304.1539.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health National Institute on Aging [Contract N01-AG31009]; and by the Foundation for Polish Science (TEAM Programme). The article was developed using the equipment purchased within the Project “The equipment of innovative laboratories doing research on new medicines used in the therapy of civilization and neoplastic diseases” within the Operational Program Development of Eastern Poland 2007–2013, Priority Axis I Modern Economy, Operations I.3 Innovation Promotion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Jozwiak K, Khalid C, Tanga MJ, Berzetei-Gurske I, Jimenez L, Kozocas JA, Woo A, Zhu W, Xiao RP, Abernethy DR, Wainer IW. J Med Chem. 2007;50:2903–2915. doi: 10.1021/jm070030d. [DOI] [PubMed] [Google Scholar]
- 2.Woo AYH, Wang TB, Zeng X, Weizgog Z, Abernethy DR, Wainer IW, Xiao RP. Mol Pharmacol. 2009;75:158–165. doi: 10.1124/mol.108.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jozwiak K, Woo AY, Tanga MJ, Toll L, Jimenez L, Kozocas JA, Plazinska A, Xiao RP, Wainer IW. Bioorg Med Chem. 2010;18:728–736. doi: 10.1016/j.bmc.2009.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toll L, Pajak K, Plazinska A, Jozwiak K, Jimenez L, Kozocas JA, Tanga MJ, Bupp JE, Wainer IW. Mol Pharmacol. 2012;81:846–854. doi: 10.1124/mol.111.077347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toll L, Jimenez L, Waleh N, Jozwiak K, Woo AY, Xiao RP, Bernier M, Wainer IW. J Pharmacol Exp Ther. 2011;336:524–532. doi: 10.1124/jpet.110.173971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim TH, Chung KY, Manglik A, Hansen AL, Dror RO, Mildorf TJ, Shaw DE, Kobilka BK, Prosser RS. J Am Chem Soc. 2013;135:9465–9474. doi: 10.1021/ja404305k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Siluk D, Mager DE, Kim HS, Wang Y, Furimsky AM, Ta A, Iyer LV, Green CE, Wainer IW. Xenobiotica. 2010;40:195–206. doi: 10.3109/00498250903434533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 10.HyperChem(TM) 6.03. Hypercube, Inc; 1115 NW 4th Street, Gainesville, Florida 32601, USA: [Google Scholar]
- 11.Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 12.Thomsen R, Christensen MH. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- 13.Lamb PB, McElhinney CJ, Sninski T, Purdom H, Carroll FI, Lewin AH. J Label Compd Radiopharm. 2009;52:457–462. [Google Scholar]
- 14.Conrow RE, Dean WD. Org Process Res Dev. 2008;12:1285–1286. [Google Scholar]
- 15.Koppenhoefer B, Schurig V. Org Synth. 1993;Coll 8:119. [Google Scholar]
- 16.Birk C, Jurgen VC. Tetrahedron. 1996;52:12745–12760. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.