

Abstract
Hard metal lung disease (HMLD) is an occupational lung disease specific to inhalation of cobalt-containing particles whose mechanism is largely unknown. Cobalt is a known hypoxia mimic and stabilizer of the alpha subunits of hypoxia-inducible factors (HIFs). Previous work revealed that though HIF1α contrib utes to cobalt toxicity in vitro, loss of HIF1α in the alveolar epithelial cells does not provide in vivo protection from cobalt-induced lung inflammation. HIF1α and HIF2α show unique tissue expression profiles, and HIF2α is known to be the predominant HIF mRNA isoform in the adult lung. Thus, if HIF2α activation by cobalt contributes to pathophysiology of HMLD, we hypothesized that loss of HIF2α in lung epithelium would provide protection from cobalt-induced inflammation. Mice with HIF2α-deficiency in Club and alveolar type II epithelial cells (ATIIs) (HIF2αΔ/Δ) were exposed to cobalt (60 µg/day) or saline using a subacute occupational exposure model. Bronchoalveolar lavage cellularity, cytokines, qRT-PCR, and histopathology were analyzed. Results show that loss of HIF2α leads to enhanced eosinophilic inflammation and increased goblet cell metaplasia. Additionally, control mice demonstrated a mild recovery from cobalt-induced lung injury compared with HIF2αΔ/Δ mice, suggesting a role for epithelial HIF2α in repair mechanisms. The expression of important cytokines, such as interleukin (IL)-5 and IL-10, displayed significant differences following cobalt exposure when HIF2αΔ/Δ and control mice were compared. In summary, our data suggest that although loss of HIF2α does not afford protection from cobalt-induced lung inflammation, epithelial HIF2α signaling does play an important role in modulating the inflammatory and repair response in the lung.
Key Words: hypoxia-inducible factor 2, doxycycline inducible knockout, lung epithelium, cobalt, inflammation.
Epithelial cells are required to play several vital roles in order to maintain proper homeostasis, including transport of nutrients and waste, responding to varying environmental stresses and protection from pathogens by interacting with resident immune cells, and participating in complex cytokine signaling. In addition to initiating innate immune responses, epithelial cells have been increasingly recognized as key players in directing adaptive immunity and regulating inflammatory processes in the lung (Kato et al., 2007). For lung diseases, such as asthma and chronic obstructive pulmonary disease (COPD), epithelial inflammation dominates the pathophysiology; accordingly, research efforts are focused on understanding the cell-specific signaling responsible for these inflammatory disease states and developing more targeted therapies to circumvent them.
Concomitant to inflammatory events is localized hypoxia or decreased availability of oxygen to nearby cells and tissues. Hypoxia has important metabolic consequences that require all cells, especially those experiencing inflammation, to adapt (Nizet et al., 2009). The hypoxic response on the cellular level is largely mediated by the hypoxia-inducible factors (HIFs), members of the Per-Arnt-Sim (PAS) superfamily of transcriptional regulators (Greer et al., 2012; Patel et al., 2008). HIFs are heterodimeric transcription factors, consisting of a cytosolic alpha subunit (HIF1α, 2α, or 3α) that is oxygen sensitive, and a ubiquitously expressed nuclear beta subunit, the aryl hydrocarbon receptor nuclear translocator (ARNT, also known as HIF1β) and ARNT2. HIF activity is regulated primarily through protein stability by the action of prolyl hydroxylase domain (PHD) proteins, which use available O2 to modify key residues of HIFα (Epstein et al., 2001). These hydroxylated proline residues target HIFα for ubiquitination by the Von-Hippel Lindau (VHL) protein, leading to proteasomal degradation during normoxic conditions (del Peso et al., 2003). In contrast, under hypoxic conditions, oxygen deficiency leads to PHD inactivity, and the HIFα subunit stabilizes, translocates to the nucleus to pair with ARNT, and coordinates transcription of hypoxia-regulated genes (for review, see Kaluz et al., 2008).
Cobalt is a transition metal with many industrial uses. Prosthetic joints and sintered carbides (also known as hard metal) utilize cobalt in their alloys which imbues superior wear resistance and strength at high temperatures. They also represent key potential toxic exposure routes for humans (Korovessis et al., 2006; Milosev et al., 2006; Nordberg, 2007; Simonsen et al. ). The most widely known cobalt-specific pathology is hard metal lung disease (HMLD) or “cobalt lung.” HMLD is caused by inhalation of cobalt-containing dust particles from hard metal grinding and drilling (Lison, 1996). Furthermore, cobalt is also a hypoxia mimic and has been used therapeutically to treat anemic patients (Fisher, 1998). Subsequent research has shown that cobalt has the ability to inhibit PHD activity, stabilize HIFs, and induce transcription of HIF target genes (Epstein et al., 2001; Maxwell et al., 2004; Vengellur et al., 2003).
Previous work in our laboratory showed that HIF1α−/− mouse embryonic fibroblasts were partially resistant to cobalt toxicity (Vengellur et al., 2004). Considering the evidence that HIF1α can contribute to cobalt toxicity, we predicted that loss of HIF1α in a lung-specific, inducible deficiency mouse model might provide protection from cobalt-induced lung inflammation. Contrary to our expectation, loss of HIF1α increased overall cellularity and significantly altered the inflammatory profile from neutrophilic to eosinophilic (Saini et al., 2010a). These results suggested that HIF1α has an important role in regulating the lung epithelium’s response to cobalt-induced inflammation.
Of the 3 HIFα subunits, HIF1α is the most ubiquitously expressed and, to date, most studied. Though HIF2α shares significant sequence homology to HIF1α, the 2 transcription factors regulate the expression of a battery of genes that only slightly overlap. In addition, the 2 HIFα subunits’ tissue-specific expression varies considerably (Loboda et al., 2012; Patel et al., 2008). In the lung, HIF2α appears to be the dominant hypoxia-induced mRNA isoform, seen in both epithelial cells and widely throughout the parenchyma (Rajatapiti et al., 2008; Wiesener et al., 2003). Considering these features, it was hypothesized that loss of HIF2α in lung epithelium would protect against cobalt-induced lung inflammation, and given the temporospatial differences in gene expression of HIF isoforms, this impact would be different than the role exhibited by epithelial HIF1α. To investigate the role of epithelial HIF2α loss on acute and subacute cobalt-induced lung inflammation, we utilized HIF2α-deficient mice which employ the SP-C-driven inducible knockout system.
MATERIALS AND METHODS
Animals.
These studies utilize the SP-C-rtTA/(tetO)7-CMV-Cre mouse model, a doxycycline (DOX)–inducible, lung epithelial–specific Cre-recombinase system, which was a kind gift from Dr Jeffrey Whitsett, Cincinnati Children’s Hospital Medical Center. Mating between these mice and HIF2αflox/flox (a kind gift from Dr M. Celeste Simon, University of Pennsylvania) resulted in the SP-C-rtTA+/tg/(tetO)7-CMV-Cre+/tg/HIF2αflox/flox (HIF2αfl/fl) mice. Recombination in the floxed HIF2α gene, specifically in the respiratory epithelium, occurs when mice are exposed to DOX (Perl et al., 2002). Throughout this article, this DOX-treated strain will be referred to as HIF2α∆/∆.
Genotyping of the mice was performed by PCR for the 2 SP-C/Cre loci as described previously (Saini et al., 2008). Genotyping primers for the HIF2α locus were as follows: 5′-CAG GCA GTA TGC CTG GCT AAT TCC AGT T-3′ (forward) and 5′-CTT CTT CCA TCA TCT GGG ATC TGG GAC T-3′ (reverse) generating a 410bp and 440 bp amplicon in WT and floxed loci, respectively. All of the mice used in this study were males and were maintained in a mixed C57BL/6 and FVB/N background. All the procedures regarding the handling, maintenance, exposure, and necropsy protocols of the mice used in this study were approved by the university laboratory animal resources (ULAR) regulatory unit at Michigan State University.
DOX treatment regimen.
DOX-induced recombination was performed using the same paradigm that was successfully used to remove HIF1α postnatally (Saini et al., 2010b). Briefly, lactating dams were exposed to DOX-containing feed (625mg DOX/kg; Harlan Teklad, Madison, Wisconsin) and drinking water (0.8mg/ml; MP Biochemicals, Solon, Ohio) ad libitum beginning on postnatal day 4 (P4) until weaning (~P21). After weaning, mice were maintained on the same DOX-containing food and water ad libitum until P30. The dose of DOX used was slightly lower than the concentration that has been used to induce recombination without any observable toxicity or impact on alveolarization (Whitsett et al., 2006). Animals used as controls in this study were triple transgenic (HIF2αfl/fl), which were given normal food and water ad libitum.
Cobalt exposure, tissue harvesting, and processing.
HIF2αΔ/Δ mice and their respective floxed controls were randomly assigned to receive either cobalt or saline vehicle. Prior to each daily aspiration starting at P42, mice were anesthetized with isoflurane using the drop jar method. Oropharyngeal aspirations of 25 μl of either 10mM cobalt chloride or sterile saline were given using an occupational exposure as follows: once daily for 5 consecutive days, followed by 2 days of no treatment, then another 5 consecutive days (Fig. 1). This dose of cobalt corresponds to a daily dose of 60 μg CoCl2 per mouse, a dose known to reliably reproduce substantial inflammation (Saini et al., 2010a, b). Mice were sacrificed at day 2 (1 dose of cobalt), 6 (5 doses of cobalt), and 15 (10 doses of cobalt) beginning with an IP injection of 1ml of a 40mg/ml avertin (2,2,2-tribromoethanol, Sigma-Aldrich, St Louis, Missouri). A midline laparotomy was performed, and mice were exsanguinated by transecting the renal artery. The lungs were exposed and trachea cannulated. Heart and lungs were removed en bloc, sterile saline was used in 2 successive 1ml lavages to make bronchoalveolar lavage (BAL), and the 2 fractions were pooled. Total cells were counted using a hemacytometer, and differential cell counts were made from CytoSpin samples of BAL stained with DiffQuick reagent (Baxter, Florida). Following lavage, right lung lobes were snap-frozen in liquid nitrogen for later RNA and protein analysis, and left lung lobes were gravity-inflation fixed at 30cm pressure with 10% phosphate-buffered formalin for histopathological and immunohistochemistry (IHC) analyses. A total of 5–8 mice were used for each treatment group in this study.
FIG. 1.
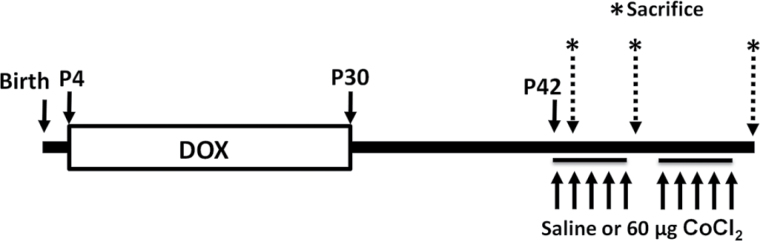
Experimental timeline and dosing scheme. Lactating dams were randomized to receive either DOX food and water (HIF2αΔ/Δ) or regular food and water (CTL) ad libitum starting at P4. Weaning occurred at P21, and mice were continued on their respective diets until P30. After a minimum of 10 days free of DOX food and water, animals began an occupational exposure of a once daily dose of either 60 μg cobalt or saline vehicle via oropharyngeal aspiration. Sacrifice of mice was made 24h after the final dose for the 1- and 5-dose groups, and 72h after the final dose for the 10-dose group (* and dashed arrows), corresponding to day 2 (1 dose), day 6 (5 doses), and day 15 (10 doses given as 5 once daily doses, 2 day break and 5 once daily doses). Abbreviations: CTL, control; DOX, doxycycline; HIF, hypoxia-inducible factor.
Histopathology and IHC.
Histopathology was assessed by using formalin-fixed left lung lobes of all samples from each treatment group. Left lobes were cut at the 5th and 11th generation (G5/11), paraffin embedded, cut into 5 μm sections, mounted on glass slides, and stained with hematoxylin and eosin (H&E) or alcian blue (pH 2.5) periodic acid Schiff (AB-PAS) to detect mucosubstances. Immunostaining was performed for major basic protein (MBP; polyclonal rabbit anti-mouse MBP, 1:500, Mayo Clinic, Scottsdale, Arizona, a kind gift from Dr James Lee) as described previously (Saini et al., 2010a). Immunostaining for HIF2α (polyclonal rabbit anti-HIF2α, 1:100, Novus Biologicals NB100-122, Littleton, Colorado) was performed using the Vectastain Elite ABC Kit (Rabbit IgG) as described previously (Saini et al., 2008). A histopathologic index was utilized to score the severity of lung inflammation in sections stained with H&E. Slides were blinded, and both the bronchial and alveolar regions of the G5 section were scored independently by 2 separate investigators (SPP and JJL) using a 6-point inflammation scale from 0 (no inflammation) to 4 (marked inflammation). Specifically, the scale included 0 (normal, no inflammation), 0.5 (sparse perivascular infiltrates only), 1 (minimal infiltrates, no lesions), 2 (mild infiltrates, few small lesions), 3 (moderate infiltrates, sizable lesions), and 4 (marked infiltrates, large lesions).
Cytokine quantitation by bead array.
Lung lysates (LL) were generated from snap-frozen right lung lobes in RIPA buffer using a Retsch MM200 bead beater system (Retsch, Haan, Germany). RIPA buffer contained protease inhibitors aprotinin, leupeptin, and pepstatin (1 μg/ml each, Sigma-Aldrich), cOmplete Mini Protease Inhibitor Cocktail (Roche) and phenylmethylsulfonyl fluoride (PMSF, 1mM), phosphatase inhibitors sodium orthovanadate (Na3VO4, 1mM) and sodium fluoride (NaF, 1mM), and EDTA (1mM). Supernatants were cleared by centrifugation at 10 000 x g for 15min at 4°C and quantitated for protein content using the Bradford Method (Bio-Rad, Hercules, California) (Bradford, 1976). Fifty microliters of each sample supernatant was used for maximum detection of cytokines using CBA Flex Sets and analyzed on a FACSCalibur flow cytometer according to manufacturer’s protocols (BD Biosciences, San Jose, California). Cytokine content was then normalized to total protein in each sample.
RNA isolation and quantitative real-time PCR analysis.
Snap-frozen lung tissue was homogenized in TRIzol reagent (Life Technologies, Carlsbad, California) using the bead beater system described above. Total RNA was quantitated spectrophotometrically (Nano-Drop ND-1000 UV-Vis Spectrophotometer). Total RNA (1 μg) was reverse transcribed using Superscript III (Life Technologies), and quantitative real-time PCR reactions were performed using gene-specific primers (listed in Supplementary Table 1) and Power SYBR Green PCR Master Mix on an ABI PRISM 7000 (Life Technologies). Gene expression was measured using the standard curve method with hypoxanthine guanine phosphoribosyl transferase (Hprt) as the housekeeping gene. Expression was normalized to both Hprt and the control saline-treated group.
Quantitative analysis.
All cell count, gene expression, and cytokine data were analyzed by 1-way ANOVA using Fisher’s LSD method (α = 0.05). All error bars are presented as SEM. Any data with p value < .05 were considered significant.
RESULTS
Postnatal Deletion of HIF2α in Lung Epithelial Cells
We have previously reported postnatal deletion of HIF1α for HIF1αfl/fl mice using the SP-C-rtTA/(TetO)7-Cre model (Saini et al., 2010b). To determine whether a similar deletion profile exists for the DOX-treated HIF2αfl/fl mice with this system, we performed IHC for HIF2α on left lobe sections at G5/11. Figure 2 shows representative images of HIF2α IHC from the HIF2αfl/fl mice. In the control (CTL) mice (given regular food and water), HIF2α staining is visualized in bronchial airway (BA) epithelial cells (arrow, Fig. 2A) and alveolar epithelial cells (arrows, Fig. 2B). In contrast, HIF2α∆/∆ mice (given DOX food and water) had marked decreased staining compared with controls in both the BA epithelial cells (arrow, Fig. 2C) and alveolar epithelial cells (arrow, Fig. 2D). This demonstrates that 26 days of DOX during the postnatal period is adequate for recombination using this system.
FIG. 2.

HIF2α IHC for CTL and P4–30 DOX-treated mice. HIF2α (NB100-122 Rabbit pIgG, 1:100) with Vectastain ABC Rabbit Kit, DAB coloring agent, and light hematoxylin counterstain was performed on control (A and B) and DOX-treated (C and D) mice. A and C show the BA, and Club cells indicated with solid arrows. B and D show the parenchymal compartment. Abbreviations: BA, bronchial airway; CTL, control; DOX, doxycycline; HIF, hypoxia-inducible factor; IHC, immunohistochemistry.
BAL Cellularity
BAL fluid was taken, total cells were counted, and differentials were obtained from control and HIF2αΔ/Δ mice after 1, 5, and 10 doses of cobalt (Fig. 1). The number of total cells significantly increased following 5 doses of cobalt treatment in CTL and HIF2αΔ/Δ mice. In contrast, after 10 doses, the number of total cells of CTL mice decreased compared with the 5-dose group, whereas the total cells remained elevated in in the HIF2αΔ/Δ mice (Fig. 3A). The number of macrophages increased after 5 doses in the cobalt-treated groups, and after 10 doses, the number of macrophages decreased compared with the 5-dose group. In all dose groups, CTL and HIF2αΔ/Δ mice showed a similar number of macrophages (Fig. 3B). Interestingly, eosinophils were elevated after 5 doses in both groups; however, only the CTL mice reached significance. After 10 doses, however, the number of eosinophils went up significantly in the HIF2αΔ/Δ mice and had returned to basal levels in the CTL mice (Fig. 3C). Neutrophil infiltration was evident following a single dose of cobalt and reached significance after 5 doses in the CTL mice. In contrast, neutrophil infiltration was not a significant portion of cellular infiltrate in the HIF2αΔ/Δ mice. After 10 doses, the number of neutrophils had returned to basal levels in the CTL mice (Fig. 3D). Finally, the numbers of lymphocytes increased after 5 doses in cobalt-treated CTL and HIF2αΔ/Δ mice, however, even at this increased level, they represented approximately 1% of the total number of cells (Fig. 3E).
FIG. 3.

BAL cellularity. Mice were given 1, 5, and 10 doses of 60 μg CoCl2 or sterile saline vehicle. BAL was taken and total cells are in (A), with differentials in (B–E). Open bars correspond to saline-treated CTL mice, diagonal hatched bars correspond to saline-treated HIF2αΔ/Δ mice, black bars indicate cobalt-treated CTL mice, and horizontal hatched bars indicate cobalt-treated HIF2αΔ/Δ mice. *, significant from saline-treated control; brackets, significant across CTL/Δ/Δ groups or same group on different number of doses, 1-way ANOVA with Fisher’s LSD, α = 0.05. Abbreviations: BAL, bronchoalveolar lavage; CTL, control; HIF, hypoxia-inducible factor.
Histopathological Analysis
Representative images from H&E stained lungs are shown in Figure 4, and histopathology scores are depicted in Figure 5. Most of the pathology was observed in the proximal airways (G5) central acinar region, with large and terminal bronchioles affected most by cobalt aspiration. Saline-treated mice had minimal pulmonary histopathology after 10 doses, and there were no histologically detectable differences between the lungs of CTL and HIF2αΔ/Δ mice (Figs. 4A, 4E, and 5). After 1 dose of cobalt, lungs from both CTL and HIF2αΔ/Δ mice showed evidence of perivascular and peribronchiolar mixed inflammatory cell infiltrates in the interstitial tissues surrounding preterminal and terminal BA (Figs. 4B and F). Histopathologic scoring showed this change was statistically significant in the bronchiolar region (Fig. 5A). After 5 doses of cobalt, the infiltrates had extended to the proximal alveolar ducts and adjacent alveoli in cobalt-treated lungs (Figs. 4C and G). The change in alveolar inflammation was significant at 5 days (Fig. 5B). Additionally, thickening of the bronchiolar epithelium (hypertrophy/hyperplasia) was noted after 5 doses in both CTL and HIF2αΔ/Δ mice (Figs. 4C and G). After 10 doses, the epithelial thickening appeared to resolve in the CTL mice but not in HIF2αΔ/Δ mice. Although airway lesions were still evident in the lungs of cobalt-treated CTL mice after 10 doses, the overall severities of these pathological lesions were attenuated compared with those in mice receiving only 5 doses (Figs. 4D and 5). This reduction in the severity of histopathological lesions in the 10-dose, cobalt-treated CTL mice (Fig. 5A) corresponded with the BAL cellularity data, as total inflammatory cells were significantly less in the animals that received 10 doses of cobalt compared with those receiving 5 doses (primarily attributed to the decreased macrophage and neutrophil counts). In contrast, the pulmonary pathology in HIF2αΔ/Δ mice receiving cobalt looked similar in severity after 5 and 10 doses (Figs. 4G, 4H, and 5), suggesting that resolution of inflammatory infiltrates and bronchial epithelial thickening was somewhat dependent on presence of epithelial HIF2α.
FIG. 4.

Hematoxylin and eosin stain. Control (A–D) and HIF2αΔ/Δ (E–H) mice are given sterile saline vehicle (A and E) or 1 dose (B and F), 5 doses (C and G), and 10 doses (D and H) of 60 μg CoCl2. All images were taken from the G5 section. *, site of mixed inflammatory cell infiltrates. Abbreviations: a, alveolus; ad, alveolar duct; BA, bronchial airway; bv, blood vessel; HIF, hypoxia-inducible factor.
FIG. 5.

Histopathology scores. Bronchial (A) and alveolar (B) regions of G5 sections stained with hematoxylin and eosin were scored, in a blinded fashion, independently by 2 investigators on a 6-point inflammation scale from 0 (no inflammation) to 4 (marked inflammation). Specifically, the scale included 0 (normal, no inflammation), 0.5 (sparse perivascular infiltrates only), 1 (minimal infiltrates, no lesions), 2 (mild infiltrates, few small lesions), 3 (moderate infiltrates, sizable lesions), and 4 (marked infiltrates, large lesions). Open bars correspond to CTL mice, whereas solid bars correspond to DOX-treated mice. *, significant from saline-treated control; brackets, significant across CTL/DOX groups or same group on a different number of doses, 1-way ANOVA with Fisher’s LSD, α = 0.05. Abbreviations: CTL, control; DOX, doxycycline.
MBP, a cell-specific cytoplasmic constituent in eosinophil secretory granules, was probed using IHC. As expected, the lungs of saline-treated mice did not have eosinophilic infiltration after 10 doses (Figs. 6A and E). After 1 dose of cobalt, only a few widely scattered eosinophils were found in the lung. These granulocytes did not make up a substantial amount of the inflammatory cells, and there were also no observable differences between CTL and HIF2αΔ/Δ mice (Figs. 6B and F). After 5 doses, many more eosinophils are seen in the tissues of both CTL and HIF2αΔ/Δ mice (Figs. 6C and G, respectively). This eosinophilic inflammatory response resolved in CTL mice after 10 doses (Fig. 6D). Interestingly, after 10 doses in the HIF2αΔ/Δ mice, the number of eosinophils increases greatly (this increase supports BAL data [Fig. 3C]), and they appear more in parenchymal areas and are closely approximated to each other within lesions (Fig. 6H).
FIG. 6.

MBP IHC. Immunostaining was performed for MBP (polyclonal rabbit anti-mouse MBP, 1:500, Mayo Clinic). Control (A–D) and HIF2αΔ/Δ (E–H) mice are given sterile saline vehicle (A and E) or 1 dose (B and F), 5 doses (C and G), and 10 doses (D and H) of 60 μg CoCl2. *, site of mixed inflammatory cell infiltrates, eosinophils in red stain. Abbreviations: a, alveolus; AA, axial airway; bv, blood vessel; HIF, hypoxia-inducible factor; MBP, major basic protein.
AB-PAS histochemical staining was performed to visually identify acidic and neutral mucosubstances in mucous goblet cells in airway epithelium. No AB-PAS–stained mucosubstances were observed in saline-treated mice after 10 doses (Figs. 7A and E), and very few were seen after 1 dose of cobalt (Figs. 7B and F). After 5 doses of cobalt, increases in AB-PAS–stained mucosubstances were found in the bronchiolar epithelium of some of the treated animals, though no histochemical differences between the CTL and HIF2αΔ/Δ groups were found (Figs. 7C and G). After 10 doses, conspicuous mucous cell metaplasia with markedly more intraepithelial AB/PAS-stained mucosubstances was apparent in the bronchioles of HIF2αΔ/Δ mice compared with CTL mice (Figs. 7D and H).
FIG. 7.

AB-PAS stain. AB-PAS staining was performed to detect mucosubstances. Control (A–D) and HIF2αΔ/Δ (E–H) mice are given sterile saline vehicle (A and E) or 1 dose (B and F), 5 doses (C and G), and 10 doses (D and H) of 60 μg CoCl2. Black arrows, mucous material visualized in goblet cells. Abbreviations: AA, axial airway; AB-PAS, alcian blue periodic acid schiff; HIF, hypoxia-inducible factor.
Whole Lung Gene Expression Profiling
For each dose group, all expression was normalized to the saline-treated CTL mice. Interleukin (IL)-4 expression did not significantly change between any of the dose groups (Fig. 8A). IL-5 expression in cobalt-treated HIF2αΔ/Δ mice increased nonsignificantly to ~1.5-fold after 1 dose (Fig. 8B). IL-6 gene expression was increased in cobalt-treated mice after 1 and 5 doses (Fig. 8C). IL-10 expression was significantly increased in HIF2αΔ/Δ mice after 5 doses and was increased compared with cobalt-treated CTL mice after 10 doses (Fig. 8D). Keratinocyte-derived chemokine (KC) expression displayed some insignificant variability between groups; however, the only observable trends were a steady decline in the cobalt-treated CTL mice as the dose number increased and an increase in KC in HIF2Δ/Δ mice after 10 doses (Fig. 8E). The only significant change in interferon gamma (IFNγ) expression was seen in the HIF2αΔ/Δ mice treated with saline (Fig. 8F).
FIG. 8.

Gene expression from LL mRNA. All samples were quantitated by the standard curve method and normalized to hypoxanthine guanine phosphoribosyl transferase. Cytokines IL-4 (A), IL-5 (B), IL-6 (C), IL-10 (D), KC (E), and IFNγ (F) are shown as fold change relative to the saline-treated control mice in each time point. *, significant from saline-treated control within genotype; a, significant between CTL and HIF2α∆/∆ within treatment (ie, saline or cobalt), 1-way ANOVA with Fisher’s LSD, α = 0.05. Abbreviations: CTL, control; HIF, hypoxia-inducible factor; IFNγ, interferon gamma; IL, interleukin; KC, keratinocyte-derived chemokine.
Cytokine Profiling
Cytokines were analyzed from LL using a cytokine bead array system. IL-4 was significantly increased in cobalt-treated CTL mice but did not show substantial changes across dosing groups and was not detectable in the 5 dose group (Fig. 9A). IL-5 increased significantly in the cobalt-treated HIF2αΔ/Δ group after 1 dose, and levels tapered off after 5 and 10 doses (Fig. 9B). Although the increase of IL-5 expression was not significant after a single dose (Fig. 8B), the increased protein after 1 dose suggests that this increased expression is relevant. IL-6 was increased in cobalt-treated HIF2αΔ/Δ mice after 1 dose, though after 5 doses the highest levels were seen in cobalt-treated CTL mice (Fig. 9C). These protein levels are similar to patterns of IL-6 expression that began to increase after a single dose and peaked after 5 doses (Fig. 8C). Similar to IL-4, IL-10 levels were not detectable in the 5-dose samples. The significance of this finding is unknown because these values were all near the limit of detection (Fig. 9D). Further, IL-10 expression patterns (Fig. 8D) did not match the protein levels. Leukocyte chemokine KC protein levels increased (nonsignificantly) after 1 dose in the HIF2αΔ/Δ mice, though after 5 doses the CTL mice showed higher levels of KC and all groups showed tapered levels after 10 doses (Fig. 9E). IFNγ was highest in cobalt-treated CTL mice after 5 doses but was very low in the 1-dose and 10-dose groups (Fig. 9F). Although the IFNγ gene expression (Fig. 8F) does not match the pattern of protein levels for most groups, IFNγ proteins were decreased or near limit of detection in all groups of HIF2αΔ/Δ mice.
FIG. 9.

LL cytokine analysis. LL was analyzed for cytokine protein levels using cytokine bead array and FACSCalibur flow cytometer (BD Biosciences). Shown are IL-4 (A), IL-5 (B), IL-10 (C), KC (D), tumor necrosis factor alpha (E), and IFNγ (F). Open bars correspond to saline-treated CTL mice, diagonal hatched bars correspond to saline-treated HIF2αΔ/Δ mice, black bars indicate cobalt-treated CTL mice, and horizontal hatched bars indicate cobalt-treated HIF2αΔ/Δ mice. *, significant from saline-treated control within genotype; a, significant between CTL and HIF2αΔ/Δ within treatment (ie, saline or cobalt), 1-way ANOVA with Fisher’s LSD, α = 0.05. Abbreviations: CTL, control; HIF, hypoxia-inducible factor; IFNγ, interferon gamma; IL, interleukin; KC, keratinocyte-derived chemokine; LL, Lung lysates.
DISCUSSION
These studies are the first to investigate the role of lung epithelial–specific HIF2α in cobalt-induced lung inflammation. Combined with previous studies of HIF1α deficiency, the studies provide evidence that loss of either HIFα subunit in ATII and Club cells resulted in similar eosinophilic inflammation, albeit at different time points (Saini et al., 2010a, b). It is also important to note that the bias toward eosinophilic inflammation seen in the HIF1α∆/∆ mice was dependent upon presence of all 3 transgenes and DOX exposure, thus eliminating the possibility that these observed inflammatory changes are due to DOX alone or Cre toxicity (Saini et al., 2010b). Although the mechanism for this induction of eosinophilic inflammation requires more study, these data provide evidence that epithelial HIF signaling has important roles in modulating the immune response and likely has important implications for targeting HIFs in inflammatory lung disease, including HMLD.
HIF2α shares substantial domain and sequence homology with HIF1α (Ema et al., 1997). Although both HIFα subunits bind identical genomic DNA sequences, the hypoxia response element 5′-A/GCGTG-3′, under hypoxic conditions, it is well established that HIF1α and HIF2α regulate divergent tissue-/cell-specific gene expression profiles. The mechanism for their unique expression profiles most likely stems from differences in protein structure (Hu et al., 2007) and cell-type/tissue-dependent HIFα activation (Kaluz et al., 2008; Zagorska and Dulak, 2004), though the differential contribution of coactivators/repressors has not been elucidated for the HIFs. The differential expression of HIF2α, notably the lung-specific presence of large amounts of HIF2α mRNA, suggests that HIF2α plays unique roles in the lung (Wiesener et al., 1998, 2003).
Many investigations have attempted to clarify the role of HIF1α and HIF2α in cobalt toxicity of lung epithelial cells. One study of A549s by Uchida et al. (2004) demonstrated that although early hypoxic or cobalt induction of HIF1α and HIF2α was nearly identical, HIF2α protein and mRNA persisted during prolonged hypoxia or cobalt treatment compared with HIF1α, due in part to upregulation of antisense HIF1α. This finding implicates a more important role for HIF2α than HIF1α in chronic hypoxia/cobalt treatment, which may explain the difference in histopathology seen between the 10-dose groups described here and in HIF1αΔ/Δ mice (Saini et al., 2010b). Toxicogenomic profiling of A549s treated with cobalt (Malard et al., 2007) revealed many metal-responsive targets; however, there was surprisingly little overlap with known HIF1α target genes (8%). These results suggest the potential for many other cobalt-responsive transcription factors, which may include HIF2α (for review, see Cummins et al., 2005). Our investigations of both HIF1α and HIF2α lung epithelial–specific deletion, revealing a similar eosinophilic inflammation phenotype, suggest that either a common target between HIF1α and HIF2α or a downstream effector pathway may be responsible for eosinophilia in the HIF-deficient mice. More specific profiling of mammalian lung epithelium could provide further insight into this mechanism (Saini et al., 2010a, b).
Eosinophilia was induced in cobalt-treated HIF2αΔ/Δ mice at day 15 of occupational exposure (10 doses of cobalt), which is a similar response to that seen at 6 days (5 doses) in HIF1αΔ/Δ mice. Explanations for this eosinophilia might lie in secretion of specific cytokines such as IL-5, IL-10, and KC. It is unlikely caused by a single cytokine but the combined effect of several acting in concert. For example, IL-5 protein levels and patterns of expression are nearly identical in the HIF1α∆/∆ and HIF2α∆/∆ mice (Fig. 9B; Saini et al., 2010a, b), yet the timing of eosinophilia is quite different. KC levels were increased in both control mice compared with HIF2αΔ/Δ mice after 5 doses, and because KC is more associated with chemotaxis of neutrophils than eosinophils, this may partially explain why neutrophils peaked after 5 doses in cobalt-treated control mice and were less numerous in BAL of cobalt-treated HIF2αΔ/Δ mice. What other cytokines or metabolites might be influencing the eosinophil infiltration remains one of the most important unanswered questions of the current studies.
Although eosinophils are classically associated with allergic disease, parasitic infections, and TH2-mediated inflammation, their function and purpose remain somewhat elusive. Current discussion in the field has postulated that eosinophils are involved in immune modulation and in assisting areas of tissue remodeling and repair (Jacobsen et al., 2012; Lee et al., 2010). With this in mind, one possible explanation for the eosinophilic phenotype in epithelial HIF-deficient mice is that epithelial cells lacking HIF2α are more susceptible to cell death, perhaps a lesser ability to adapt to hypoxia, and as a result, there is more epithelial cell turnover in HIF2αΔ/Δ mice treated with cobalt, leading eventually to eosinophil chemotaxis. Indeed, many more eosinophils were seen in the parenchymal compartment in both the cobalt-treated HIF2αΔ/Δ and HIF1αΔ/Δ mice, which is coincidentally the site of cell-specific HIF recombination (Saini et al., 2010a, b). Future analyses of cell death and turnover will be helpful in exploring whether this plays a role in eosinophilia seen with this phenotype.
According to histopathology, it appears that after 10 doses of cobalt treatment, control mice have mild recovery of overall injury compared with HIF2αΔ/Δ mice (Figs. 4D and H, respectively). This observation is supported by total cell counts (Fig. 3A) and histopathologic scoring (Fig. 5). AB-PAS staining also suggest that the amount of mucous substances decreases for control mice from the 5-doses to 10-doses groups (Figs. 7C and D) compared with HIF2αΔ/Δ mice (Figs. 7G and H). When comparing these results to previous studies of HIF1αΔ/Δ mice, we see very similar amounts of total cells after 10 doses for both controls and HIF-deficient mice, indicating that a slight recovery or normalization is achieved at the 10-dose time point which is prevented in both HIF1αΔ/Δ and HIF2αΔ/Δ mice (Saini et al., 2010b). This observation of slight histopathologic recovery in the controls suggests that HIF2α plays a role in remodeling of lung epithelium, and chronic exposures may reveal the extent to which the recovery can progress with regular exposures to cobalt that more closely mimic HMLD.
The highly variable presentation and course of patients with HMLD make modeling this disease challenging. One important feature of HMLD missing in this model system is presence of tungsten carbide particulate inhalation, which in the presence of cobalt has been hypothesized to provide an inflammatory drive for HMLD (Lison et al., 1996). It is possible that this additional feature is important in driving the pathophysiology of HMLD, and perhaps, loss of epithelial HIF2α could provide protection in this context, to which further studies may provide insight. Workers with the worst forms of HMLD typically have longer chronic exposures, opening the additional possibility that important molecular events occur in this context, which was beyond the scope of this study.
In summary, these studies show that loss of HIF2α in lung epithelial cells fails to provide protection from cobalt-induced lung inflammation, but rather is capable of modulating the inflammatory response to cobalt, and therefore may play an important role in HMLD from an immunomodulatory standpoint. Further, HIF2α seems to confer some reparative features in subacute cobalt exposure, which suggests that epithelial cells require HIF2α for optimal resolution of inflammation. Despite the usually disparate nature of HIF1α and HIF2α expression profiles, loss of either subunit in the lung epithelium causes increases in IL-5 and eosinophilic inflammation, which suggests shared HIF target(s) or their downstream effects are responsible for the similar phenotype. These studies further highlight the importance of HIFs in the epithelium and its contribution to optimal signaling during inflammatory responses. Further studies of these epithelial cells and other cell type–specific models will provide a more complete understanding of how HIFs function in HMLD and other inflammatory lung diseases such as asthma and COPD.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
FUNDING
National Institute of Environmental Health Sciences at the National Institutes of Health (R01ES-12186).
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Dr Jeffrey Whitsett for supplying the SPC-rtTA-/tg/ (tetO)7-CMV-Cretg/tg transgenic mice and Dr M. Celeste Simon for the HIF2αfl/fl mice. We also wish to thank Amy S. Porter, Kathleen A. Joseph, and Katlyn M. Welch of the Michigan State University Investigative Histopathology Lab for expertise and technical assistance with immunohistochemistry. Finally, we would like to thank Katie Kim and Christian Merrell for their help with animal experiments.
REFERENCES
- Bradford M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- Cummins E. P., Taylor C. T. (2005). Hypoxia-responsive transcription factors. Pflugers. Arch. 450, 363–371 [DOI] [PubMed] [Google Scholar]
- del Peso L., Castellanos M. C., Temes E., Martin-Puig S., Cuevas Y., Olmos G., Landazuri M. O. (2003). The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J. Biol. Chem. 278, 48690–48695 [DOI] [PubMed] [Google Scholar]
- Ema M., Taya S., Yokotani N., Sogawa K., Matsuda Y., Fujii-Kuriyama Y. (1997). A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. U.S.A. 94, 4273–4278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O’Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., et al. (2001). C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 [DOI] [PubMed] [Google Scholar]
- Fisher J. W. (1998). A quest for erythropoietin over nine decades. Annu. Rev. Pharmacol. Toxicol. 38, 1–20 [DOI] [PubMed] [Google Scholar]
- Greer S. N., Metcalf J. L., Wang Y., Ohh M. (2012). The updated biology of hypoxia-inducible factor. EMBO J. 31, 2448–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C. J., Sataur A., Wang L., Chen H., Simon M. C. (2007). The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol. Biol. Cell 18, 4528–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen E. A., Helmers R. A., Lee J. J., Lee N. A. (2012). The expanding role(s) of eosinophils in health and disease. Blood 120, 3882–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluz S., Kaluzová M., Stanbridge E. J. (2008). Regulation of gene expression by hypoxia: Integration of the HIF-transduced hypoxic signal at the hypoxia-responsive element. Clin. Chim. Acta. 395, 6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A., Schleimer R. P. (2007). Beyond inflammation: Airway epithelial cells are at the interface of innate and adaptive immunity. Curr. Opin. Immunol. 19, 711–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korovessis P., Petsinis G., Repanti M., Repantis T. (2006). Metallosis after contemporary metal-on-metal total hip arthroplasty. Five to nine-year follow-up. J. Bone Joint Surg. Am. 88, 1183–1191 [DOI] [PubMed] [Google Scholar]
- Lee J. J., Jacobsen E. A., McGarry M. P., Schleimer R. P., Lee N. A. (2010). Eosinophils in health and disease: The LIAR hypothesis. Clin. Exp. Allergy 40, 563–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lison D. (1996). Human toxicity of cobalt-containing dust and experimental studies on the mechanism of interstitial lung disease (hard metal disease). Crit. Rev. Toxicol. 26, 585–616 [DOI] [PubMed] [Google Scholar]
- Lison D., Lauwerys R., Demedts M., Nemery B. (1996). Experimental research into the pathogenesis of cobalt/hard metal lung disease. Eur. Respir. J. 9, 1024–1028 [DOI] [PubMed] [Google Scholar]
- Loboda A., Jozkowicz A., Dulak J. (2012). HIF-1 versus HIF-2–is one more important than the other? Vascul. Pharmacol. 56, 245–251 [DOI] [PubMed] [Google Scholar]
- Malard V., Berenguer F., Prat O., Ruat S., Steinmetz G., Quemeneur E. (2007). Global gene expression profiling in human lung cells exposed to cobalt. BMC Genomics 8, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell P., Salnikow K. (2004). HIF-1: An oxygen and metal responsive transcription factor. Cancer Biol. Ther. 3, 29–35 [DOI] [PubMed] [Google Scholar]
- Milosev I., Trebse R., Kovac S., Cör A., Pisot V. (2006). Survivorship and retrieval analysis of Sikomet metal-on-metal total hip replacements at a mean of seven years. J. Bone Joint Surg. Am. 88, 1173–1182 [DOI] [PubMed] [Google Scholar]
- Nizet V., Johnson R. S. (2009). Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 9, 609–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg G. (2007). Handbook on the Toxicology of Metals, 3rd ed Academic Press, Amsterdam, The Netherlands [Google Scholar]
- Patel S. A., Simon M. C. (2008). Biology of hypoxia-inducible factor-2alpha in development and disease. Cell Death Differ. 15, 628–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A. K., Wert S. E., Nagy A., Lobe C. G., Whitsett J. A. (2002). Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc. Natl. Acad. Sci. U.S.A. 99, 10482–10487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajatapiti P., van der Horst I. W., de Rooij J. D., Tran M. G., Maxwell P. H., Tibboel D., Rottier R., de Krijger R. R. (2008). Expression of hypoxia-inducible factors in normal human lung development. Pediatr. Dev. Pathol. 11, 193–199 [DOI] [PubMed] [Google Scholar]
- Saini Y., Greenwood K. K., Merrill C., Kim K. Y., Patial S., Parameswaran N., Harkema J. R., LaPres J. J. (2010). Acute cobalt-induced lung injury and the role of hypoxia-inducible factor 1alpha in modulating inflammation. Toxicol. Sci. 116, 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini Y., Harkema J. R., LaPres J. J. (2008). HIF1alpha is essential for normal intrauterine differentiation of alveolar epithelium and surfactant production in the newborn lung of mice. J. Biol. Chem. 283, 33650–33657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini Y., Kim K. Y., Lewandowski R., Bramble L. A., Harkema J. R., Lapres J. J. (2010). Role of hypoxia-inducible factor 1{alpha} in modulating cobalt-induced lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 298, L139–L147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen L. O., Harbak H., Bennekou P. (2012). Cobalt metabolism and toxicology–A brief update. Sci. Total Environ. 432, 210–215 [DOI] [PubMed] [Google Scholar]
- Uchida T., Rossignol F., Matthay M. A., Mounier R., Couette S., Clottes E., Clerici C. (2004). Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: Implication of natural antisense HIF-1alpha. J. Biol. Chem. 279, 14871–14878 [DOI] [PubMed] [Google Scholar]
- Vengellur A., LaPres J. J. (2004). The role of hypoxia inducible factor 1alpha in cobalt chloride induced cell death in mouse embryonic fibroblasts. Toxicol. Sci. 82, 638–646 [DOI] [PubMed] [Google Scholar]
- Vengellur A., Woods B. G., Ryan H. E., Johnson R. S., LaPres J. J. (2003). Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1alpha null mouse embryonic fibroblasts. Gene Expr. 11, 181–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitsett J. A., Perl A. K. (2006). Conditional control of gene expression in the respiratory epithelium: A cautionary note. Am. J. Respir. Cell Mol. Biol. 34, 519–520 [DOI] [PubMed] [Google Scholar]
- Wiesener M. S., Jürgensen J. S., Rosenberger C., Scholze C. K., Hörstrup J. H., Warnecke C., Mandriota S., Bechmann I., Frei U. A., Pugh C. W., et al. (2003). Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 17, 271–273 [DOI] [PubMed] [Google Scholar]
- Wiesener M. S., Turley H., Allen W. E., Willam C., Eckardt K. U., Talks K. L., Wood S. M., Gatter K. C., Harris A. L., Pugh C. W., et al. (1998). Induction of endothelial PAS domain protein-1 by hypoxia: Characterization and comparison with hypoxia-inducible factor-1alpha. Blood 92, 2260–2268 [PubMed] [Google Scholar]
- Zagórska A., Dulak J. (2004). HIF-1: The knowns and unknowns of hypoxia sensing. Acta Biochim. Pol. 51, 563–585 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.