

Summary
Using a large cohort study, we identify 749 CpG sites having consistent age-related methylation changes in blood and other normal tissues. Increasingly methylated aging sites are significantly overmethylated in many human cancers, perhaps explaining in part increased cancer risk with age.
Abstract
Epigenetic marks are extensively altered in cancer but may also change in normal tissues with age, which is the primary risk factor for most cancers. We conducted an epigenome-wide study to identify age-related methylation sites and examine their relationship to cancer and other underlying epigenetic marks. We analyzed 1006 blood DNA samples of women aged 35–76 years from the Sister Study and found that 7694 (28%) of the 27 578 CpGs assayed were associated with age (false discovery rate, q < 0.05). Using independent data sets, we confirmed 749 ‘high-confidence’ age-related CpG (arCpGs) sites in normal blood. Based on The Cancer Genome Atlas data, we show that these age-related changes are largely concordant in a broad variety of normal tissues and that a significantly higher (71–91%, P < 10–74) than expected proportion of increasingly methylated arCpGs (IM-arCpGs) were overmethylated in a wide variety of tumor types. IM-arCpGs sites occurred almost exclusively at CpG islands and were disproportionately marked with the repressive H3K27me3 histone modification (P < 1 × 10− 50). Genes containing these IM-arCpG sites were highly enriched for developmental and signaling pathways (P < 10− 10). Our findings suggest that as cells acquire methylation at age-related sites, they have a lower threshold for malignant transformation that may explain in part the increase in cancer incidence with age.
Introduction
Aging is the strongest risk factor for cancer and for many other diseases (1), but the mechanisms underlying age-related diseases remain largely unknown. Age-related DNA methylation changes at selected candidate genes have been reported (2–6) and several age-related DNA methylation sites have been linked to specific cancers (7–10). Large-scale identification of age-related CpG (arCpG) methylation sites may help provide understanding of both the underlying biological processes of aging and the risk of age-related diseases including cancer. A few genome-wide studies have begun to identify arCpG sites, but these studies have been based on relatively small numbers of individuals: Teschendorff et al. (11) examined blood DNA from 261 women (148 healthy and 113 with ovarian cancer) and identified a set of 589 arCpGs, 62% of which had decreasing methylation with age; Rakyan et al. (12) examined blood DNA from 93 women and identified 360 arCpGs but found the majority (59%) had increasing methylation with age. A recent study by Bell et al. (13) identified 490 arCpGs in whole blood of 172 healthy female twins, almost all of which (99%) showed increasing methylation with age. We used genome-wide methylation analysis of blood DNA samples and found that age-related methylation changes were very common across the genome. Using independent data sets, we confirmed a subset of these age-related changes and show that a special subset of age-related sites are disproportionately methylated in multiple types of human cancer. These findings suggest that with increasing age, there are an increasing proportion of cells that have undergone epigenetic switching to methylation-based gene repression and that the cells acquiring age-related changes have a lower threshold for neoplastic transformation.
Materials and methods
Study samples
Samples used to identify arCpG came from the Sister Study (www.sisterstudy.org), a nationwide prospective cohort of 50 884 women. To be eligible for the Sister Study, women should not themselves have had breast cancer but must have a sister with breast cancer (14). Participants provided extensive information via questionnaire and informed consent and blood samples were obtained during a home visit. The study was approved by the Institutional Review Boards of the National Institute of Environmental Health Sciences and the Copernicus Group. As described previously in detail elsewhere (15), we analyzed DNA methylation from whole blood for 1006 women aged 35–76 years, including 327 who developed breast cancer within 46 months of blood draw and 679 women who remained cancer free during up to 55 months of follow-up.
Three independent data sets were utilized to identify ‘high- confidence’ arCpGs from among the arCpGs identified in Sister Study samples. Phenotype data and whole blood DNA methylation profiles with Infinium HumanMethylation27 BeadChip were downloaded from National Center for Biotechnology Information Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE19711 (11), GSE20067 (11) and GSE20236 (12). Details about these samples were described previously (11,12), and a brief summary of the data sets are provided in Supplementary Table 1, available at Carcinogenesis Online.
Genome-wide DNA methylation data sets for seven different types of tumors and adjacent normal tissue pairs were obtained from The Cancer Genome Atlas (TCGA) project (http://cancergenome.nih.gov/). Sample size, age range and methylation array type are summarized in Supplementary Table 2, available at Carcinogenesis Online. For tumors with HumanMethylation450 data, we only analyzed the subset (25978 CpGs) of CpGs that were present on the HumanMethylation27. Histone modification ChIP-seq data for human embryonic stem cell (HESC), a normal lymphoblastoid cell line GM12878, and six other differentiated cells were downloaded from ENCODE web site (http://genome.ucsc.edu/cgi-bin/hgFileUi?db=hg18&g=wgEncodeBroadChipSeq) (16) and cell types listed in the figure legend of Supplementary Figure 8, available at Carcinogenesis Online. We compared CpG chromosome positions with ENCODE ChIP-seq data for histone-binding positions and counted the histone mark as present if the CpG position was within the histone-binding peak region. Genome-wide gene expression data from Affymetrix U133A/GNF1H arrays for 78 different types of human normal tissues were downloaded from bioGPS (http://biogps.org) (17). Log2-transformed gene expression value were used in our analysis.
Genome-wide DNA methylation profiling
In Sister Study samples and the three other methylation data sets for blood, genome-wide DNA methylation was profiled using the Infinium HumanMethylation27 BeadChip, which allows interrogation of methylation level at 27 578 different CpG sites around promoter region of 14 495 Human RefSeq Genes at single CpG site resolution. Of the promoters, 82.4% are represented by two or more CpG sites. Of all CpG sites, 72.5% are located within CpG island region. At each CpG site, the methylation level is assessed with two probes, one designed for the unmethylated sites (U) and one for methylated site (M). The methylation level (β value) was calculated as the ratio of fluorescent intensities between methylated and unmethylated alleles 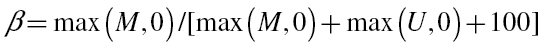 . The HumanMethylation27 BeadChip included 40 probes for sample-dependent and sample-independent internal quality controls. Of these, four probes (with one pair targeting converted and other pair targeting unconverted DNA sequences) were bisulfite conversion (BSC) controls to assess the efficiency of BSC. In each of the four methylation data sets, we excluded samples with low-quality data based on the following three filters: (i) low BSC (as determined by average intensity value < 3800 for the pair of internal BSC probes that target converted sequences); (ii) clear outliers on boxplots of total intensity I = U + M values or histograms of β values and (iii) samples with >5% of CpGs whose intensity values fell below background levels (Beadstudio detection P > 0.05). Based on these criteria, we excluded 33 samples from data set GSE19711 and 49 samples from data set GSE20067. We normalized methylation data by (i) robust multiarray average background correction and quantile normalization of U and M value; (ii) recalculation of β values from normalized U and M value; (iii) adjustment for experimental batch and BSC efficiency intensities with a linear multivariate regression model.
. The HumanMethylation27 BeadChip included 40 probes for sample-dependent and sample-independent internal quality controls. Of these, four probes (with one pair targeting converted and other pair targeting unconverted DNA sequences) were bisulfite conversion (BSC) controls to assess the efficiency of BSC. In each of the four methylation data sets, we excluded samples with low-quality data based on the following three filters: (i) low BSC (as determined by average intensity value < 3800 for the pair of internal BSC probes that target converted sequences); (ii) clear outliers on boxplots of total intensity I = U + M values or histograms of β values and (iii) samples with >5% of CpGs whose intensity values fell below background levels (Beadstudio detection P > 0.05). Based on these criteria, we excluded 33 samples from data set GSE19711 and 49 samples from data set GSE20067. We normalized methylation data by (i) robust multiarray average background correction and quantile normalization of U and M value; (ii) recalculation of β values from normalized U and M value; (iii) adjustment for experimental batch and BSC efficiency intensities with a linear multivariate regression model.
Statistical analysis
Associations between continuous chronological age and individual CpG methylation level (β value) were tested using a linear mixed model. Methylation β value was modeled as the response variable and the two BSC internal controls were adjusted as fixed effect variables in the analysis. Other covariables were adjusted as fixed effects and included two-category case status (case, non-case) for Sister Study data; three-category case status (control, pretreatment, posttreatment) for GSE19711 and two-category gender (male, female) for GSE20067. A variable for batch was adjusted as a random effect in all analyses. Association analyses were performed for each CpG separately. To assess possible effects of blood cell subtype heterogeneity, we adjusted for leukocyte subtype proportions as fixed effects. Individual leukocyte subtype proportions were estimated using a method developed by Houseman et al. (18) based on methylation profiles for eight different leukocyte subtypes (GSE39981) (19). For age methylation association analysis in normal tissues from the TCGA project, we first background corrected and normalized methylation β value within each type of normal tissues using the robust multiarray average method and then employed a robust linear regression model adjusting for experimental batch to test whether age was associated with methylation value at each CpG site for each type of normal tissue. A paired t-test was used to test the difference of methylation levels between tumor and normal tissues at each CpG site for each tumor type from the TCGA project. To correct for multiple testing, we estimated the false discovery rate (FDR) using q value framework (20).
Geneset enrichment analysis
Based on the annotation provided by Illumina for the HumanMethylation27 Beadchip, we mapped each CpG to the nearest gene. We then performed gene set enrichment analysis for genes using the hypergeometric test against the Gene Ontology pathway database. Permutation P values were calculated based on 5000 pathway analyses results for the permuted CpG sites.
Results
Genome-wide identification of arCpGs
We conducted a genome-wide methylation profiling (Illumina HumanMethylation27 Beadchips) of whole blood DNA from 1006 women who ranged in age from 35 to 76 years (Supplementary Figure 1, available at Carcinogenesis Online) who were part of a case–cohort study of breast cancer in which all women were clinically cancer free at the time of blood draw (15). Initial analysis was restricted to the 679 women who remained cancer free for up to 55 months of follow-up. Age methylation associations were substantively the same with the addition of women who later developed breast cancer (Pearson’s correlation coefficient = 0.98, Supplementary Figure 2, available at Carcinogenesis Online), so all subsequent analyses used the combined data set with adjustment for case status. Of the 27 578 CpGs on the array, 7694 (28%) were significantly associated with age at FDR threshold q < 0.05 (Figure 1). Of these significant arCpGs, 5037 (65%) were located in CpG island regions and 2657 (35%) were located in non-island regions. Among arCpGs in island regions, 3047 (60%) showed increasing methylation with age. In contrast, among arCpGs in non-island regions, there was the opposite direction of methylation changes with 2164 (81%) having decreasing methylation with age. Thus, these findings provide evidence for both increasing and decreasing methylation of arCpGs but indicate that the direction of change with age is dependent on genomic context: increasing methylation with age predominating at island sites and decreasing methylation with age predominating at non-island sites.
Fig. 1.

Manhattan plot of age methylation association P values based on 1006 women. The P values were sorted by chromosome physical position. Dashed line indicate FDR threshold of 0.05. The 749 high-confidence arCpGs verified with independent data sets are marked in red.
Validation of arCpGs with independent data sets of blood
Applying similar quality control and analytical methods, we analyzed three publically available blood DNA whole genome methylation data sets GSE19711, GSE20067 and GSE20236 with sample sizes of 507, 146 and 93 people, respectively (Supplementary Table 1, available at Carcinogenesis Online). The overall association signals in each of these three data sets were weaker than that in Sister Study data reflecting their smaller sample sizes (Supplementary Figure 3, available at Carcinogenesis Online). Using a combination of P value and direction of change with age, we sequentially filtered the 7694 arCpGs identified from the Sister Study against the results of each of the three data sets and found 749 arCpGs (459 increasingly methylated, 290 decreasingly methylated) that were consistent across all data sets, which we term ‘high-confidence arCpGs’ (Figure 1, Supplementary Figure 4 and Table 3, available at Carcinogenesis Online). The proportion of high-confidence arCpGs was correlated with the association strength in Sister Study data (Supplementary Figure 5, available at Carcinogenesis Online) with 47% of arCpGs with P ≤ 10−10 being high confidence and 70% of arCpGs with P ≤ 10−20 being high confidence. High-confidence arCpGs were distributed to island (73%) and non-island regions (27%) in the same proportions that these two regions were represented on the array but the direction of methylation change differed by genomic context: arCpGs in island regions were 82% increasingly methylated with age, whereas arCpGs in non-island regions were 95% decreasingly methylated with age (Supplementary Figure 4, available at Carcinogenesis Online). The average methylation level changes over a 10-year interval was +0.8% for increasingly methylated arCpGs (IM-arCpGs) and −0.9% for decreasingly methylated arCpGs (DM-arCpGs; Figure 2). The number of high-confidence arCpGs on each chromosome was largely proportional to the number of CpGs on the array for each chromosome, except for the X chromosome, which was underrepresented (Figure 1, Supplementary Figure 6, available at Carcinogenesis Online). A total of 690 age-related CpG sites have been identified by at least one of three prior studies of adults (11–13). Of these, 535 (78%) were among the 7694 age-related sites identified in our study, and 273 (40%) were members of the set of high-confidence arCpGs; all of which were concordant for direction of change with age. In addition, we compared the 749 high-confidence arCpGs to a set of 2078 age-associated CpGs (477 increasingly methylated, 1601 decreasingly methylated) identified from 389 boys aged 3–17 years (21) and found 342 arCpGs that were common to both sets. There was perfect concordant between the two studies for these 342 arCpGs for the direction of change with age (132 increasingly methylated, 210 decreasingly methylated), providing further validation of the high-confidence arCpGs and suggesting that the change with age for at least some of these sites starts at a young age.
Fig. 2.

DNA methylation change with age for arCpGs identified in 1006 human whole blood samples. (A) An example of arCpGs (cg06493994) that was increasingly methylated with age. Methylation beta values are plotted as a function of age at 5 year interval. (B) An example of arCpGs (cg23124451) that was decreasingly methylated with age. (C) Average methylation beta value change in 10 years for increasingly methylated arCpGs (IM-arCpG; N = 459) and decreasingly methylated arCpGs (DM-arCpG; N = 290). The horizontal bars of each box-and-whisker diagram from top to bottom represent the largest value within 1.5 interquartile range of the upper quartile, upper quartile, median and lower quartile and the smallest value within 1.5 interquartile range of lower quartile. The open circle dots are outlier values outside the whiskers.
Leukocyte subtypes
Shifts in leukocyte subtypes with age could be responsible for the observed age changes in whole blood DNA methylation profiles. If this were so, CpGs that are known to be differentially methylated in different leukocyte subpopulations would be expected to be overrepresented among arCpGs. There was no evidence of such enrichment. Among the 50 CpG set reported to be differentially methylated in leukocyte subtypes (22), only two were among the 7694 arCpGs at FDR threshold of 0.05 (Supplementary Figure 7A, available at Carcinogenesis Online) and none were members of the 749 high-confidence arCpGs. In addition, adjustment for individual leukocyte subtypes produced highly correlated P values (Pearson’s correlation coefficient = 0.98 Supplementary Figure 7B, available at Carcinogenesis Online). Therefore, we find no evidence to suggest that these age-related changes are simply due to changes in leukocyte subpopulations that might also occur with age.
Concordant methylation changes between blood and other human normal tissues
The same blood DNA methylation changes with age were also evident in multiple other normal tissues available from TCGA. There were differing number of samples and age distributions within each normal tissue category (Supplementary Table 2, available at Carcinogenesis Online), so we examined high-confidence arCpGs in two ways. First, for each arCpG, we counted the number of normal tissue types that were concordant with blood for the direction of change with age and compared that with the distribution expected by chance (Figure 3, Wilcoxon rank sum test P = 5 × 10–112). Second, we identified significant (FDR q < 0.05) age-related sites within each normal tissue type and found that high-confidence arCpGs were observed at frequencies much higher than would be expected by chance (P value range 2 × 10–3 to 4 × 10–119, Supplementary Table 4, available at Carcinogenesis Online) and depending on tissue type, we found that these were 95.4–100% concordant for the direction of change in blood (Supplementary Table 5, available at Carcinogenesis Online). These data strongly suggest that the arCpGs we identified in blood can be generalized to many types of normal human tissues and are a general feature of aging.
Fig. 3.

Cross-tissue concordance of direction of methylation change with age for arCpGs. For each high-confidence arCpG, the number of other normal tissues that were concordant with findings from blood for the direction of methylation change with age (maximum of seven possible) are shown for the 651 high-confidence arCpGs that had data for all seven normal tissues. Normal tissue methylation data are from The Cancer Genome Atlas project (see Supplementary Table 2, available at Carcinogenesis Online). The concordance was significantly higher than expected by chance (Wilcoxon rank sum test P = 5×10–112). Dotted line indicates expected distribution by chance.
Functional pathways enrichment
Genes containing high-confidence IM-arCpGs at island sites were enriched for developmental and signaling pathways, with 13 pathways having FDR q < 10−10 (Supplementary Table 6, available at Carcinogenesis Online). Forty-two percent of IM-arCpG were located in developmental genes including the promoters of key developmental regulator genes for different tissue types: Cdx2 (Trophectoderm), Gata4 (primitive endoderm), Sox17 (endoderm), T (mesoderm), MyoD1 (muscle), Sox1 (neuroectoderm) and Hox genes (A, C and D clusters, embryo body plan). In contrast, there was little evidence of pathway enrichment for genes containing DM-arCpGs at islands or for genes containing DM-arCpGs at non-island sites (data not shown). Since increasing DNA methylation might be thought to interfere with proper regulation of genes, our data suggest a role for age-related methylation in determining the gene expression profile of key developmental and signaling genes that might in turn be associated with age-related diseases.
Histone modifications around aging-related CpGs
We investigated histone modifications patterns near high-confidence arCpGs using ENCODE ChIP-seq data (16) for a HESC and a normal lymphoblastoid cell line GM12878. The frequency of histone modifications differed between island and non-island CpGs represented on the array and between HESC and GM12878, so these groups were analyzed separately (Figure 4). DM-arCpGs at island sites had significantly higher frequency of H3K4me1 in both embryonic and differentiated cell lines but otherwise were similar to other island sites represented on the array (Figure 4). DM-arCpGs at non-island sites had similar histone modification patterns in both cell lines, where they showed significantly higher frequency of permissive H3K4me1-3 and H3K9ac marks.
Fig. 4.

Histone marker enrichment around arCpGs. Percentage of CpGs overlapped with histone marker binding peak region based on ENCODE histone modification ChIP-seq data. For specific histone markers, we used Fisher’s exact test to compare the percentages between arCpG and corresponding island/ non-island CpGs on array. The levels of statistical significance were denoted as *P < 0.001; **P < 1×10− 6; ***P < 1×10−50.
The remaining analysis focused on the 449 high-confidence IM-arCpGs at island sites and compared the frequency of selected histone modification to that of the 20 006 CpG island sites represented on the array. HESC and GM12878 cells had similar frequency of polycomb-mediated H3K27me3 marks (Figure 4). For both cell lines, the mark was very common (~80%) at IM-arCpG island sites but much less common (~40%) at CpG island sites on the array. In addition, the identity of H3K27me3 marking at specific sites was maintained across the two cell lines: 87% of the IM-arCpG island sites that carried the H3K27me3 mark in HESC cells also carried that mark in GM12878 cells. The enrichment of H3K27me3 at IM-arCpGs in islands was surprisingly consistent across other differentiated cell types (Supplementary Figure 8, available at Carcinogenesis Online), suggesting that this repressive mark is a common, although not universal, mark of IM-arCpG sites.
In contrast, the frequency of the permissive H3K4me3 histone mark (as well as other permissive marks, see Figure 4) was different across the two cell lines: in HESC cells, both IM-arCpG and all island CpG sites on the array were marked by H3K4me3 >90% of the time. In GM12878 cells, although all island CpGs sites still had high frequency (78%) of H3K4me3 marks, at IM-arCpG island sites, there was much lower frequency (30%) of H3K4me3 marks. In HESC cells, IM-arCpG island sites had a much higher proportion (71%) of bivalent sites (carrying both H3K27me3 and H3K4me3 marks) than all island sites on the array (37%). We note, however, that the frequency of bivalent sites is entirely consistent with the expectation from random assortment (i.e. is exactly predicted by the product of the frequency of H3K4me3 and H3K27me3 marks) and represents no statistical enrichment of bivalency at IM-arCpG island sites (Supplementary Table 7, available at Carcinogenesis Online).
Given that the differentiated GM12878 cell line had a high frequency of repressive H3K27me3 marks at IM-arCpG sites, we hypothesized that expression of genes with these sites should be low in differentiated tissues. We examined publicly available gene expression data for 78 normal tissues and found that average expression of genes with IM-arCpG sites were significantly lower in all tissues (Wilcoxon rank sum test P < 2 × 10− 8 to 3 × 10− 44) than genes represented by other sites on the array in all tissues (Figure 5, Supplementary Table 8, available at Carcinogenesis Online). In contrast, there was no evidence for altered expression of genes that contained DM-arCpGs at either island or non-island sites (data not shown).
Fig. 5.

Gene expression values for genes near increasingly methylated arCpGs in human normal tissues. In all 78 human normal tissues, the median gene expression values for genes with increasingly methylated arCpG at CpG island were significantly lower (Wilcoxon rank sum test P < 2×10− 8 to 3×10−44) than that for other genes with CpGs at island sites represented on array. See Supplementary Table 8, available at Carcinogenesis Online, for a list of normal tissues and Wilcoxon rank sum test P values. Gene expression data were obtained from bioGPS (http://biogps.org) (17).
Methylation in tumor tissue
We tested the hypothesis that aging sites would be associated with cancer by examining methylation data for tumor–normal tissue pairs for seven types of cancer in the TCGA database. Considering all CpGs represented on the array, the proportion of significantly (FDR q < 0.05) overmethylated sites in tumor tissue varied somewhat by tumor type ranging between 19.4 and 33.6%, whereas significantly undermethylated sites varied between 14.5 and 47% (Supplementary Figure 9, available at Carcinogenesis Online). There was little evidence that DM-arCpG sites were disproportionately undermethylated in tumor tissue (data not shown). However, IM-arCpG sites were disproportionately much more likely to be overmethylated in all tumor types, with frequencies ranging from 70.9 to 91.4% (P values from 1×10− 74 to 1×10− 163; Figure 6A). In addition, the magnitude of overmethylation in tumor tissue was significantly higher for IM-arCpG sites compared with other significantly overmethylated sites in tumors (Figure 6B). Even after excluding IM-arCpG sites in developmental genes, the proportion of IM-arCpG sites that were overmethylated in tumors remained significantly higher than for other sites on the array, suggesting that the relationship between aging and tumor overmethylation was not being driven by developmental genes alone. These data suggest that increasingly methylated aging sites are strongly associated with a broad variety of cancer and are much more likely to be methylated in tumor tissue and to have a larger increase in methylation than other sites that become overmethylated in cancer.
Fig. 6.

Proportion of increasingly methylated arCpGs that were significantly overmethylated in tumor and the corresponding magnitude of methylation changes in tumors. (A) High-confidence, increasingly methylated arCpGs were disproportionately overmethylated in all seven different types of tumors. Chi-square test (compared with array): *P < 1×10− 50, **P < 1×10− 100. (B) The magnitude of methylation difference between tumor and adjacent normal tissue for the IM-arCpGs overmethylated in tumor is significantly larger than other overmethylated CpG sites in tumors. Wilcoxon rank sum test: *P < 0.001; **P < 1×10− 20.
Discussion
Our study of blood DNA from a national sample of adult women is the largest genome-wide examination to date of age-related methylation. The panel of CpGs on the array provides basic coverage of >14 000 gene promoter regions, with 88% of all CpGs located within 750 bp of transcription start sites and 73% of CpGs located within CpG islands. We find that almost a third of the 27 578 CpGs evaluated showed association with age (at FDR q ≤ 0.05), a proportion that is similar to that reported in mice (23). We studied in more detail a subset of high-confidence arCpGs that were consistent in their associations with age across three additional public data sets where methylation was measured in blood samples. There were dramatic differences between arCpGs at island and non-island sites for the direction of methylation change with age: 80% of arCpGs at island sites were increasingly methylated with age, whereas 95% of arCpGs at non-island sites became progressively decreasingly methylated with age.
The same age-related changes found in blood also exist in multiple types of normal human tissues. Although the number of samples within each category of normal tissue was limited, seven TCGA data sets of different normal tissues showed highly significant concordance for the direction of association with age. In analysis restricted to only those CpGs that had statistically significant associations with age within each normal tissue, high-confidence arCpG sites were significantly overrepresented in each tissue. A recent study of blood and brain tissue supports the argument that blood can serve as a surrogate for other tissues when studying the effects of age on DNA methylation profiles (24). There was no evidence that the association between methylation and age was driven simply by changes in leukocyte subpopulation shifts with age as none of the published leukocyte subpopulation methylation markers (22) had significant associations with age. Thus, the methylation-aging associations that we describe appear to apply broadly across a range of human tissues.
Decreasingly methylated sites
There is a complex interplay of reversible histone modifications and of more permanent DNA methylation for short- and long-term control of gene repression (25) and in the differentiation of stem cells (26). In order to explore the biologic basis of arCpGs, we used ENCODE data to examine the underlying pattern of histone modifications at arCpG sites relative to all CpG sites represented on the array. Consistent with Mikkelsen et al. (27) and Ernst et al. (28), we found that the overall histone modification patterns differ between CpG island and non-island sites on the array and between the embryonic stem (ES) cell line HESC and a differentiated lymphoblast cell line GM12878. Although non-island CpGs had high absolute levels of methylation (66% in our data), we found that arCpGs at non-island sites became progressively demethylated with age and were more likely (compared with all non-island sites on the array) to have the permissive H3K4me1-3 histone marks in both HESC and GM12878 cells. Decreasing methylation at non-island sites has been described in a pediatric study of aging, where they comprised the majority of the age-related sites (21). Three-quarters of the high-confidence DM-arCpG non-island sites that we identified in adults had been previously identified in the pediatric population, suggesting that these decreasingly methylated sites start their demethylation early in childhood and continue through adulthood. This same feature holds for decreasingly methylated island sites, where we found that 65% of the high-confidence sites that we identified in adults had been reported previously in children. Like DM-arCpGs at non-island sites, these DM-arCpGs at island sites were frequently marked by H3k4me1 but were otherwise similar to other island sites on the array in their pattern of histone modifications.
Increasingly methylated sites, developmental genes, histone modifications and gene expression
The largest class of arCpGs that we identified (n = 449), comprising 60% of the high-confidence arCpGs, involved increasing methylation at island sites. Only 28% of the IM-arCpGs were previously identified in the pediatric population (21), which may suggest that methylation changes at these sites starts later in life. Unlike genes with DM-arCpGs where there was little evidence of pathway enrichment, genes with IM-arCpGs were strongly enriched for developmental and signaling pathways. Cells that have acquired methylation at these sites could have altered differentiation and signaling programs that may in turn affect risk of developing cancer and other age-associated diseases.
IM-arCpGs were similar to developmental genes for histone and expression characteristics. In ES cells, developmental genes are reported to often have both (bivalent) permissive H3K4me3 and repressive H3K27me3 marks, suggesting that these genes are transcriptionally repressed but poised for activation during ES cell differentiation (29,30). Many island sites on the array were bivalently marked in ES cells, but IM-arCpG sites had nearly double the frequency of these marks. We note that because almost all of island sites on the array had the permissive H3K4me3 mark in ES cells, bivalency was largely determined by the less frequent H3K27me3 mark. Two smaller cross-sectional studies of age-related methylation changes in blood using the Illumina 27K array have both noted the relationship between increasingly methylated age-related CpG sites and H3K27me3 histone marks in ES cells (11,12) and some of these previously identified age-related sites were confirmed as high-confidence sites in our analysis.
In differentiated cells, developmental genes are reported to lose H3K4me3 but to maintain the H3K27me3 mark, so that these genes continue to be transcriptionally repressed after differentiation has taken place (31). We found that IM-arCpG sites followed this same pattern of histone modifications in differentiated lymphoblasts, having a low frequency of H3K4me3, but maintaining a high frequency of H3K27me3 marks and, as might be expected based on these histone marks, we also found that genes with IM-arCpGs were transcriptionally repressed in a broad array of 78 normal differentiated human tissues.
Aging-related sites and methylation changes in tumor tissue
Most importantly, we found that a disproportionate number of IM-arCpG sites were overmethylated in all the tumor types we examined and that the magnitude of tumor overmethylation at these sites was greater than at other sites that were overmethylated in tumors. In contrast, DM-arCpG sites were not disproportionately undermethylated in tumors, suggesting that considered together as a class, these latter sites do show an association with cancer. Although many IM-arCpGs occur in developmental genes, the association with cancer remained even after sites in developmental genes were excluded, suggesting that the aging-cancer methylation association was not driven by the effect of developmental genes alone. Developmental genes and other genes marked by H3K27me3 in ES cells appear to have a higher likelihood of being overmethylated in cancer (32), representing an epigenetic switch from less stable histone-based gene repression in stem and differentiated cells to permanent DNA methylation-based gene repression in cancer cells (33). Despite already being transcriptionally repressed in both stem cells and differentiated tissues, this epigenetic switching to permanent DNA methylation repression may reflect a selection within a tumor for cells that can proliferate but are permanently blocked from differentiation (32,34,35). Genes with IM-arCpG sites may represent a special subset of genes associated with increased cancer incidence with age. The increase in methylation per decade at most arCpG sites was small, but given the large number of arCpG sites and a long human lifespan, most adult cells would be expected to be methylated at multiple IM-arCpG sites or alternatively, a small number of cells might be methylated at a large number of IM-arCpG sites.
Our study provides the strongest evidence to date that age-related methylation changes are widespread throughout the genome, that they occur across a wide array of human tissues and that the subgroup of increasingly methylated sites are associated with a broad variety of cancers. We suggest that age-related methylation at critical developmental and signaling genes results in an increasing pool of cells that have a lower threshold for neoplastic transformation that explain in part the increased incidence with age observed for many types of human cancer.
Supplementary material
Supplementary Tables 1–8 and Figures 1–9 can be found at http://carcin.oxfordjournals.org/
Funding
The Intramural Research Program of the National Institutes of Health; National Institute of Environmental Health Sciences (Z01 ES044005, Z01 ES044032, Z01 ES049033).
Supplementary Material
Acknowledgements
We thank the women who volunteered to participate in the Sister Study and Dale Sandler who leads that study. We also thank K.Adelman, S.Peddada, G.Hu, R.Jothi, P.Wade and D.Umbach for useful discussions and comments in developing the manuscript.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- BSC
bisulfite conversion
- ES
embryonic stem
- FDR
false discovery rate
- HESC
human embryonic stem cell
- TCGA
The Cancer Genome Atlas.
References
- 1. Sahyoun N.R., et al. (2001). Trends in causes of death among the elderly. Aging Trends, 1, 1–10 [DOI] [PubMed] [Google Scholar]
- 2. Christensen B.C., et al. (2009). Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet., 5, e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fernandez A.F., et al. (2012). A DNA methylation fingerprint of 1628 human samples. Genome Res., 22, 407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boks M.P., et al. (2009). The relationship of DNA methylation with age, gender and genotype in twins and healthy controls. PLoS One, 4, e6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Madrigano J., et al. (2012). Aging and epigenetics: longitudinal changes in gene-specific DNA methylation. Epigenetics, 7, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hernandez D.G., et al. (2011). Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet., 20, 1164–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Euhus D.M., et al. (2008). DNA methylation in benign breast epithelium in relation to age and breast cancer risk. Cancer Epidemiol. Biomarkers Prev., 17, 1051–1059 [DOI] [PubMed] [Google Scholar]
- 8. Issa J.P., et al. (2001). Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res., 61, 3573–3577 [PubMed] [Google Scholar]
- 9. Ahuja N., et al. (1998). Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res., 58, 5489–5494 [PubMed] [Google Scholar]
- 10. Nakagawa H., et al. (2001). Age-related hypermethylation of the 5’ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res., 61, 6991–6995 [PubMed] [Google Scholar]
- 11. Teschendorff A.E., et al. (2010). Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res., 20, 440–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rakyan V.K., et al. (2010). Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res., 20, 434–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bell J.T., et al. ; MuTHER Consortium (2012). Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet., 8, e1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weinberg C.R., et al. (2007). Using risk-based sampling to enrich cohorts for endpoints, genes, and exposures. Am. J. Epidemiol., 166, 447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu Z., et al. (2013). Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst., 105, 694–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ernst J., et al. (2011). Mapping and analysis of chromatin state dynamics in nine human cell types. Nature, 473, 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Su A.I., et al. (2004). A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. U. S. A., 101, 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Houseman E.A., et al. (2012). DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics, 13, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Accomando W.P., et al. (2012). Decreased NK cells in patients with head and neck cancer determined in archival DNA. Clin. Cancer Res., 18, 6147–6154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Storey J.D., et al. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A., 100, 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alisch R.S., et al. (2012). Age-associated DNA methylation in pediatric populations. Genome Res., 22, 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koestler D.C., et al. (2012). Peripheral blood immune cell methylation profiles are associated with nonhematopoietic cancers. Cancer Epidemiol. Biomarkers Prev., 21, 1293–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maegawa S., et al. (2010). Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res., 20, 332–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Horvath S., et al. (2012). Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol., 13, R97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cedar H., et al. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet., 10, 295–304 [DOI] [PubMed] [Google Scholar]
- 26. Meissner A., et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature, 454, 766–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mikkelsen T.S., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature, 448, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ernst J., et al. (2010). Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol., 28, 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bernstein B.E., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell, 125, 315–326 [DOI] [PubMed] [Google Scholar]
- 30. Lee T.I., et al. (2006). Control of developmental regulators by Polycomb in human embryonic stem cells. Cell, 125, 301–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ku M., et al. (2008). Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet., 4, e1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Widschwendter M., et al. (2007). Epigenetic stem cell signature in cancer. Nat. Genet., 39, 157–158 [DOI] [PubMed] [Google Scholar]
- 33. Gal-Yam E.N., et al. (2008). Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. U. S. A., 105, 12979–12984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohm J.E., et al. (2007). A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet., 39, 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schlesinger Y., et al. (2007). Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat. Genet., 39, 232–236 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.