

Abstract
Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) delivers high resolving power, mass measurement accuracy, and the capabilities for unambiguously sequencing by a top-down MS approach. Here, we report isotopic resolution of a 158 kDa protein complex - tetrameric aldolase with an average absolute deviation of 0.36 ppm and an average resolving power of ~520,000 at m/z 6033 for the 26+ charge state in magnitude mode. Phase correction further improves the resolving power and average absolute deviation by 1.3 fold. Furthermore, native top-down electron capture dissociation (ECD) enables the sequencing of 149 C-terminal amino acid (AA) residues out of 463 total AAs. Combining the data from top-down MS of native and denatured aldolase complexes, a total of 58% of the backbone cleavages efficiency is achieved. The observation of complementary product ion pairs confirms the correctness of the sequence and also the accuracy of the mass fitting of the isotopic distribution of the aldolase tetramer. Top-down MS of the native protein provides complementary sequence information to top-down ECD and CAD MS of the denatured protein. Moreover, native top-down ECD of aldolase tetramer reveals that ECD fragmentation is not limited only to the flexible regions of protein complexes and that regions located on the surface topology are prone to ECD cleavage.
INTRODUCTION
“Native” mass spectrometry (MS) is an emerging technique that has been successfully used to characterize intact, noncovalently-bound protein complexes, providing stoichiometry and structural information that is complementary to data supplied by conventional structural biology techniques.1–3 To confidently characterize protein complexes, electrospray ionization (ESI)-MS measurements acquired with isotopic resolving power (RP) and high mass accuracy and capabilities for deriving primary structure, i.e., sequence, information would be ideal. Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR MS) is prominent for its superior resolving power and mass accuracy and its utility for tandem MS (MS/MS) with a variety of fragmentation techniques; FT-ICR MS is noted for characterizating posttranslational modifications (PTMs) and protein-ligand and protein-protein interactions.4–9 However, it remains challenging to isotopically resolving large biomolecules over 100 kDa due to sample heterogeneity, cation/solvent/buffer addition, space charge effects, and electric and magnetic field inhomogeneity (for FT-ICR).10–13 Unit mass resolution has been achieved for a few denatured proteins, including a 112 kDa protein with 3 Da mass error using a 9.4 T FT-ICR MS,14 a 115 kDa protein by a 7 T instrument with a mass error of 5 ppm,4 and a 148 kDa protein with a mass error of 1 Da by a 9.4 T FTMS.10
Compared to denatured proteins, it is more difficult to achieve isotopic resolution for inherently lower charged (and thus, higher m/z) native protein complexes because (1) the peak height is proportional to its charge state, (2) the resolving power is inversely proportional to mass-to-charge ratio for FT-ICR MS, and (3) the broader isotope distribution of large biomolecules reduces overall signal-to-noise ratio.15 However, the introduction of a new FT-ICR analyzer cell – the ParaCell, by Nikolaev and coworkers has significantly increased the resolving power of FT-ICR MS.16, 17 By dynamically harmonizing the electric field potential at any radius of cyclotron motion in the entire cell volume, a resolving power of 39 M has been achieved for the alkaloid, resperine (m/z 609), using a 7 T system.18 In addition, a few native protein complexes, including enolase dimer (93 kDa, RP ~ 800,000 at m/z 4250), alcohol dehydrogenase tetramer (147 kDa, RP ~ 500,000 at m/z 5465), and enolase tetramer (186 kDa), have been isotopically resolved with a 12 T FT-ICR system with the new ICR cell.18 Although Mitchell and Smith reported that cyclotron phase locking due to Coulombic interactions limits the highest mass that unit mass resolution can be achieved by FT-ICR MS (Mmax ≈ 1×104B, where B is magnetic field strength),19 the ParaCell has made it significantly easier and promising to measure high resolution mass spectra for large native protein complexes.
Another advantage of FT-ICR MS is its capability to employ a variety of fragmentation techniques, especially for top-down MS analysis, including in-source dissociation (ISD), collisionally activated dissociation (CAD), electron transfer dissociation (ETD), electron capture dissociation (ECD), electron detachment dissociation (EDD), and infrared multiphoton dissociation (IRMPD). Therefore, for the characterization of large proteins and protein complexes, beyond accurately measuring molecular masses (and thereby providing information on complex stoichiometry), further structural information (e.g., amino acid sequence, point mutations, metal binding sites, identification and quantification of subunit variants) can be obtained.5, 6, 8
Here, we report sub-parts-per-million (ppm) mass accuracy with isotopic resolution of a 158 kDa protein complex (aldolase homotetramer) using FT-ICR MS with a dynamically harmonized cell. Top-down MS analyses confirm the sequence and also the accuracy of the mass fitting of the isotopic distribution of the protein tetramer. In addition, native top-down ECD analysis of the protein complex reveals that ECD fragmentation is not limited to only the flexible regions of protein structure, and it also demonstrates that native top-down ECD provides complementary sequence information to top-down ECD and CAD of denatured proteins.
EXPERIMENTAL METHODS
High resolution measurements for the native protein solutions were acquired using a 12 T Bruker solariX™ XR FT-ICR MS (Bruker Daltonics, Bremen, Germany) with the ParaCell. Other experiments were performed using a 15 T Bruker solariX FT-ICR MS with an infinity cell. (See Supporting Information for more details.)
RESULTS AND DISCUSSION
Isotopically Resolved Aldolase Tetramer (~ 160 kDa)
Fructose-1,6-bisphosphate aldolase (aldolase; EC 4.1.2.13) catalyzes the cleavage of fructose 1,6-bisphosphate to yield dihydroxyacetone phosphate (DHAP) and glyceraldehyde 3-phosphate in glycolysis. Aldolases are homotetrameric complexes, with each subunit having an architecture with a (α/β)8-barrel fold. The aldolase subunit from rabbit muscle is composed of 363 amino acids and an average mass of ~ 39211 Da, and its corresponding homotetramer complex has a calculated mass of 156,845 Da.
Figure 1A shows the MS spectrum of aldolase tertramer under native nanoESI conditions using a 12 T FT-ICR MS with the ParaCell analyzer. A relatively narrow charge state distribution (29+ to 25+) was observed between m/z 5400 – 6300. Five isotopic “beats” were observed for the time-domain acquisition period (36 s) with a beat period of 5.1 s (Figure 1B). The relatively long beat period makes the achievement of ultra-high resolving power very challenging because of space charge effects, and electric and magnetic field inhomogeneity can induce frequency shifts during a long transient period, leading to signal decay.11, 12 Using our 15 T FT-ICR MS with an infinity cell, only 3 isotopic “beats” were observed for measurements of the isolated 27+ charge state ions at m/z 5820 (isolated with a mass window of ± 50 Da to minimize ion cloud interactions), which is insufficient for achieving isotopic resolution (Figure S-1). The ParaCell significantly improves the ability to collect long transients, however, by dynamically harmonizing the electric field potential at any radius of cyclotron motion in the cell, thus making isotopic resolution of larger molecules achievable.16, 20
Figure 1.

Native ESI-mass spectrum of aldolase tetramer with a 12 T FT-ICR MS with a ParaCell. A) Full mass spectrum of aldolase tetramer, B) corresponding time-domain transient acquired for aldolase tetramer, C) the expanded MS spectrum of the 26+ charge state of aldolase tetramer with the theoretical isotopic distribution for the 26+ non-adducted non-adducted aldolase tetramer [(C1733H2773N489O525S11)4+26H]26+ in red, D) expanded spectrum of Figure 1C, and E) expanded mass spectra in magnitude mode, and F) in absorption mode.
Figure 1C shows the expanded mass spectrum for the 26+ charge state ions and it can be seen that the analyte peaks span over 20–100 Da, likely due to a combination of sample heterogeneity, neutral losses, and cation/buffer/solvent adduction, which makes the isotopic mass fitting of the aldolase tetramer challenging. To confirm the accurate molecular weight, aldolase was denatured with acetonitrile-H2O-formic acid (49.95/49.95/0.10) (See Figure S-2) and measured by nanoESI-MS. Taking the 26+ charged aldolase monomer as an example, the peak at m/z 1509 fits the theoretical mass of aldolase A (from rabbit muscle) with an mass error of 0.7 ppm and a series of sodium adduct peaks were also observed at higher m/z. Top-down CAD and ECD experiments were further performed to confirm the sequence of aldolase from rabbit (Oryctolagus cuniculus) (UniProtKB/Swiss-Prot: P00883/ALDOA_RABIT) (Figure S-3). The observation of a series of b/y and c/z• product ions, particularly the complementary ion pairs, b18315+/y18111+ (Figure S-3B inset) and c21714+/z14611+• (Figure S-3A inset), confirms that there are no PTMs or mutations present (at least above 5% relative abundance) and the elementary composition of the monomer is C1733H2773N489O525S11. The isotopic distribution of the 26+ non-adducted aldolase tetramer [(C1733H2773N489O525S11)4+26H]26+ was then simulated to fit the experimental result (Figure 1C and 1D). As the molecular mass increases, the isotopic distribution widens and the peak intensity of the monoisotope (i.e., all light isotopes) is reduced;21 for the ~158 kDa aldolase tetramer, the monoisotopic peak is no longer observable. Therefore, the mass of the peak with the highest intensity is reported here, which is off by 0.08189 Da compared to the calculation of the theoretical mass. An average absolute deviation of 0.36 ppm (Table S-1) was achieved for the entire isotopic peak distribution of non-adducted aldolase tetramer.
It has been demonstrated that phase correction can greatly improve resolving power, mass accuracy, and signal-to-noise for FT-ICR MS.22–24 By correcting the phase using Bruker solariX™ XR software, an average mass resolving power of ~680,000 (Figure 1F) was achieved for the 26+ ions at m/z 6033 in absorption mode, which is 1.3 fold higher than the resolving power in magnitude mode (Figure 1E). In addition, peak shape becomes narrower and smoother after phase correction and the average absolute mass deviation improved by 1.3 fold (to 0.28 ppm).
Top-Down ECD of Aldolase Complex
One of the unique features of ECD is the preservation of labile bonds, including bonds associated with PTMs and noncovalent interactions, while cleaving backbone N-Cα bonds.25 Thus, potentially ECD is a powerful tool to locate the noncovalent binding sites or interfaces of metal, ligand, or protein to their protein target.6, 8, 26–28 Recently, Gross and coworkers proposed that the regions of a protein complex that fragment by native top-down ECD correlate with the “B-factor” of that protein, namely ECD products originate from the flexible regions of a protein.6
The entire charge state envelope was subjected to ECD without prior ion selection due to the upper mass isolation limit (m/z ~6000) of the mass selective quadrupole. Skimmer 1 voltage was varied to pre-activate ions and to improve ECD fragmentation efficiency. With a skimmer potential of 90 V, native ECD of aldolase tetramer starts to induce fragments from the AA 280–360 region (see Figure S-4). Further increasing the skimmer potential to 120 V improves the ECD backbone cleavage efficiency. However, by increasing the skimmer potential to 150 V or even higher, although more fragments from the middle region of the sequence are generated, many of the fragments from AA 280–330 disappear, which suggests that a conformational change of the aldolase tetramer may have been induced. Figure 2A shows the top-down ECD mass spectrum of the aldolase tetramer with the skimmer potential of 120 V. Products ions resulting from cleavage of N-Cα bonds are found at m/z 3500 and below, and are assigned with mass errors within ± 5 ppm (Figure S-5). No b/y product ions were induced during the pre-activation and most of the fragment ions are z•-ions from amino acid (AA) residues 211–363, except for two c-ions (c7 and c22) (shown in cyan, Figure 2B).
Figure 2.

A) Native top-down ECD mass spectrum of the aldolase tetramer and B) backbone cleavage coverage of aldolase by top-down MS/MS. c/z• ions from the top-down ECD of the 26+ charge state of denatured aldolase monomer are in red, b/y ions from the top-down CAD of the 26+ charge state of denatured aldolase are in green, and the c/z• ions from the native top-down ECD of aldolase tetramer (entire charge state envelope without isolation) are in cyan.
Native top-down CAD and ISD were also performed for the aldolase tetramer; dissociation of the tetramer to yield monomer was observed in both approaches and no sequence information was obtained. The cleavage sites from ECD (colored in red) and CAD (colored in green) of the denatured aldolase monomer (26+) are overlaid with the native ECD results for aldolase tetramer (Figure 2B). As shown in Figure 2B, in contrast to the limited number of c-ion fragments observed in the ECD of aldolase tetramer, ECD of denatured aldolase monomer induces extensive c-ion fragments in the N-terminal region and enables the assignment of first 156 N-terminal AA residues. Surprisingly, the number of z•-ions observed from ECD of the denatured aldolase monomer is much less compared to the ECD of the native aldolase tetramer. Although it may be possible that the z•-ions may undergo secondary fragmentation due to excess available energy, electrons, or long ion-electron reaction times during the ECD experiment, ECD experiments with reduced reaction time and bias voltages were performed and the results argue against this assumption. Overall, 58% of the total number of backbone bonds are cleaved from combining top-down MS of native aldolase complex and denatured aldolase monomer (20% for native ECD of aldolase tetramer, 37% for ECD of denatured aldolase, and 5% for CAD of denatured aldolase).
The three dimensional structure of the aldolase tetramer is shown in Figure 3. To compare the flexibility of the structure to the data from ECD of the aldolase tetramer, one of the subunits (B-chain) is presented as B-factor putty and the D-chain is shown with its native ECD backbone cleavage regions colored in red. The remaining A- and C-chains are shown in grey. Although the C-terminal region (AA 340–363) of each subunit is highly flexible based on the crystallography B-factor (see B-chain in Figure 3A), only 4 out of 75 backbone cleavage sites are from the AA 340–363 region. Instead, the native ECD fragments largely originate from surface regions of the protein structure (see D-chain in Figure 3A). The N-terminal regions are not directly involved in the interfaces between subunits, but they are located in regions that are partially buried, which is consistent with the limited c-ions observed. To better show the native ECD backbone cleavage regions, the D-chain is rotated 90 degrees clockwise (Figure 3B). It is clear that, although protein structure flexibility might play a role in the native top-down ECD fragmentation pattern, for aldolase the ECD cleavage sites are not limited to the flexible region. In addition, backbone cleavage regions from CAD (yellow) and ECD (cyan) of denatured aldolase are complementary with the native ECD results.
Figure 3.
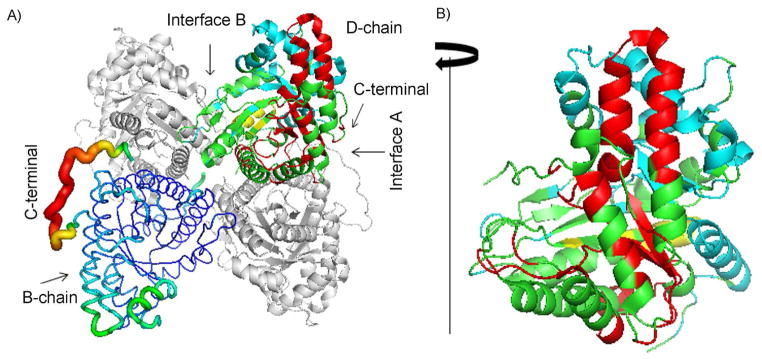
A) Structure of tetrameric aldolase (1ZAH)29. A- and C-chains are shown as grey ribbons, the B-chain is shown in B-factor putty, and the D-chain is in cartoon with native ECD cleavage sites colored in red, CAD cleavage sites of denatured aldolase in yellow, and ECD cleavage sites of the N-terminal region from ECD of denatured aldolase in cyan. B) The D-chain is rotated 90 degrees clockwise to show the outer surface region of the subunit structure.
In summary, unit mass resolution measurements for a 158 kDa protein complex under native MS with high mass accuracy are demonstrated. The results demonstrate that with the superior high resolving power, accurate mass accuracy, and versatile fragmentation techniques, rich information related to the three dimensional structure of protein complexes can be gathered by FT-ICR MS.
Supplementary Material
Acknowledgments
Support from the US National Institutes of Health (R01 GM103479 and S10 RR028893) and the US Department of Energy (UCLA Institute of Genomics and Proteomics; DE-FC03-02ER63421) are acknowledged.
Footnotes
Experimental details and additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Heck AJR. Nat Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- 2.Lemaitre JM, Danis E, Pasero P, Vassetzky Y, Méchali M. Cell. 2005;123:787–801. doi: 10.1016/j.cell.2005.08.045. [DOI] [PubMed] [Google Scholar]
- 3.Hilton GR, Benesch JLP. J Royal Soc Interface. 2012;9:801–816. doi: 10.1098/rsif.2011.0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ge Y, Rybakova IN, Xu Q, Moss RL. Proc Natl Acad Sci U S A. 2009;106:12658–12663. doi: 10.1073/pnas.0813369106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao Y, Valeja SG, Rouse JC, Hendrickson CL, Marshall AG. Anal Chem. 2013;85:4239–4246. doi: 10.1021/ac303525n. [DOI] [PubMed] [Google Scholar]
- 6.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Anal Chem. 2011;83:5598–5606. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geels RBJ, van der Vies SM, Heck AJR, Heeren RMA. Anal Chem. 2006;78:7191–7196. doi: 10.1021/ac060960p. [DOI] [PubMed] [Google Scholar]
- 8.Xie Y, Zhang J, Yin S, Loo JA. J Am Chem Soc. 2006;128:14432–14433. doi: 10.1021/ja063197p. [DOI] [PubMed] [Google Scholar]
- 9.Clarke D, Murray E, Hupp T, Mackay CL, Langridge-Smith PR. J Am Soc Mass Spectrom. 2011;22:1432–1440. doi: 10.1007/s13361-011-0155-3. [DOI] [PubMed] [Google Scholar]
- 10.Valeja SG, Kaiser NK, Xian F, Hendrickson CL, Rouse JC, Marshall AG. Anal Chem. 2011;83:8391–8395. doi: 10.1021/ac202429c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boldin IA, Nikolaev EN. Rapid Commun Mass Spectrom. 2009;23:3213–3219. doi: 10.1002/rcm.4231. [DOI] [PubMed] [Google Scholar]
- 12.Boldin IA, Nikolaev EN. Eur J Mass Spectrom. 2008;14:1–5. doi: 10.1255/ejms.904. [DOI] [PubMed] [Google Scholar]
- 13.Nakata MT, Hart G, Peterson B. J Am Soc Mass Spectrom. 2010;21:1712–1719. doi: 10.1016/j.jasms.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Kelleher NL, Senko MW, Siegel MM, McLafferty FW. J Am Soc Mass Spectrom. 1997;8:380–383. [Google Scholar]
- 15.Marshall AG, Hendrickson CL, Jackson GS. Mass Spectrom Rev. 1998;17:1–35. doi: 10.1002/(SICI)1098-2787(1998)17:1<1::AID-MAS1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 16.Boldin IA, Nikolaev EN. Rapid Commun Mass Spectrom. 2011;25:122–126. doi: 10.1002/rcm.4838. [DOI] [PubMed] [Google Scholar]
- 17.Nikolaev EN, Boldin IA, Jertz R, Baykut G. J Am Soc Mass Spectrom. 2011;22:1125–1133. doi: 10.1007/s13361-011-0125-9. [DOI] [PubMed] [Google Scholar]
- 18.Nikolaev EN, Vladimirov GN, Jertz R, Baykut G. Mass Spectrometry. 2013;2:1–6. doi: 10.5702/massspectrometry.S0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell DW, Smith RD. J Mass Spectrom. 1996;31:771–790. [Google Scholar]
- 20.Nikolaev E, Boldin I, Jertz R, Baykut G. J Am Soc Mass Spectrom. 2011;22:1125–1133. doi: 10.1007/s13361-011-0125-9. [DOI] [PubMed] [Google Scholar]
- 21.Yergey J, Heller D, Hansen G, Cotter RJ, Fenselau C. Anal Chem. 1983;55:353–356. [Google Scholar]
- 22.Qi Y, Barrow MP, Li H, Meier JE, Van Orden SL, Thompson CJ, O’Connor PB. Anal Chem. 2012;84:2923–2929. doi: 10.1021/ac3000122. [DOI] [PubMed] [Google Scholar]
- 23.Qi Y, Thompson CJ, van Orden SL, O’Connor PB. J Am Soc Mass Spectrom. 2011;22:138–147. doi: 10.1007/s13361-010-0006-7. [DOI] [PubMed] [Google Scholar]
- 24.Xian F, Hendrickson CL, Blakney GT, Beu SC, Marshall AG. Anal Chem. 2010;82:8807–8812. doi: 10.1021/ac101091w. [DOI] [PubMed] [Google Scholar]
- 25.Zubarev RA. Mass Spectrom Rev. 2003;22:57–77. doi: 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- 26.Wen J, Zhang H, Gross ML, Blankenship RE. Biochemistry. 2011;50:3502–3511. doi: 10.1021/bi200239k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yin S, Xie Y, Loo JA. J Am Soc Mass Spectrom. 2008;19:1199–1208. doi: 10.1016/j.jasms.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin S, Loo J. J Am Soc Mass Spectrom. 2010;21:899–907. doi: 10.1016/j.jasms.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 29.St-Jean M, Lafrance-Vanasse J, Liotard B, Sygusch J. J Biol Chem. 2005;280:27262–27270. doi: 10.1074/jbc.M502413200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.