

Abstract
Rationale
1) Despite intense interest in strategies to predict which kinase inhibitor (KI) cancer therapeutics may be associated with cardiotoxicity, current approaches are inadequate. 2) Sorafenib is a KI of concern since it inhibits growth factor receptors and Raf-1/B-Raf, kinases that are upstream of ERKs and signal cardiomyocyte survival in the setting of stress.
Objectives
1) Explore the potential use of zebrafish as a pre-clinical model to predict cardiotoxicity. 2) Determine whether sorafenib has associated cardiotoxicity and, if so, define the mechanisms.
Methods and Results
We find that the zebrafish model is readily able to discriminate a KI with little or no cardiotoxicity (gefitinib) from one with demonstrated cardiotoxicity (sunitinib). Sorafenib, like sunitinib, leads to cardiomyocyte apoptosis, a reduction in total myocyte number per heart, contractile dysfunction and ventricular dilatation in zebrafish. In cultured rat cardiomyocytes, sorafenib induces cell death. This can be rescued by adenovirus-mediated gene transfer of constitutively active MEK1 which restores ERK activity even in the presence of sorafenib. While growth factor-induced activation of ERKs requires Raf, α-adrenergic agonist-induced activation of ERKs does not. Consequently, activation of α-adrenergic signaling markedly decreases sorafenib-induced cell death. Consistent with these in vitro data, inhibition of α-adrenergic signaling with the receptor antagonist prazosin worsens sorafenib-induced cardiomyopathy in zebrafish.
Conclusions
1) Zebrafish may be a valuable pre-clinical tool to predict cardiotoxicity. 2) The α-adrenergic signaling pathway is an important modulator of sorafenib cardiotoxicity in vitro and in vivo and appears to act via a here-to-fore unrecognized signaling pathway downstream of α-adrenergic activation that bypasses Raf to activate ERKs.
Keywords: zebrafish, kinase inhibitors, cancer, cardiotoxicity, ERK
Introduction
Cardiotoxicity of cancer therapeutics has become a significant problem and will likely continue to be so with the explosion in drugs targeting kinases that are mutated or over-expressed in cancer. Cardiotoxicity with these agents will continue to plague drug development until reliable pre-clinical screening strategies are developed. Unfortunately, at this point, there are few if any pre-clinical models that can accurately predict cardiotoxicity, leading on occasion to unfortunate surprises1, 2. Cell lines, which are typically non-contractile and glycolytic, bear little relationship to cardiomyocytes and do not appear to be reliable models for predicting cardiotoxicity. In the future, induced pluripotent stem (iPS) cell-derived cardiomyocytes from patients with demonstrated cardiotoxicity might provide insights into mechanisms of cardiotoxicity, but this is not a practical screening approach at the present time. Primary cardiomyocytes have been used successfully to examine mechanisms of toxicity, but the general consensus is that a reliable in vivo model is needed. Rodents have been used for this purpose but can be insensitive, particularly when endpoints are based on measurements of left ventricular contractile function2. This may be due, at least in part, to the ability of rodents to compensate for loss of myocytes by recruiting compensatory mechanisms, and to the fact that rodents, unlike the typical cancer patient, have no co-morbidities (e.g. coronary artery disease or hypertension). Indeed we have found that even with agents known to have associated cardiotoxicity (e.g. sunitinib), LV function can be maintained in rodents, even in the setting of an additional stressor (i.e. moderate hypertension)2, 3. Transmission electron microscope (TEM) may be the most sensitive technique but quantification of abnormalities on TEM is very difficult.
Over the past decade, the zebrafish (Danio rerio) has gained popularity as a model organism for human disease research. Zebrafish possess several advantages over other models for cardiovascular research4, 5. Most importantly, they have a closed cardiovascular system that can readily be studied during development because the fish are transparent. In addition, techniques for detailed and quantitative phenotyping of zebrafish heart mutants are available. Since zebrafish can survive in the absence of cardiac output and in the presence of major vascular defects for several days, abnormalities can be studied that would be rapidly fatal in mammals. Finally, zebrafish may be useful for cardiovascular drug discovery since the fish are readily permeable to small molecule drugs when they are added to incubation medium6, 7.
Given the above, we asked whether zebrafish might serve as a model to predict cardiotoxicity of small molecule kinase inhibitors. The zebrafish kinome is very similar to human, especially in the ATP pocket where most inhibitors interact with the kinase8. Herein we employ 1) morphometric analysis, including evidence of pericardial edema, a marker of cardiac dysfunction in fish embryos, 2) staining of whole fish for cardiomyocyte apoptosis, 3) determination of total cardiomyocyte number per heart, employing a fish in which cardiomyocytes are readily identified in vivo, and 4) videomicroscopy to quantify wall thickness and contractile function of the fish. We employ three KIs: one with well-documented cardiotoxicity (sunitinib)2, 9, 10, one with minimal to no signal for LV dysfunction or heart failure (gefitinib), and one with questionable cardiotoxicity (sorafenib). To our knowledge, there are only two studies that have examined sorafenib cardiotoxicity. One reported significant drug-related cardiac abnormalities but the vast majority of these were ECG abnormalities or minor increases in cardiac injury bio-markers (creatine kinase-MB or troponin T)10. Left ventricular ejection fraction (LVEF) was found to be below the lower limit of normal at the time of “cardiac events” in 21.4% of patients but the significance of this is entirely unclear since baseline LVEF was not determined. The only study that examined baseline and serial LVEF determinations in patients on sorafenib reported that mean LVEF declined only 0.8 to 1.2 EF percent11. The authors concluded that the effects on LVEF were modest and were unlikely to be of clinical significance, but 13% of patients experienced significant declines in EF (≥ 10 EF points).
Herein, we ask whether the zebrafish model can 1) discriminate cardiotoxicity vs. none (comparing sunitinib vs. gefitinib) and 2) predict whether sorafenib is likely to have significant cardiotoxicity. Our findings demonstrate feasibility and support the ability of the model to predict cardiotoxicity. We support these studies with studies in isolated, contracting neonatal rat cardiomyocytes as a confirmatory approach. We then identify inhibition of Raf-1/B-Raf as a key mechanism of cardiotoxicity of sorafenib by partially rescuing sorafenib-induced cardiomyocyte death with adenovirus-mediated gene transfer of a constitutively active MEK1, a kinase immediately downstream of Raf-1/B-Raf and upstream of ERK1/2 in the ERK/MAP kinase cascade. We then identify a novel Raf-independent pathway from α-adrenergic receptors to ERK activation that appears to play a role in limiting cardiotoxicity of sorafenib. Finally, we demonstrate the importance of this α-adrenergic pathway in the fish in vivo in limiting sorafenib cardiotoxicity. We believe these studies identify a strategy to screen for cardiotoxicity and describe an approach that can be used to both confirm cardiotoxicity in mammalian cardiomyocytes and to identify the key pathways mediating cardiotoxicity of specific agents in NRVMs in culture and in fish in vivo.
Methods
Zebrafish use and handling at the Thomas Jefferson University (TJU) Zebrafish Facility was approved by the Institutional Animal Care and Use Committee at TJU. Wild-type or transgenic adult fish lines were mated in embryo collection tanks. Viable embryos were washed with embryo medium (EM) and sorted (30 embryos per 60-mm dish in 10 ml EM) at the one- to two-cell developmental stage (approximately 0.5–1 h post fertilization [hpf]), and then were maintained under normoxic conditions at 28.5°C. EM was changed after dechorionation at 24–48hpf and again at 72–96 hpf. For zebrafish to be examined by videomicroscopy or fluorescence microscopy, embryo medium containing 1-phenyl-2-thiourea (PTU, 50 μM) was used to suppress pigmentation. Zebrafish were treated with the various KIs at the concentrations and for the times noted in the figure legends. Unless otherwise noted, treatment occurred at 2 dpf. Toxicity analyses were conducted by monitoring survival and morphology of zebrafish for up to 7 dpf.
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Results
Sorafenib and sunitinib are cardiotoxic in the zebrafish model
To examine whether sorafenib or sunitinib induced cardiotoxicity in zebrafish, we treated zebrafish at 2dpf with various concentrations of sorafenib, sunitinib and, as negative controls, gefitinib or vehicle. Treatment with sorafenib and sunitinib, but not gefitinib, increased mortality in the fish but only at a high concentration (5 μM; Fig. 1A). Both agents (but not gefitinib) at 5 μM also induced noticeable body malformations that included a curved body shape and un-inflated swim bladder. Pericardial edema, a marker of cardiac dysfunction in the fish, was particularly pronounced (Supplemental Fig. I).
Figure 1. Sorafenib and sunitinib are cardiotoxic in zebrafish.

A. Survival rate in fish treated with vehicle or increasing concentrations of KIs.
Fish at 2 dpf were placed in embryo media (EM), EM plus vehicle (Veh; DMSO), or EM plus sorafenib (Sor), sunitinib (Sun), or gefitinib (Gef) at increasing concentrations of drug (0.5 μM, 1 μM, 2 μM, or 5 μM). At 7 dpf the number of viable zebrafish were counted and percent survival was determined. The data are based on five independent experiments with n=30 fish per treatment group. *p<0.05 vs. EM and vs. Veh.
B. Lack of noticeable body malformations or pericardial edema in fish at 7 dpf treated as above with vehicle or 0.5 μM sorafenib, sunitinib, or gefitinib.
Based on previous reports examining pharmacokinetics in phase I clinical trials11, 12, the maximum plasma concentration of sorafenib after a 28-day cycle in patients receiving 400mg two times per day was 8.5 μM and the trough concentration was 6.4 μM. As for sunitinib, trough levels have been reported to be in the range of 125–250nM13–16. However, given the large volume of distribution of sunitinib (~2230 L; www.pfizer.com/files/products/uspi_sutent.pdf), the tissue levels are predicted to be in the 1–3 μM range. Thus, in the following experiments we chose what we believed to be a conservative concentration of 0.5 μM. At this concentration we did not observe any obvious body malformations or pericardial edema in the fish (Fig. 1B), nor did we observe any malformations of the vasculature in KI-treated TG: VEGFR2-GRCFP transgenic zebrafish (Supplemental Fig. II).
We next employed videomicroscopy to quantify cardiac function in the drug-treated zebrafish heart. A representative cardiac image at end-systole in a fish at 5 dpf that had been treated with sorafenib (0.5 μM) at 2dpf is shown in Fig. 2. We quantified end-diastolic dimension (EDD) and end-systolic dimension (ESD) in both long and short axes, and ventricular wall thickness in long axis. From these values we also calculated fractional shortening (FS) as a measure of contractile function. We found that sorafenib and sunitinib, but not gefitinib (all at 0.5 μM), significantly reduced ventricular wall thickness (p<0.01 for sorafenib and p<0.001 for sunitinib vs. vehicle or gefitinib) (Table 1). Contractile function, as expressed by fractional shortening, was markedly reduced by both drugs (p<0.0001 for sorafenib and sunitinib vs. vehicle or gefitinib) (Table 1). Cardiac dilatation, as determined by EDD, was also pronounced in sunitinib-treated fish (p<0.01 vs. vehicle or gefitinib; Table 1). In summary, both sorafenib and sunitinib, but not gefitinib, led to reduced cardiac wall thickness, ventricular dilatation, and markedly impaired contractile function.
Figure 2. Imaging the zebrafish heart.

A representative heart image at end-systole in a fish at 5 dpf that had been treated with 0.5 μM sorafenib at 2 dpf. The ventricle is outlined by a solid line; the heart and outflow tract (pointed by solid arrow) together are delineated with a dashed line.
Table 1.
Videomicroscopic measurements in fish 5dpf that were treated at 2dpf with vehicle or 0.5 μM KIs.
| n | Long-axis
|
Short-axis
|
||||||
|---|---|---|---|---|---|---|---|---|
| EDD | ESD | FS=(EDD-ESD)/EDD | Wall thickness | EDD | ESD | FS=(EDD-ESD)/EDD | ||
| vehicle | 6 | 98.7 ±1.8 | 40.7±2.9 | 0.59±0.03 | 40.4±1.0 | 62.4±1.9 | 24.0±1.9 | 0.61±0.04 |
|
| ||||||||
| gefitinib | 6 | 98.0±5.0 | 39.2±1.3 | 0.59±0.03 | 39.9±3.5 | 55.5±2.5 | 22.4±1.6 | 0.59±0.03 |
|
| ||||||||
| sorafenib | 12 | 102.9±5.4 | 69.7±6.4* | 0.33±0.03* | 27.1±2.2* | 61.3±3.7 | 37.7±2.8* | 0.39±0.02* |
|
| ||||||||
| sunitinib | 12 | 117.1±4.1*# | 78.6±4.5* | 0.33±0.03* | 23.9±1.0* | 60.8±2.5 | 42.6±2.1* | 0.30±0.03*# |
Ventricular wall thickness along the long axis, end-diastolic dimension (EDD), and end-systolic dimension (ESD) in both long and short axes were quantified and expressed as arbitrary units. Fractional shortening (FS) was then calculated as a measure of contractile function. For all video images, 100 arbitrary units equal 0.268 mm.
p<0.01 vs. vehicle or gefitinib;
p<0.05 vs. sorafenib.
Sorafenib induces cell death in NRVMs and fish in vivo
We then explored possible mechanisms underlying the contractile dysfunction. Mechanisms of sunitinib-induced cardiotoxicity have been the subject of prior reports2, 3 and, therefore, those studies were not repeated here. To elucidate the mechanisms underlying the cardiotoxicity of sorafenib, we employed isolated NRVMs. We found that sorafenib dose-dependently induced cell death in NRVMs as determined by two different approaches, TUNEL staining and the ToxiLight assay (Fig 3).
Figure 3. Sorafenib dose-dependently induces cell death in NRVMs.
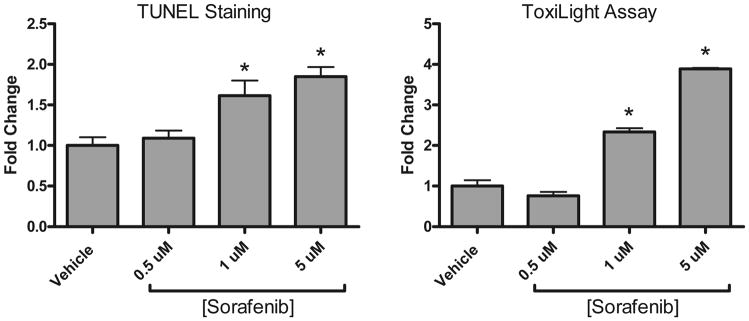
Cells were plated in 8-well chamber slides and treated with vehicle or 0.5–5 μM sorafenib for 24 hours. Apoptosis was assessed with terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. Cell death was also assessed based on the release of adenylate kinase from damaged cells into culture supernatants (ToxiLight).
*p<0.05 vs. vehicle; n=3 independent experiments
We then asked whether the death of NRVMs translated to findings in the fish in vivo by asking whether the thinning of the ventricular walls following sorafenib treatment might be due in part to cardiomyocyte loss. To address this question, we employed Cmlc2::dsRed-nuc transgenic fish in which the cardiomyocyte nucleus is red, allowing quantification of total cardiomyocyte number. We found a highly significant reduction in number of ventricular myocytes in the sorafenib-treated fish at all doses (Fig 4A).
Figure 4. Sorafenib induces ventricular cardiomyocyte loss and cardiomyocyte apoptosis in zebrafish.


A. Upper panel: a representative image of the heart taken in a Cmlc2::dsRed-nuc transgenic fish at 4 dpf. Lower panel: Cmlc2::dsRed-nuc fish were treated with vehicle, gefitinib, sunitinib or increasing concentrations of sorafenib at 2 dpf. Images of the heart were taken at 4 dpf and the number of ventricular myocytes per heart were quantified.
*p<0.01 vs. vehicle; n=12–16 fish per condition
B. Representative Grayscale images of acridine orange (AO)-positive and AO-negative ventricles in fish at 3dpf that had been treated at 2dpf with vehicle or 0.5 μM KIs. The ventricle is outlined with ovals. AO positive cells were observed as “beating” green dots in the ventricular myocardium. The percentage of AO-positive ventricles is quantified in the inset graph.
**p<0.01 and *p<0.05 vs. vehicle; #p<0.05 vs gefitinib; n=153–167 fish per condition
We next asked, based on the findings in NRVMs exposed to sorafenib, if this reduction in cardiomyocyte number might be due in part to apoptosis. We employed acridine orange (AO) staining, a widely used method to detect apoptosis in zebrafish17–19. We treated fish with 0.5 μM drug at 2 dpf and then stained whole fish at 3 dpf with the vital dye acridine orange (AO). AO positive cells were observed in the myocardium of the fish. Representative images of AO-positive ventricles are shown in Fig. 4B. With sorafenib treatment, 38.9% of fish demonstrated AO-positive myocardium, and this was significantly higher than the percent of AO-positive hearts treated with vehicle (23.1%) or gefitinib (26.1%) (p<0.01 and 0.05, respectively; Fig. 4B inset graph). Of note, the percent of fish with AO-positive hearts treated with sunitinib (32.2%) reached statistical significance when compared to vehicle (p=0.046) but not when compared to gefitinib (p=0.14; Fig. 4B inset graph). These data suggest that apoptosis plays a role in sorafenib (and sunitinib)-mediated cardiotoxicity.
Molecular mechanisms of sorafenib-induced cell death
We next examined signaling pathways regulating sorafenib cardiotoxicity. In a quantitative analysis of kinase inhibitor selectivity across a panel of 317 kinases, sorafenib binds to more than 15 kinases with nanomolar potency20. This makes it difficult to pinpoint the specific target(s) mediating toxicity. That said, Raf-1 (and B-Raf) are targets of sorafenib, and have been implicated in survival signaling in the heart21 (though controversies remain22). Therefore, we asked whether sorafenib-induced cardiotoxicity is mediated, at least in part, by the inhibition of Raf/ERK signaling. We first confirmed that sorafenib inhibited ERK activation in zebrafish (Fig. 5A). We then confirmed that sorafenib inhibited ERK activation in NRVMs (Fig. 5B). Treatment with sorafenib led to a persistent decrease in the basal level of pERK and this was dose-dependent, consistent with inhibition of Raf by sorafenib. Furthermore, pretreatment with sorafenib blocked ERK phosphorylation induced by growth factors such as IGF-1 and insulin, as well as oxidative stress induced by hydrogen peroxide (H2O2) (Fig. 5C). Surprisingly, sorafenib failed to block activation of ERK by phenylephrine (PE), identifying a novel Raf-independent pathway mediating α-adrenergic agonist-induced ERK activation that will be addressed further below.
Figure 5. Sorafenib inhibits ERK activity in zebrafish and in NRVMs.



A. Zebrafish. Zebrafish were treated for 24-hours with 1 μM sorafenib, and then lysates were made from the fish as described in Methods. Quantification of phospho-ERK normalized to total-ERK (tERK) is shown below the immunoblots from two independent experiments.
*p<0.05 vs. vehicle treatment
B. NRVMs. NRVMs were treated with vehicle vs. sorafenib (1 μM) for the times shown (left panel) and for 18h at the concentrations shown (right panel). Quantification is shown below the immunoblots. *p<0.05 vs. vehicle
C. Pretreatment with sorafenib (1 μM) for 1 hr. blocks the activation of ERK by IGF-1 (100ng/ml), insulin (5ug/ml), and hydrogen peroxide (H2O2; 50 μM), (but does not block phenylephrine (PE, 10 μM)-induced ERK activation. The level of pERK was normalized to GAPDH, and quantification is shown below the immunoblots.
*p<0.05 *is for the comparison of each stimulus vs. placebo control (ctr) (i.e. in absence of sorafenib treatment); #p<0.05 # is for the comparison of stimulus without sorafenib vs. stimulus with sorafenib pretreatment.
We next asked whether sorafenib-induced cardiomyocyte apoptosis is at least partially mediated by inhibition of ERK. We employed three distinct and selective inhibitors of MEK1/2, kinases that are immediately downstream of Raf-1/B-Raf and are responsible for the phosphorylation and activation of ERK1/2. Twenty-four-hour treatment of NRVMs with MEK1/2 inhibitors (PD184352, UO126, or PD98059) decreased the level of pERK and induced apoptosis by 2–3 fold (Fig. 6A). These data confirm that three distinct (and selective) MEK1/2 inhibitors could recapitulate the pro-apoptotic effect of sorafenib on cardiomyocytes, suggesting that sorafenib-induced cell death is mediated by an on-target effect: inhibition of the Raf/MEK/ERK pathway.
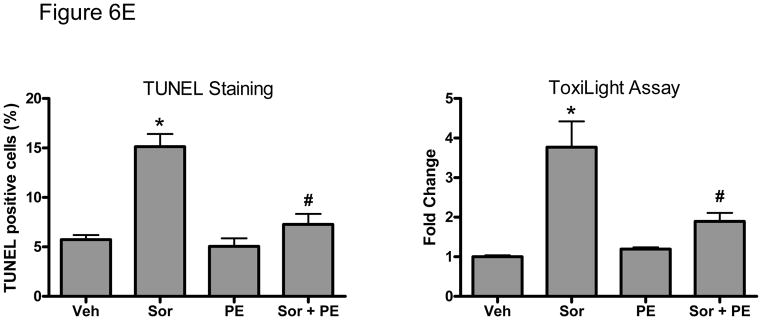
Figure 6. Inhibition of the MEK-ERK signaling pathway modulates sorafenib-induced cardiomyocyte apoptosis.




A. Inhibition of MEK1/2 induces cardiomyocyte apoptosis. NRVMs were subjected to 24h treatment with three distinct MEK1/2 inhibitors (PD184352 (10 μM), UO126 (50 μM), and PD98059 (50 μM)) and then apoptosis was determined by TUNEL assay. *p<0.05 vs. vehicle.
B. Gene transfer of constitutively active MEK-DD increases ERK activity. NRVMs were transduced for 24h with an adenovirus encoding MEK-DD at the multiplicity of infection (MOI) shown. phospho-ERK was quantified by immunoblot after normalization to GAPDH.
*p < 0.05 vs. no virus control and vs. control adenovirus expressing GFP (Ad-GFP).
C. Gene transfer of constitutively active MEK-DD rescues sorafenib-induced apoptotis. NRVMs were transduced with MEK-DD vs Ad-GFP adenoviruses at the noted MOIs. After 24h, NRVMs were treated with sorafenib at the doses shown for an additional 24h. Gene transfer of MEK-DD significantly reduced sorafenib-induced apoptosis compared to control adenovirus.
*p<0.05 vs. respective Ad-GFP control treated with sorafenib.
#p<0.05 vs respective Ad-MEK-DD treated with vehicle.
D. Phenylephrine (PE)-induced activation of ERK is Raf-independent but MEK1/2-dependent. NRVMs were treated with various combinations of PE (10 μM), sorafenib (5 μM), or PD183452 (5 μM) vs. respective vehicle controls (ctr) as shown in the figure. While sorafenib does not block PE-induced activation of ERKs, the MEK1/2 inhibitor, PD183452, does.
* p <0.05 vs vehicle; #p<0.05 vs sorafenib.
E. PE (10 μM) abrogates sorafenib (5 μM)-induced cell death. NRVMs were treated with vehicle vs. sorafenib, in the presence or absence of PE for 48h, and then apoptotic cell death by TUNEL (left panel) and cell death by ToxiLight assay (right panel) were quantified.
*p<0.05 vs vehicle; #p<0.05 vs sorafenib.
To explore this further, we employed an adenovirus that expresses a constitutively active form of MEK1/2 (MEK-DD) to attempt to rescue sorafenib cardiotoxicity. We found that transduction of NRVMs at 10–40 MOI of MEK-DD adenovirus increased pERK by 3–7 fold (Fig. 6B) and markedly reduced sorafenib-induced apoptosis (Fig. 6C). These data suggest that inhibition of ERK contributes significantly to sorafenib-induced cardiomyocyte apoptosis in mammalian cardiomyocytes just as it appears to do in zebrafish and is likely responsible for the increased apoptosis that we observed in the myocardium of sorafenib-treated fish.
Finally we examined the role of the novel Raf-independent signaling pathway that activates ERK downstream of α-adrenergic agonists (Fig. 5C). We first found that although this pathway bypassed Raf to activate ERK, activation of ERK remained entirely dependent on MEK1/2 since the MEK inhibitors abrogated PE-induced ERK activation (Fig. 6D). More importantly, we found that treatment of cardiomyocytes with the α-adrenergic agonist, PE, markedly reduced sorafenib-induced cell death (Fig. 6E). These data suggest that α-adrenergic signaling may effectively protect against sorafenib-induced cardiotoxicity. To evaluate the importance of this pathway in vivo, we employed a loss-of-function approach in the fish by treating them with the α-adrenergic receptor antagonist, prazosin. Prazosin alone had no effect on ventricular function, but when given together with sorafenib, prazosin significantly exacerbated sorafenib-induced cardiac dysfunction (Table 2).
Table 2.
Videomicroscopic measurements in fish 5dpf that were treated at 2dpf with vehicle, 0.5μM sorafenib only, or 0.5μM sorafenib supplemented with prazocin (PZ).
| n | Long-axis
|
Short-axis
|
||||||
|---|---|---|---|---|---|---|---|---|
| EDD | ESD | FS=(EDD-ESD)/EDD | Wall thickness | EDD | ESD | FS=(EDD-ESD)/EDD | ||
| veh | 9 | 100.5±5.8 | 47.8±1.3 | 0.53±0.02 | 15.0±0.3 | 63.3±4.0 | 27.4±1.1 | 0.57±0.01 |
|
| ||||||||
| sor only | 12 | 101.7±4.8 | 69.2±4.6* | 0.32±0.03* | 12.3±0.4* | 60.2±3.2 | 42.7±2.3* | 0.29±0.02* |
|
| ||||||||
| sor +5 μM PZ | 10 | 96.4±6.1 | 62.1±4.2* | 0.35±0.03* | 10.7±0.4*# | 61.7±2.9 | 39.6±2.4* | 0.35±0.04* |
|
| ||||||||
| sor +25 μM PZ | 11 | 97.6±4.1 | 77.5±3.2* | 0.20±0.02*# | 8.0±0.4*# | 60.3±2.4 | 47.6±1.1* | 0.20±0.02*# |
|
| ||||||||
| 25 μM PZ only | 10 | 89.0±3.2 | 47.0±3.5 | 0.48±0.03 | 13.1±0.7 | 59.5±2.2 | 29.8±1.1 | 0.50±0.02 |
p<0.01 vs. vehicle;
p<0.05 vs. sorafenib.
Discussion
Here we introduce zebrafish as a model to examine cardiotoxicity, or lack thereof, of three FDA-approved kinase inhibitors that are being used to treat patients with various malignancies (reviewed in23). We believe this is the first use of zebrafish for this purpose, and, based on our findings, suggest that the use of this model organism should be further evaluated for pre-clinical testing. We also identify the Raf/MEK/ERK pathway as a key target of sorafenib, and demonstrate that inhibition of this pathway likely mediates, at least in part, the cardiotoxicity associated with this agent. Finally, we identify a novel Raf-independent pathway downstream of α-adrenergic receptors that leads to ERK activation and, in isolated cardiomyocytes and fish in vivo, protection against sorafenib cardiotoxicity.
It seems to be abundantly clear that pre-clinical models of kinase inhibitor-induced cardiotoxicity are inadequate since a number of kinase inhibitors have been withdrawn in the late stages of development, at great expense to the companies developing them. Attempts to utilize various cell lines and primary cells (including NRVMs and adult VMs) to predict cardiotoxicity have been helpful, but the consensus seems to be that an in vivo system is needed for optimal detection. That said, the problems with rodent models have been mentioned above. Although rodent models have been used successfully in studies with anthracyclines, results with KIs have been less consistent24–26. For example, we were unable to demonstrate LV dysfunction in mice treated with sunitinib even when we added an additional pressure stress on the heart3. Similar findings have been reported in rats with sunitinib27.
Because of these issues, we examined whether zebrafish might be a viable model. Zebrafish have been employed extensively in embryology and genetics, establishing a well-conserved linkage to mammalian genetics. They have also been used to predict drugs that may cause teratogenic effects and conduction defects including QT interval prolongation. Although the zebrafish heart has only two chambers, zebrafish cardiomyocytes, as in mammals, express voltage-gated sodium channels, L-type and T-type calcium channels, and potassium channels.
With the zebrafish system, we were able to differentiate an agent with clear evidence of cardiotoxicity in the clinic (sunitinib) from one with no signal for cardiotoxicity (gefitinib). The system also predicts cardiotoxicity of sorafenib. It should be noted that the increased death and abnormal body habitus in zebrafish treated with sorafenib only occurred at high concentrations of drug, and because these fish had multiple abnormalities, it is impossible to differentiate cardiac from non-cardiac mortality. At lower concentrations, neither death nor abnormal body habitus was seen, but cardiotoxicity was obvious. To our knowledge, no prospective studies of sorafenib cardiotoxicity that have carefully examined LV function have been performed. Our findings, particularly given the relatively low concentrations of sorafenib we employed, suggest that post-marketing surveillance for sorafenib cardiotoxicity via registries might be prudent, especially as use of the agent broadens into cancer patients with cardiovascular co-morbidities.
Although our findings might suggest a gradient of cardiotoxicity between sorafenib and sunitinib (more cardiac dilatation and reduced fractional shortening with sunitinib; Table 1), these comparisons may not be valid given differences in pharmacokinetics and pharmacodynamics between the drugs. Until concentrations of the agents in the target tissues of interest in patients and model organisms are known, one cannot reliably compare cardiotoxicity of two agents in the model organism. One can only say whether cardiotoxicity is present or not.
One additional caveat to the use of zebrafish (or any model organism) for predicting cardiotoxicity include the possibility of amino acid sequence differences between fish and human at the ATP pocket of targeted kinases, where most KIs interact. In this scenario, cardiotoxicity could be either under- or over-estimated based on the avidity of binding of the compound to the pocket. This will obviously necessitate careful sequence comparisons between zebrafish and human genomes, and projections as to how specific sequence differences may impact kinase inhibitor binding.
To further develop the zebrafish model for cardiotoxicity testing of KIs, we believe that the next step will be to validate this model with other approved agents for which cardiotoxicity profiles are known. The next step after that could be to prospectively examine new agents in early phase clinical trials and then follow the patients in those trials for signals of cardiotoxicity. Thus more likely we will be left with examining approved agents, since it is very rare to gain access to agents while they are in development.
Finally, we identified a protective effect of α-adrenergic signaling against sorafenib-induced cardiotoxicity that is mediated by a novel signaling pathway, bypassing Raf-1/B-Raf but acting via MEK1/2, to activate ERKs. This pathway appears to be uniquely activated by α-adrenergic stimuli since Raf-1/B-Raf was necessary for activation of ERKs downstream of growth factor receptors and oxidative stress. Our findings of a protective role of α-adrenergic signaling against toxicity of sorafenib in cardiomyocytes are consistent with earlier studies demonstrating a protective role of α-adrenergic signaling in the heart exposed to severe pressure stress induced by banding of the aorta in the mouse28.
Collectively, our data raise potential concerns about the simultaneous use of α-adrenergic antagonists such as prazosin (used in patients with benign prostatic hypertrophy) and sorafenib. Given the common usage of α-adrenergic antagonists in clinical practice (~50% of men over 60 years of age have benign prostatic hypertrophy and many require treatment)29, the potential consequences of the concomitant use of these agents with sorafenib should be considered.
Supplementary Material
Novelty and Significance.
What Is Known?
Small molecule kinase inhibitors (KIs) have demonstrated success in treating cancers; however, some of them are cardiotoxic.
The current preclinical models of KI-induced cardiotoxicity, including rodents, are inadequate.
The zebrafish has been adopted as a vertebrate model for human cardiovascular disease research and drug discovery, the latter mostly focused on QT prolongation.
What New Information Does This Article Contribute?
Zebrafish may be a valuable model to predict possible KI-induced cardiotoxicity prior to human use
Inhibition of Raf/MEK/ERK pathway is a key mechanism of sorafenib-induced cardiotoxicity
A novel Raf-independent pathway from α-adrenergic receptors to ERK activation appears to protect against sorafenib-induced cardiotoxicity.
With the explosion in development of KIs targeting kinases that are mutated or overexpressed in cancer, cardiotoxicity with KIs will continue to plague drug development until reliable pre-clinical screening strategies are developed. Unfortunately, current pre-clinical models of KI-induced cardiotoxicity are inadequate. Here we introduce zebrafish as a model for this purpose. We found that the zebrafish can discriminate a KI with little or no cardiotoxicity (gefitinib) from one with demonstrated cardiotoxicity (sunitinib). Sorafenib, like sunitinib, leads to cardiomyocyte death, contractile dysfunction and ventricular dilatation in zebrafish. Furthermore, with studies in neonatal rat cardiomyocytes, we identified inhibition of the Raf/MEK/ERK pathway as a key mechanism of sorafenib-induced cardiotoxicity. We also identified a novel Raf-independent pathway from α-adrenergic receptors to ERK activation that appears to protect against sorafenib-induced cardiotoxicity both in cardiomyocytes in culture and in zebrafish. This study is the first use of zebrafish to examine KI-induced cardiotoxicity, and our findings suggest that the use of this model organism should be further evaluated for preclinical testing. Collectively, our data raise concerns about the simultaneous use of α-adrenergic antagonists and sorafenib in clinical practice.
Acknowledgments
The authors wish to acknowledge the support of the zebrafish facility of Thomas Jefferson University.
Sources of Funding
This work was supported by National Heart, Lung, and Blood Institute grants HL061688 (to T.F.) and HL091799 (to W.J.K and T.F.).
Non-standard abbreviations and acronyms
- Raf
Rapidly Accelerated Fibrosarcoma
- ERK
extracellular signal-regulated kinase
- MAPK
MAP kinase, mitogen-activated protein kinase
- MEK
mitogen-activated protein kinase/extracellular signal-regulated kinase kinase
- LV
left ventricular
- IGF
insulin-like growth factor
- KI
kinase inhibitor
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
Footnotes
Disclosures
None.
References
- 1.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 2.Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, Woulfe K, Pravda E, Cassiola F, Desai J, George S, Morgan JA, Harris DM, Ismail NS, Chen JH, Schoen FJ, Van den Abbeele AD, Demetri GD, Force T, Chen MH. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007 Dec 15;370(9604):2011–2019. doi: 10.1016/S0140-6736(07)61865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kerkela R, Woulfe KC, Durand JB, Vagnozzi R, Kramer D, Chu TF, Beahm C, Chen MH, Force T. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin Transl Sci. 2009;(2):1. doi: 10.1111/j.1752-8062.2008.00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dahme T, Katus HA, Rottbauer W. Fishing for the genetic basis of cardiovascular disease. Dis Model Mech. 2009 Jan-Feb;2(1–2):18–22. doi: 10.1242/dmm.000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chico TJ, Ingham PW, Crossman DC. Modeling cardiovascular disease in the zebrafish. Trends Cardiovasc Med. 2008 May;18(4):150–155. doi: 10.1016/j.tcm.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Rocke J, Lees J, Packham I, Chico T. The zebrafish as a novel tool for cardiovascular drug discovery. Recent Pat Cardiovasc Drug Discov. 2009 Jan;4(1):1–5. doi: 10.2174/157489009787260043. [DOI] [PubMed] [Google Scholar]
- 7.Kari G, Rodeck U, Dicker AP. Zebrafish: an emerging model system for human disease and drug discovery. Clin Pharmacol Ther. 2007 Jul;82(1):70–80. doi: 10.1038/sj.clpt.6100223. [DOI] [PubMed] [Google Scholar]
- 8.Langheinrich U. Zebrafish: a new model on the pharmaceutical catwalk. Bioessays. 2003 Sep;25(9):904–912. doi: 10.1002/bies.10326. [DOI] [PubMed] [Google Scholar]
- 9.Khakoo AY, Kassiotis CM, Tannir N, Plana JC, Halushka M, Bickford C, Trent J, 2nd, Champion JC, Durand JB, Lenihan DJ. Heart failure associated with sunitinib malate: a multitargeted receptor tyrosine kinase inhibitor. Cancer. 2008 Jun;112(11):2500–2508. doi: 10.1002/cncr.23460. [DOI] [PubMed] [Google Scholar]
- 10.Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, Ruhsam M, Hejna M, Schmidinger H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008 Nov 10;26(32):5204–5212. doi: 10.1200/JCO.2007.15.6331. [DOI] [PubMed] [Google Scholar]
- 11.Tolcher AW, Appleman LJ, Shapiro GI, Mita AC, Cihon F, Mazzu A, Sundaresan PR. A phase I open-label study evaluating the cardiovascular safety of sorafenib in patients with advanced cancer. Cancer Chemother Pharmacol. 2010 Jun 3; doi: 10.1007/s00280-010-1372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strumberg D, Clark JW, Awada A, Moore MJ, Richly H, Hendlisz A, Hirte HW, Eder JP, Lenz HJ, Schwartz B. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist. 2007 Apr;12(4):426–437. doi: 10.1634/theoncologist.12-4-426. [DOI] [PubMed] [Google Scholar]
- 13.Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand JP, Scigalla P, Raymond E. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24:25–35. doi: 10.1200/JCO.2005.02.2194. [DOI] [PubMed] [Google Scholar]
- 14.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams YJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan ED, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, MIller TA, Shirazian S, McMahon G, Cherrington JM. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–337. [PubMed] [Google Scholar]
- 15.Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O'Farrell AM, Bello CL, Allred R, Manning WC, Cherrington JM, Louie SG, Hong W, Brega NM, Massimini G, Scigalla P, Berdel WE, Hossfeld DK. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105:986–993. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- 16.Roskoski R., Jr Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem Biophys Res Commun. 2007;356(2):323–328. doi: 10.1016/j.bbrc.2007.02.156. [DOI] [PubMed] [Google Scholar]
- 17.Parng C, Seng WL, Semino C, McGrath P. Zebrafish: a preclinical model for drug screening. Assay Drug Dev Technol. 2002 Nov;1(1 Pt 1):41–48. doi: 10.1089/154065802761001293. [DOI] [PubMed] [Google Scholar]
- 18.Tucker B, Lardelli M. A rapid apoptosis assay measuring relative acridine orange fluorescence in zebrafish embryos. Zebrafish. 2007 Summer;4(2):113–116. doi: 10.1089/zeb.2007.0508. [DOI] [PubMed] [Google Scholar]
- 19.Shepard JL, Stern HM, Pfaff KL, Amatruda JF. Analysis of the cell cycle in zebrafish embryos. Methods Cell Biol. 2004;76:109–125. doi: 10.1016/s0091-679x(04)76007-0. [DOI] [PubMed] [Google Scholar]
- 20.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, Lockhart DJ, Milanov ZV, Morrison MJ, Pallares G, Patel HK, Pritchard S, Wodicka LM, Zarrinkar PP. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008 Jan;26(1):127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 21.Harris IS, Zhang S, Treskov I, Kovacs A, Weinheimer C, Muslin AJ. Raf-1 kinase is required for cardiac hypertrophy and cardiomyocyte survival in response to pressure overload. Circulation. 2004 Aug 10;110(6):718–723. doi: 10.1161/01.CIR.0000138190.50127.6A. [DOI] [PubMed] [Google Scholar]
- 22.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010 Oct;90(4):1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng H, Force T. Molecular mechanisms of cardiovascular toxicity of targeted cancer therapeutics. Circ Res. 2010 Jan 8;106(1):21–34. doi: 10.1161/CIRCRESAHA.109.206920. [DOI] [PubMed] [Google Scholar]
- 24.Fernandez A, Sanguino A, Peng Z, Ozturk E, Chen J, Crespo A, Wulf S, Shavrin A, Qin C, Ma J, Trent J, Lin Y, Han HD, Mangala LS, Bankson JA, Gelovani J, Samarel A, Bornmann W, Sood AK, Lopez-Berestein G. An anticancer C-Kit kinase inhibitor is reengineered to make it more active and less cardiotoxic. J Clin Invest. 2007 Dec;117(12):4044–4054. doi: 10.1172/JCI32373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kerkela R, Grazette L, Yacobi R, Iliescu C, Patten R, Beahm C, Walters B, Shevtsov S, Pesant S, Clubb FJ, Rosenzweig A, Salomon RN, Van Etten RA, Alroy J, Durand JB, Force T. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat Med. 2006 Aug;12(8):908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 26.Wolf A, Couttet P, Dong M, Grenet O, Heron M, Junker U, Laengle U, Ledieu D, Marrer E, Nussher A, Persohn E, Pognan F, Riviere GJ, Roth DR, Trendelenburg C, Tsao J, Roman D. Imatinib does not induce cardiotoxicity at clinically relevant concentrations in preclinical studies. Leuk Res. 2010 Sep;34(9):1180–1188. doi: 10.1016/j.leukres.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 27.French KJ, Coatney RW, Renninger JP, Hu CX, Gales TL, Zhao S, Storck LM, Davis CB, McSurdy-Freed J, Chen E, Frazier KS. Differences in effects on myocardium and mitochondria by angiogenic inhibitors suggest separate mechanisms of cardiotoxicity. Toxicol Pathol. 2010;38(5):691–702. doi: 10.1177/0192623310373775. [DOI] [PubMed] [Google Scholar]
- 28.Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, O'Connell TD. An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation. 2007;115:763–772. doi: 10.1161/CIRCULATIONAHA.106.664862. [DOI] [PubMed] [Google Scholar]
- 29.Kojima Y, Kubota Y, Sasaki S, Hayashi Y, Kohri K. Translational pharmacology in aging men with benign prostatic hyperplasia: molecular and clinical approaches to alpha1-adrenoceptors. Curr Aging Sci. 2009 Dec;2(3):223–239. doi: 10.2174/1874609810902030223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.