

Abstract
Hepatocellular carcinoma (HCC) is one of the deadliest forms of human liver cancer and does not respond well to conventional therapies. Novel effective treatments are urgently in need. G-protein-coupled kinase 2 (GRK2) is unique serine/threonine kinase that involves in many signaling pathways and regulates various essential cellular processes. Altered levels of GRK2 have been linked with several human diseases including cancer. In this study, we investigated a novel approach for HCC treatment by inducing overexpression of GRK2 in human HCC cells. We found that overexpression of GRK2 through recombinant adenovirus transduction inhibits the growth of human HCC cells. BrdU incorporation assay showed that the growth inhibition caused by elevated GRK2 level was due to reduced cell proliferation but not apoptosis. To examine the anti-proliferative function of increased GRK2 level, we performed cell cycle analysis using propidium iodide staining. We found that the proliferation suppression was associated with G2/M phase cell cycle arrest by the wild-type GRK2 but not its kinase-dead K220R mutant. Furthermore, increased levels of wild-type GRK2 induced upregulation of phosphor-Ser15 p53 and cyclin B1 in a dose-dependent manner. Our data indicate that the anti-proliferative function of elevated GRK2 is associated with delayed cell cycle progression and is GRK2 kinase activity-dependent. Enforced expression of GRK2 in human HCC by molecular delivery may offer a potential therapeutic approach for the treatment of human liver cancer.
Hepatocellular carcinomas (HCC) are a complicated human disease in terms of etiology and molecular carcinogenic mechanisms. HCC is globally the fifth most common malignancy and the third largest cause of cancer deaths [Bosch et al., 2004). The frequency of HCC in Southeast Asia and sub-Saharan Africa is greater than that in North America and Western Europe. However, recent data show that the overall frequency of HCC in developed countries is rising [Lau and Lai, 2008]. This increase is primarily due to persistent infection with hepatitis C and chronic alcohol abuse that causes cirrhosis [Kern et al., 2002; Bruix and Sherman, 2005]. HCC is a potentially curable at early stage through surgical resection and liver transplantation. Unfortunately, the majority of patients with HCC are usually in the advanced-stage with severe background liver disease which is not suitable for such treatments [Wang et al., 2002]. Moreover, HCC is a type of tumor highly resistant to conventional medical treatment such as chemotherapy and radiation. There is a critical need to develop novel strategies for effective prevention and therapy of this disease.
G-protein-coupled receptor kinase 2 (GRK2) is a ubiquitously expressed serine/threonine kinase. It is the unique member of GRK family with diverse functions [Metaye et al., 2005; Ribas et al., 2007]. The role of GRK2 was first discovered in the desensitization of G-protein-coupled receptors (GPCR) signaling by phosphorylating agonist-activated 7-transmembrane receptors. The phosphorylated receptor enhances the binding of β-arrestins to form a molecular complex which prevents further coupling of the receptor from its G-protein, leading to attenuation of the receptor-mediated signalings [Aragay et al., 1998; Ribas et al., 2007]. Despite of its traditional function as a kinase in receptor desensitization, a growing body of evidence has been documented that GRK2 interacts with a variety of other cytosolic proteins involved in signaling pathways relevant to essential cellular processes, such as proliferation/apoptosis, migration, trafficking, cell cycle, and development [Penela et al., Penela et al., 2008, 2010b; Guo et al., 2009; Jiang et al., 2009; Kahsai et al., 2010]. Some of these physiological functions of GRK2 are achieved through kinase-independent mechanisms by directly binding to other proteins [Cipolletta et al., 2009; Jiang et al., 2009; Namkung et al., 2009; Chen et al., 2010]. Altered expression levels of GRK2 have been reported in many human diseases including heart failure, hypertension, rheumatoid arthritis, cystic fibrosis, and cancer [Lombardi et al., 1999; Mak et al., 2002; Vroon et al., 2004, 2005; Metaye et al., 2005; Lymperopoulos et al., 2007].
Overexpression of GRK2 has been reported to reduce cell proliferation in smooth muscle cells and thyroid cancer cells [Peppel et al., 2000; Metaye et al., 2008]. Interestingly, some thyroid tumors actually have higher GRK2 level as compared with its surrounding normal tissues. The mechanism underlying this growth inhibition is still largely unclear, given the fact that GRK2 has a complex interactome and lies in the crossroad of many signaling pathways. In this report, we tested the inhibitory effects of GRK2 overexpression on the growth of human HCC cells and examined the molecular mechanism by which GRK2 overexpression causes this growth inhibition.
Materials and Methods
Cell culture
Human HCC cell lines, Mahlavu, HepG2, Hep3B, Huh7, and PLC/PRF/5 were maintained in DMEM (Mediatech, Inc., Manassas, VA) supplemented with 10% FBS (Sigma-Aldrick, St. Louis, MO) and 2 nmol/L L-glutamine and penicillin–streptomycin. Cells were cultured in an incubator with humidified air at 37°C with 5% CO2.
Adenovirus transduction
Recombinant adenoviruses expressing bovine wild-type and K220R mutant GRK2 were obtained from the Center for translational medicine, Thomas Jefferson University. Overexpression of GRK2 was achieved by transducing HCC cells with an MOI of 50 as standard for the rest of experiments. Equal expression levels of wild-type and mutant GRK2 were determined by Western blot. For dose-dependent experiments, HCC cells were transduced with corresponding adenovirus at 2×, 3×, and 4× of the amount used for achieving standard GRK2 overexpression.
Cell viability assays
The viability of HCC cells were examined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method. Equal numbers of cells in a volume of 200 μl were seeded in a 48-well plates (2 × 104 cells/well). The plates were incubated for 4 days. For each measurement, 50 μl MTT (5 mg/ml) was added into each well and incubated at 37°C with 5% CO2 for 2 h. The wells were then decanted and the purple formazan crystals formed were dissolved in 200 μl DMSO. The absorbance of the plate was taken at 595 nm in an ELISA plate reader. All assays were done in triplicate. Hemocytometer was also used for the viable cell counting.
Cell cycle analysis
HCC cells (2 × 105) seeded in 6-well plates were transduced with adenoviruses expressing WT GRK2 and K220R mutant, respectively. After 2 days of culture, cells were harvested and propidium iodide (PI) staining was performed as standard method. Cell cycle progression was determined using flow cytometry (FACS Calibur) and data analysis was performed using FlowJo software.
Cell proliferation assay
5-Bromo-2′-deoxy-uridine Labeling and Detection Kit I (Roche, Indianapolis, IN) was used for the detection of cell proliferation. Briefly, cells were transduced using adenovirus and cultured on a coverslip for 2 days. Cells were fed with BrdU (final concentration 10 μM) for 20 min at 37°C with 5% CO2. The coverslip was then washed three times and fixed with ethanol. After another washing for three times, anti-BrdU solution was added and the coverslip was incubated for 30 min at 37°C. The secondary fluorescent antibody (Alexa fluor 555) was used for the detection of BrdU positive cells. Coverslips were placed on slides using a DAPI containing Vectashield mounting medium (Vector Laboratories, Inc., Burlingame, CA). Counting was based on randomly chosen five fields for each coverslip.
Tunel assay
Cells (5 × 104) were seeded on a coverslip/well in 6-well plates. After adding adenovirus, the transduced cells were cultured for another 2 days before Tunel assay. Briefly, cells on coverslips were fixed in room temperature 4% paraformaldehyde for 20 min followed by 30 min in PBS and 2 min in 100% ethanol. Tunel enzymatic labeling was performed using the In situ Cell Death Detection kit (Roche).
Western blot analysis
The whole cell lysate was prepared using Glo lysis buffer (Promega, Madison, WI) containing protease inhibitor cocktail and phosphotase inhibitor. Protein concentration was determined using Bradford assay (BioRad, Hercules, CA). Twenty-five micrograms of total cell lysates were loaded and separated by gel electrophoresis using NuPage 4–12% Bis–Tris precast gel (Invitrogen, Carlsbad, CA). The proteins were electrotransferred onto a PVDF membrane. The membranes were blocked in Odyssey Licor Blocking Buffer for 1 h at room temperature and then incubated with primary antibodies at 4°C overnight. Membranes were washed three times for 5 min each time and probed with secondary antibodies for 1 h at room temperature. Proteins were visualized with the Licor Odyssey scanner. All primary antibodies were diluted 1:1,000 and all secondary antibodies were diluted 1:5,000 in Odyssey Licor Blocking Buffer.
Results
Expression of GRK2 in HCC cell lines
To gain a profile of basal GRK2 levels in HCC cells, we examined GRK2 expression in five HCC cell lines by Western blot analysis (Fig. 1). As described in the introduction, GRK2 is ubiquitously expressed serine/threonine kinase and mainly distributed in the cytosol (Fig. 1A). We detected GRK2 expression in all five cell lines tested, with the highest level in the Huh7 cell line and the lowest level in the Mahlavu cell line (Fig. 1B). We thus selected Huh7 and Mahlavu cell lines for further GRK2 overexpression experiments.
Fig. 1.
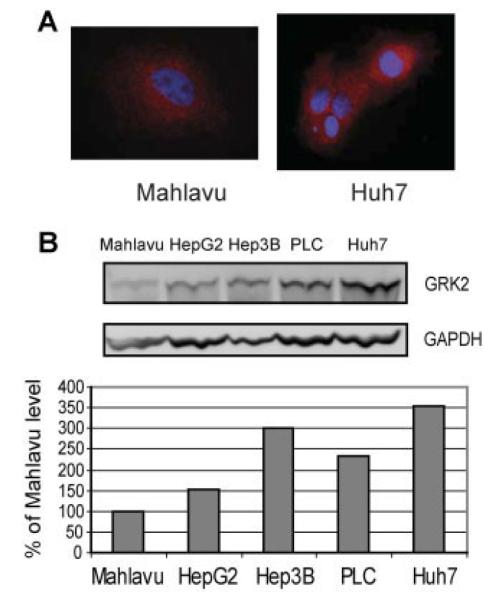
Basal GRK2 protein levels in HCC cell lines. A: Cytosolic distribution of GRK2. B: Basal GRK2 protein levels in all five HCC cell lines detected by Western blotting. Cell lysates (25 μg total protein) were subjected to immunoblotting with antibodies as indicated. GRK2 levels were normalized to GAPDH. The immunoblot shown is a representative of two independent experiments.
GRK2 overexpression inhibits cell growth in HCC cells
We then examined the growth inhibitory effects of GRK2 overexpression on Huh7 and Mahlavu HCC cell lines, using both hemocytometer and MTT assays. As shown in Figure 2B, overexpression of GRK2 but not the kinase dead K220R mutant inhibited the growth of Mahlavu and Huh7 cells. This is in consistent with previously published results that overexpression of GRK2 inhibits growth of smooth muscle cells and thyroid cancer cells [Peppel et al., 2000; Metaye et al., 2008]. Our result further demonstrated that the inhibitory effect of GRK2 overexpression on cell growth is GRK2 kinase activity-dependent.
Fig. 2.

Overexpression of GRK2 reduces cell viability in HCC cells. A: Western blots showing overexpression of wild-type GRK2 and its kinase-dead K220R mutant in Mahlavu and Huh7 cells transduced with recombinant adenovirus containing WT GRK2 or GRK2 K220R mutant. β-Actin was used as total protein loading control. B: MTT method was used to detect cell viability with overexpression of GRK2 and K220R mutant during 4-day growth period in 10% FBS medium. [Color figure can be seen in the online version of this article, available at http://wileyonlinelibrary.com/journal/jcp]
GRK2 overexpression suppresses cell proliferation in HCC cells
To determine whether the inhibitory effect of GRK2 overexpression was associated with induction of apoptosis or reduction of cell proliferation in HCC cells, we carried out TUNEL assay and BrdU assay, respectively. We did not detect any difference of apoptosis between wild-type GRK2 and K220R mutant overexpression (data not shown). However, we found that there was a significant difference between wild-type GRK2 and K220R mutant overexpression in HCC cell proliferation (Fig. 3A). For the Huh7 cell line, the BrdU positive cells under GRK2 overexpression condition were 37.4±5.4%, while the untreated control and K220R mutant overexpression were 54.4±4.7% and 56.8±3.3%, respectively (Fig. 3B). As expected, there was no significant difference of proliferation between the untreated control and K220R mutant overexpression. Since the overexpression levels of wild-type GRK2 and K220R mutant were almost equivalent (Fig. 2A), the cell proliferation reduction was obviously GRK2 kinase activity-dependent. Moreover, the proliferation reduction induced by GRK2 overexpression was dose-dependent in Mahlavu, with 42.7±6.4% BrdU positive cells in lower GRK2 overexpression and only 29.2±6.2% in higher GRK2 overexpression, as compared with untreated control 53.2±5.8% (Fig. 3C).
Fig. 3.

Overexpression of GRK2 suppresses cell proliferation in HCC cells. A: Immunofluorescent staining of BrdU incorporation in Huh7 cells. DAPI was used for staining nucleus. Antibody against BrdU was immunoblotted with a secondary antibody labeled with Tritc. Overexpression of GRK2 and K220R mutant was achieved by adenovirus transduction as indicated in Figure 2. B: BrdU incorporation in Huh7 cells as shown in A. C: BrdU incorporation in Mahlavu cells. GRK2 H represents at least twofold higher of MOI of adenovirus used for achieving GRK2 overexpression. *P < 0.01.
GRK2 overexpression induces G2/M phase cell cycle arrest
To investigate the mechanism by which GRK2 overexpression induces the HCC cell proliferation reduction, we performed cell cycle progression analysis by fluorescence-activated cell sorter scanning using PI staining method. In both Huh7 and Mahlavu cells, GRK2 overexpression caused a significant increase in the proportion of cells in G2/M phase as compared with the untreated WT control and K220R mutant overexpression (Fig. 4). Specifically, the G2/M phase of Huh7 cells increased from 21.5% to 41.8% and 44.3% (higher GRK2 overexpression), while the control and K220R mutant overexpression remained almost the same, 21.5% versus 21.4%, respectively. In Mahlavu cells, the G2/M phase increased from 27.4% to 48.4% and 62% (higher GRK2 overexpression), while the proportion of the untreated control and K220R mutant overexpression cells were similar, 27.4% versus 25.9%, respectively. These results indicated that overexpression of wild-type GRK2 leads to arrest in the G2/M phase of the cell cycle in HCC cells, and this cell cycle arrest is GRK2 kinase activity-dependent, as overexpression of the kinase dead K220R mutant did not cause any phase change.
Fig. 4.

Overexpression of GRK2 leads to G2/M phase arrest in HCC cells. A: Representative cell cycle his to grams with over expression of GRK2 and K220R mutant 2 days post-adenovirus transduction. Cells were analyzed by FACS for DNA content by PI staining. GRK2 H represents at least twofold higher of MOI of adenovirus used for achieving GRK2 overexpression. B: Cells distributed in different phases as shown in A.
GRK2 overexpression upregulates phosphor-Ser15 p53 and cyclin B1
To further determine the molecular events that might be involved in the G2/M phase cell cycle arrest, we screened several cell cycle relevant proteins for changes in expression level and phosphorylation status. As shown in Figure 5, we detected increased levels of phosphor-Ser15 p53 and cyclin B1. These increases were dose-dependent with regarding to the increased GRK2 levels (Fig. 5A). In contrast, we did not see the above changes with the increased expression of the kinase dead K220R mutant (Fig. 5B). Surprisingly, the phosphor-Ser473 Akt level was also increased with the increased level of wild-type GRK2 but not the kinase dead K220R mutant. Again, these results indicated that all these changes were GRK2 kinase activity-dependent.
Fig. 5.

Overexpression of GRK2 induces upregulation of phosphor-p53, cyclin B1, and phosphor-Akt. A. Overexpression of GRK2 in Huh7 was achieved by increasing MOI of adenovirus for examining dose-dependent effects. Cell lysates were subjected to immunoblotting with antibodies as indicated on the penal. In Mahlavu cells, Western blotting was compared between overexpression of GRK2 and K220 R mutant. B: Western blotting with overexpression of GRK2 K220R mutant in a dose-dependent manner in Huh7 and Mahlavu cells.
Discussion
GRK2 has been intensively studied for more than two decades, largely related with GPCR and other receptors. Recently, emerging evidence has demonstrated that this protein kinase plays a critical role in many cellular processes by interacting with various cytosolic proteins and may have much wider range of functions than thought before [Aragay et al., 1998; Ho et al., 2005; Ribas et al., 2007; Penela et al., Penela et al., 2009, 2010a]. In this report, we have demonstrated that overexpression of GRK2 attenuated human HCC cell proliferation through inducing G2/M phase cell cycle arrest. Our data suggested that a possible mechanism involving the elevated levels of p53 phosphorylation at serine 15 and cyclin B1.
GRK2 is mainly distributed in cytosol and its expression in different cell/tissue is varied. It appears that the level of GRK2 in a specific cell/tissue adapts to the unique physiological function of the cell/tissue, and thus the endogenous GRK2 protein level is tightly controlled. Some reports have shown that GRK2 protein level is molecularly regulated by mechanisms through mitogen-activated protein kinase (MAPK), Mdm2-mediated ubiquitination and degradation, and mTOR pathway involved translation [Elorza et al., 2003; Salcedo et al., 2006; Cobelens et al., 2007]. No matter which mechanism is involved, the GRK2 protein level in a specific cell is also affected by different extracellular stimuli. Such cases have been reported with GRK2 upregulation in mouse liver FL83B cells under chronic treatment of insulin [Shahid and Hussain, 2007], vascular smooth muscle cells, and human HCC cells under TGFβ treatment [Ho et al., 2005; Guo et al., 2009], and human thyroid cells under the treatment of thyrotropin plus insulin or insulin-like growth factor 1 [Metaye et al., 2008]. Some other cases with GRK2 downregulation have also been documented in human mononuclear leucocytes and rat smooth muscle cells under the influence of proinflammatory cytokines, such as interleukin 6 and interferon γ [Lombardi et al., 1999; Ramos-Ruiz et al., 2000; Vroon et al., 2005]. Despite extracellular stimuli, a recent report demonstrated that GRK2 protein level in HeLa cells was transiently downregulated during G2 progression by a mechanism involving CDK2-mediated phosphorylation of GRK2 at Serine 670, indicating an intracellular stimulus during cell cycle [Penela et al., 2010b]. Taken together, alternation of GRK2 protein levels, either through extracellular or intracellular stimuli, would dramatically change the normal cellular processes, leading to pathophysiological disorders and diseases [Gros et al., 1997; Iaccarino et al., 2005; Vroon et al., 2005; Nijboer et al., 2008].
Enforced overexpression of GRK2 reduced cell proliferation in human smooth muscle cells and in two poorly differentiated human thyroid cancer cells [Peppel et al., 2000; Metaye et al., 2008]. In this study, we demonstrated that overexpression of GRK2 by adenovirus transduction suppressed HCC cell proliferation and growth. Although we found that the basal protein levels in our HCC cell lines were varied, enforced overexpression of GRK2 still impaired normal cell cycle progression and led to cell cycle arrest at G2/M phase in Huh7 (endogenous GRK2 high) and Mahlavu (endogenous GRK2 low) cells. Cells that arrest at G2/M phase could be induced by many different compounds or proteins. For instance, gambogic acid can induce G2/M phase cell cycle arrest in human gastric carcinoma cells [Yu et al., 2007], and the human cytomegalovirus (HCMV) IE86 protein can cause G2/M phase arrest in p53 null cells [Song and Stinski, 2005]. In addition, it is reported that the MAPK pathway is involved in the G2/M progression [Astuti et al., 2009; Dumesic et al., 2009]. Our study did not detect any changes of Erk1/2 phosphorylation (data not shown), however, we found the increased levels of phosphor-Ser15 p53 and cyclin B1 were associated with G2/M phase arrest induced by GRK2 overexpression, a phenomenon similar to the effect of HCMV IE86 protein [Song and Stinski, 2005]. Since both Huh7 and Mahlavu cells have loss of function mutation in p53, the mechanism by which cell cycle arrests at G2/M phase through the overexpression of GRK2 and HCMV IE86 protein appears to be similar. Moreover, a recent report suggested that GRK2 participates in the control of cell cycle progression by transiently downregulated during the G2/M phase transition [Penela et al., 2010b]. This finding implies that overexpression of GRK2 would impair the normal G2/M phase transition which is consistent with our data. Although GRK2 kinase activity independent regulation of cell cycle in zebrafish has been reported [Jiang et al., 2009], our data showed that the suppression of cell proliferation through G2/M phase arrest was kinase activity dependent, since the kinase dead K220R mutant GRK2 did not exert the inhibitory effect.
It is interesting to note that elevated levels of GRK2 have been reported in some cancer cells, which seems to be contradictory to our result and the other published data [Metaye et al., 2008]. For example, thyroid carcinoma, transformed mammary cells, and primary prostate tumors actually exhibit higher GRK2 protein levels than that of the corresponding normal tissue or cells [Salcedo et al., 2006; Prowatke et al., 2007; Metaye et al., 2008]. One possible explanation of these observations is that there may be a threshold of GRK2 levels in different cells. Cancer cells are usually mutated from normal cells that display uncontrolled growth. It is possible that endogenously elevated level of GRK2 in cancer cells seems to be a rescue result for desensitization of the cancerous transformation according to the traditional function of GRK2. It is also possible that higher endogenous GRK2 level favors other cellular processes relevant to tumor formation and progression such as migration and anchorage independent growth. However, if the level of GRK2 is reached over the threshold such as the enforced overexpression of GRK2 by transfection or transduction, cancer cells start to die due to the impaired cell cycle progression as we indicated in this study.
Similarly, the increased phosphor-Ser473 Akt level has been observed with the increased GRK2 level in mammary transformed cells [Salcedo et al., 2006]; the same is true in HCC cells as we found in this study. However, one study reported that the level of GRK2 is conversely correlated with the phosphorylation of Akt in portal hypertension [Liu et al., 2005]. Another study reported that the rictor–mTOR complex directly phosphorylates Akt on Ser473 [Sarbassov et al., 2005]. It appears that the relationship between GRK2 protein level and phosphorylation of Akt is complex. Further examination of this relationship will be very useful for defining the role of GRK2 in cancer cells, given the fact that Akt is a key mediator of multiple signaling pathways.
The present study showed growth inhibitory effects of GRK2 overexpression in HCC cells. This anti-proliferative role of elevated GRK2 level is a kinase-dependent function. The molecular mechanisms by which altered levels of GRK2 regulate and orchestrate cellular processes deserve to be explored further for a better understanding of the role of GRK2 in cancer cells. Overall, our results provide a possible therapeutic approach for the treatment of human liver cancer by increasing the level of GRK2 in HCC cells.
Acknowledgments
We are grateful to Renato Baserga (Kimmel Cancer Center) for critical review of the article. This study was supported by the Department of Surgery and the Dean’s Office of Jefferson Medical College.
Abbreviations
- GRK2
G-protein-coupled receptor 2
- HCC
hepatocellular carcinoma
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BrdU
bromodeoxyuridine
Literature Cited
- Aragay AM, Ruiz-Gomez A, Penela P, Sarnago S, Elorza A, Jimenez-Sainz MC, Mayor F., Jr G protein-coupled receptor kinase 2 (GRK2): Mechanisms of regulation and physiological functions. FEBS Lett. 1998;430:37–40. doi: 10.1016/s0014-5793(98)00495-5. [DOI] [PubMed] [Google Scholar]
- Astuti P, Pike T, Widberg C, Payne E, Harding A, Hancock J, Gabrielli B. MAPK pathway activation delays G2/M progression by destabilizing Cdc25B. J Biol Chem. 2009;284:33781–33788. doi: 10.1074/jbc.M109.027516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: Worldwide incidence and trends. Gastroenterology. 2004;127:S5–S16. doi: 10.1053/j.gastro.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Bruix J, Sherman M. Management of hepatocellular carcinoma. Hepatology. 2005;42:1208–1236. doi: 10.1002/hep.20933. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li S, Tong C, Zhao Y, Wang B, Liu Y, Jia J, Jiang J. G protein-coupled receptor kinase 2 promotes high-level Hedgehog signaling by regulating the active state of Smo through kinase-dependent and kinase-independent mechanisms in Drosophila. Genes Dev. 2010;24:2054–2067. doi: 10.1101/gad.1948710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolletta E, Campanile A, Santulli G, Sanzari E, Leosco D, Campiglia P, Trimarco B, Iaccarino G. The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovasc Res. 2009;84:407–415. doi: 10.1093/cvr/cvp252. [DOI] [PubMed] [Google Scholar]
- Cobelens PM, Kavelaars A, Heijnen CJ, Ribas C, Mayor F, Jr., Penela P. Hydrogen peroxide impairs GRK2 translation via a calpain-dependent and cdk1-mediated pathway. Cell Signal. 2007;19:269–277. doi: 10.1016/j.cellsig.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Dumesic PA, Scholl FA, Barragan DI, Khavari PA. Erk1/2 MAP kinases are required for epidermal G2/M progression. J Cell Biol. 2009;185:409–422. doi: 10.1083/jcb.200804038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elorza A, Penela P, Sarnago S, Mayor F., Jr MAPK-dependent degradation of G protein-coupled receptor kinase 2. J Biol Chem. 2003;278:29164–29173. doi: 10.1074/jbc.M304314200. [DOI] [PubMed] [Google Scholar]
- Gros R, Benovic JL, Tan CM, Feldman RD. G-protein-coupled receptor kinase activity is increased in hypertension. J Clin Invest. 1997;99:2087–2093. doi: 10.1172/JCI119381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Chen H, Ho J, Mancini J, Sontag T, Laporte SA, Richard DE, Lebrun JJ. TGFbeta-induced GRK2 expression attenuates AngII-regulated vascular smooth muscle cell proliferation and migration. Cell Signal. 2009;21:899–905. doi: 10.1016/j.cellsig.2009.01.037. [DOI] [PubMed] [Google Scholar]
- Ho J, Cocolakis E, Dumas VM, Posner BI, Laporte SA, Lebrun JJ. The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J. 2005;24:3247–3258. doi: 10.1038/sj.emboj.7600794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino G, Barbato E, Cipolletta E, De Amicis V, Margulies KB, Leosco D, Trimarco B, Koch WJ. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur Heart J. 2005;26:1752–1758. doi: 10.1093/eurheartj/ehi429. [DOI] [PubMed] [Google Scholar]
- Jiang X, Yang P, Ma L. Kinase activity-independent regulation of cyclin pathway by GRK2 is essential for zebrafish early development. Proc Natl Acad Sci USA. 2009;106:10183–10188. doi: 10.1073/pnas.0812105106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahsai AW, Zhu S, Fenteany G. G protein-coupled receptor kinase 2 activates radixin, regulating membrane protrusion and motility in epithelial cells. Biochim Biophys Acta. 2010;1803:300–310. doi: 10.1016/j.bbamcr.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern MA, Breuhahn K, Schirmacher P. Molecular pathogenesis of human hepatocellular carcinoma. Adv Cancer Res. 2002;86:67–112. doi: 10.1016/s0065-230x(02)86003-1. [DOI] [PubMed] [Google Scholar]
- Lau WY, Lai EC. Hepatocellular carcinoma: Current management and recent advances. Hepatobiliary Pancreat Dis Int. 2008;7:237–257. [PubMed] [Google Scholar]
- Liu S, Premont RT, Kontos CD, Zhu S, Rockey DC. A crucial role for GRK2 in regulation of endothelial cell nitric oxide synthase function in portal hypertension. Nat Med. 2005;11:952–958. doi: 10.1038/nm1289. [DOI] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. FASEB J. 1999;13:715–725. doi: 10.1096/fasebj.13.6.715. [DOI] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Funakoshi H, Eckhart AD, Koch WJ. Adrenal GRK2 upregulation mediates sympathetic overdrive in heart failure. Nat Med. 2007;13:315–323. doi: 10.1038/nm1553. [DOI] [PubMed] [Google Scholar]
- Mak JC, Chuang TT, Harris CA, Barnes PJ. Increased expression of G protein-coupled receptor kinases in cystic fibrosis lung. Eur J Pharmacol. 2002;436:165–172. doi: 10.1016/s0014-2999(01)01625-9. [DOI] [PubMed] [Google Scholar]
- Metaye T, Gibelin H, Perdrisot R, Kraimps JL. Pathophysiological roles of G-protein-coupled receptor kinases. Cell Signal. 2005;17:917–928. doi: 10.1016/j.cellsig.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Metaye T, Levillain P, Kraimps JL, Perdrisot R. Immunohistochemical detection, regulation and antiproliferative function of G-protein-coupled receptor kinase 2 in thyroid carcinomas. J Endocrinol. 2008;198:101–110. doi: 10.1677/JOE-07-0562. [DOI] [PubMed] [Google Scholar]
- Namkung Y, Dipace C, Urizar E, Javitch JA, Sibley DR. G protein-coupled receptor kinase-2 constitutively regulates D2 dopamine receptor expression and signaling independently of receptor phosphorylation. J Biol Chem. 2009;284:34103–34115. doi: 10.1074/jbc.M109.055707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijboer CH, Kavelaars A, Vroon A, Groenendaal F, van Bel F, Heijnen CJ. Low endogenous G-protein-coupled receptor kinase 2 sensitizes the immature brain to hypoxia-ischemia-induced gray and white matter damage. J Neurosci. 2008;28:3324–3332. doi: 10.1523/JNEUROSCI.4769-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P, Ribas C, Aymerich I, Eijkelkamp N, Barreiro O, Heijnen CJ, Kavelaars A, Sanchez-Madrid F, Mayor F., Jr G protein-coupled receptor kinase 2 positively regulates epithelial cell migration. EMBO J. 2008;27:1206–1218. doi: 10.1038/emboj.2008.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P, Ribas C, Aymerich I, Mayor F., Jr New roles of G protein-coupled receptor kinase 2 (GRK2) in cell migration. Cell Adhes Migr. 2009;3:19–23. doi: 10.4161/cam.3.1.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P, Murga C, Ribas C, Lafarga V, Mayor F., Jr The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010a;160:821–832. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penela P, Rivas V, Salcedo A, Mayor F., Jr G protein-coupled receptor kinase 2 (GRK2) modulation and cell cycle progression. Proc Natl Acad Sci USA. 2010b;107:1118–1123. doi: 10.1073/pnas.0905778107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppel K, Jacobson A, Huang X, Murray JP, Oppermann M, Freedman NJ. Overexpression of G protein-coupled receptor kinase-2 in smooth muscle cells attenuates mitogenic signaling via G protein-coupled and platelet-derived growth factor receptors. Circulation. 2000;102:793–799. doi: 10.1161/01.cir.102.7.793. [DOI] [PubMed] [Google Scholar]
- Prowatke I, Devens F, Benner A, Grone EF, Mertens D, Grone HJ, Lichter P, Joos S. Expression analysis of imbalanced genes in prostate carcinoma using tissue microarrays. Br J Cancer. 2007;96:82–88. doi: 10.1038/sj.bjc.6603490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Ruiz R, Penela P, Penn RB, Mayor F., Jr Analysis of the human G protein-coupled receptor kinase 2 (GRK2) gene promoter: Regulation by signal transduction systems in aortic smooth muscle cells. Circulation. 2000;101:2083–2089. doi: 10.1161/01.cir.101.17.2083. [DOI] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, Garcia-Hoz C, Jurado-Pueyo M, Aymerich I, Mayor F., Jr The G protein-coupled receptor kinase (GRK) interactome: Role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta. 2007;1768:913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Salcedo A, Mayor F, Jr., Penela P. Mdm2 is involved in the ubiquitination and degradation of G-protein-coupled receptor kinase 2. EMBO J. 2006;25:4752–4762. doi: 10.1038/sj.emboj.7601351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor–mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Shahid G, Hussain T. GRK2 negatively regulates glycogen synthesis in mouse liver FL83B cells. J Biol Chem. 2007;282:20612–20620. doi: 10.1074/jbc.M700744200. [DOI] [PubMed] [Google Scholar]
- Song YJ, Stinski MF. Inhibition of cell division by the human cytomegalovirus IE86 protein: Role of the p53 pathway or cyclin-dependent kinase 1/cyclin B1. J Virol. 2005;79:2597–2603. doi: 10.1128/JVI.79.4.2597-2603.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroon A, Heijnen CJ, Lombardi MS, Cobelens PM, Mayor F, Jr., Caron MG, Kavelaars A. Reduced GRK2 level in T cells potentiates chemotaxis and signaling in response to CCL4. J Leukoc Biol. 2004;75:901–909. doi: 10.1189/jlb.0403136. [DOI] [PubMed] [Google Scholar]
- Vroon A, Kavelaars A, Limmroth V, Lombardi MS, Goebel MU, Van Dam AM, Caron MG, Schedlowski M, Heijnen CJ. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Immunol. 2005;174:4400–4406. doi: 10.4049/jimmunol.174.7.4400. [DOI] [PubMed] [Google Scholar]
- Wang XW, Hussain SP, Huo TI, Wu CG, Forgues M, Hofseth LJ, Brechot C, Harris CC. Molecular pathogenesis of human hepatocellular carcinoma. Toxicology. 2002;181–182:43–47. doi: 10.1016/s0300-483x(02)00253-6. [DOI] [PubMed] [Google Scholar]
- Yu J, Guo QL, You QD, Zhao L, Gu HY, Yang Y, Zhang HW, Tan Z, Wang X. Gambogic acid-induced G2/M phase cell-cycle arrest via disturbing CDK7-mediated phosphorylation of CDC2/p34 in human gastric carcinoma BGC-823 cells. Carcinogenesis. 2007;28:632–638. doi: 10.1093/carcin/bgl168. [DOI] [PubMed] [Google Scholar]