

Abstract
Studies of AMCase inhibition in mouse models of lung eosinophilic inflammation have produced conflicting results with some studies demonstrating inhibition of eosinophilic inflammation and others not. No studies have investigated the role of AMCase inhibition in eosinophilic esophagitis (EoE). We have used a mouse model of egg (OVA) induced EoE to determine whether pharmacologic inhibition of AMCase with allosamidin reduced eosinophilic inflammation and remodeling in the esophagus in EoE. Administration of intra-esophageal OVA for 6 weeks to BALB/c mice induced increased levels of esophageal eosinophils, mast cells, and features of esophageal remodeling (fibrosis, basal zone hyperplasia, deposition of the extracellular matrix protein fibronectin). Administration of intraperitoneal (ip) allosamidin to BALB/c mice significantly inhibited AMCase enzymatic activity in the esophagus. Pharmacologic inhibition of AMCase with ip allosamidin inhibited both OVA induced increases in esophageal eosinophilic inflammation and OVA induced esophageal remodeling (fibrosis, epithelial basal zone hyperplasia, extracellular matrix deposition of fibronectin). This inhibition of eosinophilic inflammation in the esophagus by ip allosamidin was associated with reduced eotaxin-1 expression in the esophagus. Oral allosamidin inhibited eosinophilic inflammation in the epithelium but did not inhibit esophageal remodeling. These studies suggest that pharmacologic inhibition of AMCase results in inhibition of eosinophilic inflammation and remodeling in the esophagus in a mouse model of egg induced EoE partially through effects in the esophagus on reducing chemokines (i.e. eotaxin-1) implicated in the pathogenesis of EoE.
Keywords: Allosamidin, eosinophil, IL-13, eotaxin-1, eosinophilic esophagitis
1. INTRODUCTION
Chitinases (such as acid mammalian chitinase the focus of this study) are a family of evolutionary conserved enzymes present not only in lower life forms but also in mammals that can cleave chitin a naturally occurring polysaccharide composed of N-acetylglucosamine repeats [1,2]. Chitin is highly expressed in insects and crustacean exoskeletons, fungal cell walls, and microfilarial nematode sheaths [3]. Levels of chitin are regulated by enzymes synthesizing chitin (i.e. chitin synthase) or degrading chitin (i.e. chitinases). Although mammals do not synthesize chitin (as they do not express enzymes such as chitin synthase), they do express enzymes that can degrade chitin such as acid mammalian chitinase (AMCase) [4]. Studies in mice and humans suggest that chitinases may modulate the innate immune response by either interacting with chitin [5, 6], or by exerting effects on the immune system independent of actions on chitin [7,8]. AMCase is a member of the mammalian chitinase family that has been studied in the context of eosinophilic inflammation and asthma [2, 7, 9-13], but not in eosinophilic esophagitis (EoE) which is the focus of this study. In studies of mouse models of asthma, expression of AMCase can be induced in the lung by IL-13 [7] and allergens [10-13]. IL-13 transgenic mice have increased AMCase expression in the lung (in macrophages and epithelial cells), associated with increased eosinophilic lung inflammation [7]. Administration of a pharmacologic inhibitor of AMCase (allosamidin) to IL-13 transgenic mice inhibited AMCase activity and also inhibited eosinophilic lung inflammation [7] suggesting an important role for AMCase in mediating the effect of IL-13 in inducing eosinophilic lung inflammation. As IL-13 regulates AMCase expression [7], is highly expressed in the esophagus in eosinophilic esophagitis (EoE)[14], and the IL-13 transcriptome is implicated in the pathogenesis of EoE [14], in this study we have used a mouse model of egg (OVA) induced EoE to examine whether AMCase is expressed in the esophagus and whether blocking AMCase activity with a pharmacologic inhibitor inhibits esophageal eosinophilic inflammation and remodeling of the esophagus.
EoE is a disease defined by combined clinical and histological criteria (>15 esophageal eosinophils/hpf)[15]. Increased numbers of mast cells are also noted in the esophagus in EoE [16]. In addition to the presence of inflammation of the esophagus, EoE may be complicated by the development of remodeling of the esophagus [17] and stricture formation which may necessitate repeated esophageal dilation to prevent food impactions [15]. Features of esophageal remodeling in EoE include esophageal fibrosis, epithelial basal zone thickening, and angiogenesis [15-17]. Both eosinophils and mast cells likely contribute to esophageal remodeling in EoE as increased numbers of eosinophils expressing the pro-remodeling cytokine TGF-β1 are present in the remodeled esophagus in EoE [15-17]. In addition to increased eosinophils in EoE, increased numbers of tryptase positive mast cells are present in the esophageal smooth muscle in EoE, express TGF-β1, and increase the contractility of human esophageal smooth muscle cells in vitro [16]. As such, mast cells localized to the smooth muscle in patients with EoE might modulate esophageal contractility and contribute to symptoms in EoE.
As EoE is associated with significant symptoms (nausea, vomiting, abdominal pain) and complications from remodeling such as stricture formation and food impaction, therapeutic options are needed that not only reduce esophageal eosinophilic inflammation, but also mast cells, and remodeling. In this regard current therapeutic options include elemental diet which eliminates symptoms and underscores the food dependence of EoE [15]. However, difficulties with diet adherence result in this not readily being acceptable as a long term option for most patients with EoE. Topical corticosteroids reduce esophageal eosinophils, mast cells, and features of remodeling in many but not all subjects with EoE [18]. Novel therapies such as anti-IL-5 significantly reduce eosinophilic inflammation but have not reduced symptoms in studies of EoE [19]. In this study we have used a mouse model of food (i.e. egg) induced EoE previously described in this laboratory [20] to demonstrate that allosamidin a pharmacologic inhibitor of AMCase [21] inhibits AMCase activity in the esophagus and reduces esophageal levels of eosinophils, and features of esophageal remodeling suggesting that allosamidin may be a novel therapy to investigate in EoE.
2. MATERIAL AND METHODS
2.1 Oral egg (OVA) allergen induced eosinophilic esophagitis
Eight- to ten week-old female BALB/c mice (12 mice/group; Charles River Labs Inc, Wilmington, MA) were sensitized intraperitoneally with 50μg OVA adsorbed to 1mg of aluminum hydroxide adjuvant (OVA; grade V: Sigma- Aldrich, St. Louis, MO) on day 0 and day 14 and then challenged intra-esophageally three times/week for four weeks with 10 mg OVA suspended in 100 μl PBS on days 28, 30, 32, 35, 37, 39, 42, 44, 46, 49, 51, 53 as previously described in this laboratory [20]. OVA was administered through an intragastric feeding needle (20-gauge, 1.5-inch; Pepper and Sons, Inc, New Hyde Park, NY). Mice were sacrificed 24 hrs after the last administration of intra-esophageal OVA (day 54). Control BALB/c mice were neither sensitized nor challenged. The esophagus was removed in its entirety and fixed with 4% paraformaldehyde solution (Electron Microscopy Sciences, Hatfield, PA) for 24 hrs., oriented and embedded in 1% agarose (Invitrogen, Carlsbad, CA), and subsequently five μm sections (upper, middle and lower) embedded in paraffin. Esophageal sections from each layer were included for analysis of esophageal inflammation and esophageal remodeling. In each experiment in which stained and immunostained slides were quantified by image analysis, identical light microscope conditions, magnification, gain, camera position, and background illumination were utilized. The analysis of slides was performed by an investigator blinded to the study group. All animal experimental protocols were approved by the University of California, San Diego Animal Subjects Committee.
2.2 Pharmacologic intervention with chitinase inhibitor allosamidin in mouse model of OVA induced eosinophilic esophagitis
Three different groups of BALB/c mice (n=12 mice/group) were studied: Group 1, no OVA; Group 2, OVA+ chitinase inhibitor allosamidin [21]; Group 3, OVA + control diluent. Allosamidin (1 mg/kg, a kind gift from Dr. Sakuda, Tokyo University, Japan) or PBS diluent were administered in a volume of 100μL intraperitonealy 1 hr before each of the twelve intraesophageal OVA challenges (administered between day 28-53) as described above. In selected experiments allosamidin (1 mg/kg) or PBS diluent were administered using an intragastric feeding needle (as described above for OVA challenges) in a volume of 100μL intraesophageally 1 hr before each of the twelve intra-esophageal OVA challenges.
2.3 Eosinophil quantitation in esophagus
Eosinophils were detected in esophageal tissue by immunohistochemistry using an anti-mouse major basic protein (MBP) antibody (kindly provided by James Lee PhD, Mayo Clinic, Scottsdale, AZ). Quantitation of the number of eosinophils was performed using a light microscope attached to an image-analysis system with the entire cross section of the esophagus visualized [20]. The area of the esophageal lamina propria, epithelium, or esophageal muscle layer was outlined and this area determined by the image analysis software (Image-Pro Plus; Media Cybernetics). Results are expressed as either the number of eosinophils per mm2 of lamina propria, the number of eosinophils per mm2 of esophageal epithelium, or the number of eosinophils per mm2 of esophageal muscle layer.
2.4 Mast cell quantitation in esophagus
As increased esophageal mast cells are also a prominent feature of EoE [16], esophageal sections were also stained for mast cells using chloroacetate esterase as previously described in this laboratory [22]. Results are expressed as the number of mast cells per mm2 of lamina propria.
2.5 Quantitation of esophageal remodeling
We quantitated features of esophageal remodeling characteristic of EoE including esophageal fibrosis, epithelial basal zone hyperplasia, and extracellular deposition of matrix proteins [17,20].
Esophageal trichrome staining
The area of esophageal trichrome staining (as an index of collagen deposition) in paraffin embedded esophagus was outlined and quantified using a light microscope (Leica DMLS, Leica Microsystems Inc., NY) attached to an image analysis system (Image-Pro plus, Media Cybernetics, MI) as previously described [17,20]. Results are expressed as the area in μm2 of trichrome staining per μm2 total area of lamina propria.
Esophageal collagen
The amount of esophageal collagen was measured with a collagen assay that uses a dye reagent that selectively binds to the [Gly-X-Y]n tripeptide sequence of mammalian collagens (Biocolor) as previously described in this laboratory [23]. In all experiments, a collagen standard was used to calibrate the assay. Results are expressed as μg collagen.
Deposition of extracellular matrix protein
The deposition of the extracellular matrix protein fibronectin is associated with tissue remodeling [22]. We used two methods (ELISA and immunohistochemistry) to quantitate fibronectin deposition in the esophagus. Levels of fibronectin in esophagus lysates were assayed by ELISA (Assaypro, St. Charles, MO). The sensitivity of the fibronectin assay is 50ng/ml. Esophageal lysates were prepared by homogenizing esophageal tissue in lysis buffer as previously described in this laboratory [23]. Esophageal supernatants (obtained by centrifugation of lysates at 10,000 g for 20 min) were passaged through 0.8 μm pore size filter and frozen at −80°C in polypropylene tubes until used in ELISA assays.
The area of fibronectin immunostaining in the esophagus was quantitated by immunohistochemistry using an anti-fibronectin Ab (Abcam, Cambridge, MA) and quantified using a light microscope attached to an image analysis system. Results are expressed as the area of fibronectin staining per total esophageal LP area.
Esophageal epithelial basal zone thickness
Basal zone hyperplasia is a feature of EoE [17, 20]. The epithelial basal zone thickness was quantitated in esophageal sections stained with hematoxylin and eosin using a light microscope attached to an image-analysis system. The thickness of the basal layer was recorded in μm in four randomly selected areas of esophageal epithelium in each slide.
Esophageal TGF-β1+ cells
Esophageal tissue sections were processed for immunohistochemistry using a primary mAb directed against either TGF-β1 (Santa Cruz Biotechnology Inc, Santa Cruz, CA) as described [17,20]. Results are expressed as the number of TGF-β1 positive cells per mm2 of lamina propria.
2.6 Esophageal levels of chemokines/cytokines that regulate eosinophilic inflammation
Levels of the eosinophil chemoattractant eotaxin-1, and Th2 cytokines (IL-5, IL-13) were measured in esophageal lysates by ELISA (R&D System Inc, Minneapolis, Minn). The IL-5, IL-13, and eotaxin-1 assays each had a sensitivity of 15 pg/ml.
2.7 Chitinase bioactivity assay
Levels of AMCase enzymatic activity was measured in the homogenized esophageal lysates of mice treated with either allosamidin or diluent control. AMCase enzyme activity was determined using the AMCase fluorogenic substrate 4MU-chitobiose (4-methyllumbelliferyl-β-D-N,N’-diacetylchitobiose; Sigma, St Louis, MO)[4] in an acidic environment (pH~4.5) at 37°C. The florescence of liberated 4MU (4-methyllumbelliferone) was measured using a fluorimeter with excitation at 360nm and emission at 450nm [4].
2.8 Statistical analysis
Results were compared by a Mann-Whitney test using a statistical software package (GraphPad Prism, San Diego, CA). P values <0.05 were considered statistically significant. Results are presented as mean ± SEM.
3. RESULTS
3.1 Allosamidin ip inhibits oral OVA induced eosinophilic inflammation in the esophagus
Oral OVA challenge induced a significant increase in esophageal eosinophils (Fig 1 A-B). Eosinophils were infiltrated into the esophageal epithelium, lamina propria, and muscle layer. OVA challenge increased the total number of MBP+ eosinophils in the esophagus including the epithelium (OVA vs no OVA)(p=0.0001)(Fig 1C), the lamina propria (OVA vs no OVA)(p =0.0001)(Fig 1D), and the muscle layer (OVA vs no OVA)(p= 0.0001)(Fig 1E).
FIGURE 1. Effect of allosamidin on oral OVA induced eosinophilic inflammation in esophagus.

The esophagus from non-OVA challenged (Fig 1A) and OVA challenged (Fig 1B) were immunostained with an anti-MBP Ab. The number of MBP+ eosinophils in the esophagus were quantitated per mm2 of epithelium (Fig 1C), per mm2 of lamina propria (Fig 1D), and per mm2 of muscle layer (Fig 1E) in three different groups (no OVA; OVA+ diluent; OVA+ allosamidin)(n=12 mice/group).
IP administration of the AMCase inhibitor allosamidin induced a significant decrease in esophageal eosinophils (Fig 1). Allosamidin ip induced a 63 % decrease in eosinophil numbers in esophageal epithelium (OVA vs Allosamidin ip + OVA)(p=0.03)(Fig 1C), a 50 % decrease in eosinophils in the lamina propria (OVA vs Allosamidin ip + OVA)(p=0.04)(Fig 1D), and a statistically insignificant trend of fewer in eosinophils in the muscle layer (OVA vs Allosamidin ip + OVA)(p=0.8)(Fig 1E).
3.2 Allosamidin ip inhibits oral OVA induced esophageal remodeling
Esophageal fibrosis
Oral OVA challenge induced a significant increase in esophageal fibrosis as assessed by either the area of esophageal trichrome staining (OVA vs no OVA)(p=0.0001)(Fig 2A) or the amount of esophageal collagen (OVA vs no OVA)(p=0.001)(Fig 2B). Allosamidin ip decreased the area of esophageal trichrome staining by 18% (OVA vs Allosamidin ip + OVA)(p=0.004)(Fig 2A). Although allosamidin ip also decreased the amount of esophageal collagen by a similar amount to that detected with trichrome staining (i.e. 18%), this decrease in collagen approached but did not reach statistical significance (OVA vs Allosamidin ip + OVA)(p=0.08)(Fig 2B).
FIGURE 2. Effect of allosamidin on oral OVA induced esophageal remodeling.

The esophagus from mice belonging to three different groups (no OVA; OVA+ diluent; OVA+ allosamidin)(n=12 mice/group) were processed to detect features of esophageal remodeling including, the area of esophageal trichrome staining (Fig 2A), the amount of esophageal collagen (Fig 2B), the area of esophageal fibronectin staining (Fig 2C), the amount of esophageal fibronectin quantitated by Elisa (Fig 2D), and the thickness of the esophageal epithelial basal zone layer (Fig 2E), and the number of esophageal TGFβ1 positive cells (Fig 2F).
Esophageal deposition of fibronectin
Levels of deposition of the extracellular matrix protein fibronectin were significantly increased in the esophagus following oral OVA challenge as assessed by either the area of eosophageal fibronectin staining (OVA vs no OVA)(p=0.0001)(Fig 2C) or the amount of esophageal fibronectin as quantitated by ELISA (OVA vs no OVA)(p=0.02)(Fig 2D). Allosamidin ip decreased the area of esophageal fibronectin staining in oral OVA challenged mice (OVA vs Allosamidin ip + OVA)(p=0.0001)(Fig 2C), and the amount of esophageal fibronectin quantitated by ELISA (OVA vs Allosamidin ip + OVA)(p=0.002)(Fig 2D).
Esophageal epithelial basal zone hyperplasia
The thickness of the esophageal epithelial basal zone was significantly increased following OVA challenge (OVA vs no OVA)(p=0.0001)(Fig 2E). Allosamidin ip decreased the thickness of the esophageal epithelial basal zone (OVA vs Allosamidin ip + OVA)(p<0.0001)(Fig 2E),
Esophageal TGF-β1+ cells
The number of esophageal TGF-β1+ cells increased significantly in OVA challenged mice (p=0.01)(Fig 2F). Allosamidin ip decreased the number of esophageal TGF-β1+ cells (OVA vs Allosamidin ip + OVA)(p=0.05)(Fig 2F).
3.3 Allosamidin ip inhibits oral OVA induced expression of eotaxin-1 in the esophagus
Oral OVA challenge induced a significant increase in esophageal eotaxin-1 (OVA vs no OVA)(p=0.01)(Fig 3A), IL-13 (OVA vs no OVA)(p=0.004)(Fig 3B), but not IL-5 (OVA vs no OVA)(p=ns)(Fig 3C) as assessed by ELISA. Administration of allosamidin ip to oral OVA challenged mice significantly reduced levels of esophageal eotaxin-1 (OVA vs Allosamidin ip + OVA)(p=0.05)(Fig 3A). Allosamidin ip induced a trend for reduction in IL-13 which was not significant (OVA vs Allosamidin ip + OVA)(p=0.11)(Fig 3B), and no reduction in IL-5 (OVA vs Allosamidin ip + OVA)(p=ns)(Fig 3C).
FIGURE 3. Effect of allosamidin on oral OVA induced esophageal levels of IL-13, eotaxin-1, and IL-5.

The esophagus from mice belonging to three different groups (no OVA; OVA+ diluent; OVA+ allosamidin)(n=12 mice/group) were processed for ELISA to quantitate levels of eotaxin-1(Fig 3A), IL-13 (Fig 3B), and IL-5 (Fig 3C).
3.4 Effect of allosamidin ip on oral OVA induced mast cell inflammation in the esophagus
Oral OVA challenge induced a significant increase in mast cells in the lamina propria (p= 0.001)(Fig 4), but not the epithelium (data not shown). Administration of allosamidin ip to oral OVA challenged mice resulted in a trend for reduced numbers of mast cells that approached but did not reach statistical significance (OVA vs Allosamidin ip + OVA)(p=0.07)(Fig 4).
FIGURE 4. Effect of allosamidin on oral OVA induced mast cell inflammation in esophagus.
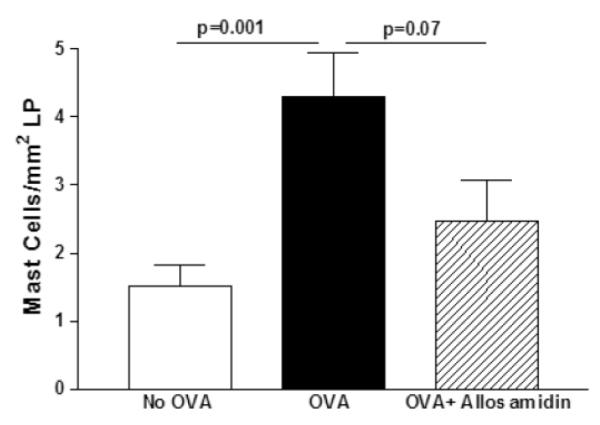
The esophagus from mice belonging to three different groups (no OVA; OVA+diluent; OVA+ allosamidin)(n=12 mice/group) were stained with chloroacetate esterase to detect mast cells which were quantitated per mm2 of lamina propria.
3.5 Effect of allosamidin ip on AMCase activity in the esophagus
The esophagus from non-OVA challenged mice expressed constitutive AMCase enzymatic activity (Fig 5). Following oral OVA challenge there was a slight but statistically significant increase in AMCase enzymatic activity in the esophagus (OVA vs no OVA)(p=0.03) (Fig 5). Administration of allosamedin ip significantly reduced AMCase activity in the esophagus (p=0.001 )(Fig 5).
FIGURE 5. Effect of allosamidin on oral OVA induced AMCase enzymatic activity in esophagus.
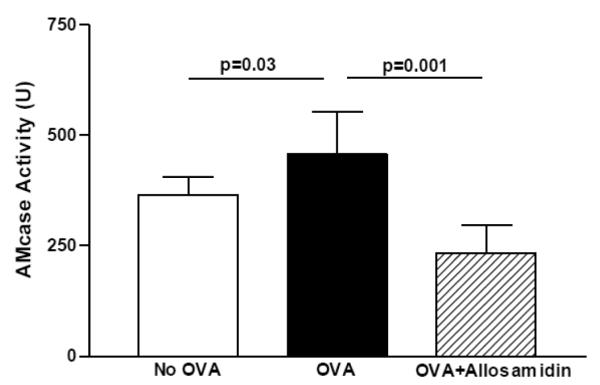
The esophagus from mice belonging to three different groups (no OVA; OVA+ diluent; OVA+ allosamidin)(n=12 mice/group) were processed to detect AMCase enzymatic activity using the substrate 4MU-chitobiose.
3.6 Effect of oral allosamidin on OVA induced esophageal eosinophilic inflammation
Oral administration of allosamidin significantly reduced the number of eosinophils in the esophageal epithelium of OVA challenged mice (OVA vs oral allosamidin + OVA)(p=0.03)(Fig 6A). While there was a trend for oral allosamidin to decrease the number of eosinophils in the lamina propria (Fig 6B), and smooth muscle (Fig 6C) this was not statistically significant. Oral administration of allosamedin did not inhibit basal layer thickness, or esophageal fibrosis as assessed by the area of trichrome staining (data not shown).
FIGURE 6. Effect of oral allosamidin on oral OVA induced eosinophilic inflammation in esophagus.

The number of MBP+ eosinophils in the esophagus were quantitated per mm2 of epithelium (Fig 6A), per mm2 of lamina propria (Fig 6B), and per mm2 of muscle layer (Fig 6C) in three different groups (no OVA; OVA+ diluent; OVA+ oral allosamidin)(n=5 mice/group).
4. DISCUSSION
In this study we have demonstrated in a mouse model of egg induced EoE that administration of a pharmacologic inhibitor of AMCase (i.e. allosamidin) ip significantly inhibited levels of esophageal AMCase activity, esophageal eosinophilic inflammation, as well as levels of esophageal remodeling including fibrosis, epithelial basal zone hyperplasia, and deposition of extracellular matrix proteins. The mechanism by which ip allosamidin inhibits esophageal eosinophilic inflammation involves effects in the esophagus to reduce key chemokines (i.e. eotaxin-1) regulating esophageal eosinophilic inflammation. Although previous studies of ip allosamidin in mouse models of asthma have demonstrated that it can inhibit eosinophilic lung inflammation [7,10,11], this is the first study to demonstrate that ip allosamidin can inhibit eosinophilic inflammation in the esophagus and describe potential mechanisms mediating this effect. For example, we have made the novel observation that ip allosamidin exerts inhibitory effects on key eosinophil chemokines expressed in the esophagus. Moreover, this is the first study to demonstrate that ip allosamidin can inhibit tissue remodeling which in EoE is important to the subset of subjects who develop esophageal strictures and food impactions. As eosinophils express TGF-β1 [17], the ability of allosamidin to inhibit the accumulation of eosinophils in the esophagus may result in reduced levels of TGF-β1 expression with resultant reduced levels of esophageal remodeling.
Insight into the mechanism by which allosamidin inhibits eosinophilic inflammation in the esophagus may be derived from studies of allosamidin, and its target AMCase. Allosamidin is a pseudo-trisaccharide consisting of two N-acetyl-D-allosamine sugars, linked to a novel moiety termed allosamizoline [21,24]. It was originally isolated from Streptomyces cultures [24] but has also been totally synthesized [25]. On the basis of sequence homologies, chitinases belong to a subset of the large family of glycosyl hydrolases (family 18 of which AMCase is a member; and family 19) [26]. Members of family 18 such as AMCase employ a substrate-assisted reaction mechanism [27], whereas those of family 19 adopt a fold-and-reaction mechanism similar to that of lysozyme [28], suggesting that these families of chitinases evolved independently to deal with chitin. Allosamidin inhibits all family 18 chitinases including AMCase (but not family 19 chitinases), with Ki in the nm to μm range [25]. The crystal structure of allosamidin complexed with human chitinase has also been described demonstrating that allosamidin binds in a groove on the chitinase [29].
Two mammalian chitinases have been cloned (AMCase and chitotriosidase)[4,30]. AMCase has been implicated in the pathogenesis of asthma [7,10,11], while increased expression of chitotriosidase is observed in lysosomal lipid storage disorders like Gaucher disease [31]. AMCase is an enzyme characterized by an acidic isoelectric point and therefore named acidic mammalian chitinase [4]. The enzyme is extremely acid stable and its constitutive expression is relatively abundant in the gastrointestinal tract and to a lesser extent in the lung [32]. AMCase is synthesized as a 50-kDa protein containing a 39-kDa N-terminal catalytic domain, a hinge region, and a C-terminal chitin-binding domain [4]. AMCase is expressed in alveolar macrophages and in the gastrointestinal tract where it has been hypothesized to play a role in digestion and/or host defense [4,32 ].
Studies in mouse models of asthma have provided conflicting evidence as to whether targeting AMCase inhibits eosinophilic lung inflammation [7,10,11], or not [12,13]. The studies in mouse models of asthma demonstrating that targeting AMCase inhibits eosinophilic lung inflammation include studies using pharmacologic inhibitors of AMCase (i.e. ip allosamidin)[7,10] and siRNA approaches to knockdown AMCase [11], while the studies that have not shown an inhibitory effect of targeting AMCse have also used pharmacologic inhibitors of AMCase (i.e. ip allosamidin)[12], and AMCase mutant mice [12,13]. It is unclear why these studies have resulted in differing results. Potential explanations include difference in protocols including the nature of the allergens used (fungal allergen Aspergillus which contains high levels of chitin vs OVA or dust mite), the allergen challenge protocol used, potential off target effects of pharmacologic inhibitors, and potential upregulation of compensatory pathways in studies using AMCase mutant mice.
We also performed studies to determine whether orally administered allosamidin could inhibit eosinophilic inflammation in the esophagus. As all prior in vivo studies with allosamidin in mice have used ip administration, we were unable to be guided by prior studies as to what might be an optimal oral allosamidin dose and dosing schedule. We selected an oral dose of allosamidin (1 mg/kg) that was the same as the ip dose used in this and other studies [7, 21]. This oral dose of allosamidin significantly inhibited esophageal epithelial eosinophilic inflammation. However, this oral dose of allosamidin was not effective in inhibiting eosinophilic inflammation in the LP, or in inhibiting esophageal fibrosis. Future studies in which the daily administration of oral allosamidin (only administered once prior to each OVA challenge in this study), and the dose of allosamidin is varied, will define the maximal inhibitory effect of oral allosamidin in this mouse model of EoE.
In summary, our study is the first study examining the effect of using ip allosamidin to inhibit AMCase and reduce eosinophilic inflammation in an organ other than the lung. Using a mouse model of egg induced EoE we have demonstrated that ip allosamidin significantly inhibited eosinophilic inflammation in the esophageal epithelium and lamina propria, but did not inhibit eosinophilic inflammation in smooth muscle. The reduction in esophageal expression of eotaxin-1 in allosamidin treated mice may be one mechanism by which allosamidin reduces esophageal eosinophilic inflammation. Furthermore, we demonstrate that ip allosamidin significantly reduces esophageal remodeling (fibrosis, extracellular deposition of extracellular matrix protein fibronectin, basal zone hyperplasia) a key finding in subjects with EoE and esophageal stricture formation [16,17]. Thus, targeting AMCase may be a novel therapeutic option in EoE, especially in the subset of EoE subjects with esophageal remodeling who may be at higher risk for esophageal stricture formation and food impactions.
Highlights.
Allosamidin inhibited AMCase activity in the esophagus
Allosamidin inhibited OVA induced esophageal eosinophilic inflammation
Allosamidin inhibited OVA induced esophageal remodeling
Allosamidin reduced OVA induced eotaxin-1 expression in the esophagus
ACKNOWLEDGEMENT
This research was supported by DOD Grant W81XWH-10-1-0705 to DB.
Dr Sakuda (Tokyo University, Japan) kindly provided allosamidin
Dr Broide is also supported by grants from the NIH AI 38425, AI 107779 AI 70535, and AI 72115.
Dr Aceves is supported by NIH AI 092135
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. REFERENCES
- 1.Bleau G, Massicotte F, Merlen Y, Boisvert C. Mammalian chitinase-like proteins. EXS. 1999;87:211–21. doi: 10.1007/978-3-0348-8757-1_15. [DOI] [PubMed] [Google Scholar]
- 2.Ober C, Chupp GL. The chitinase and chitinase-like proteins: a review of genetic and functional studies in asthma and immune-mediated diseases. Curr Opin Allergy Clin Immunol. 2009;9:401–8. doi: 10.1097/ACI.0b013e3283306533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee CG, Da Silva CA, Lee JY, Hartl D, Elias JA. Chitin regulation of immune responses: an old molecule with new roles. Curr Opin Immunol. 2008;20:684–9. doi: 10.1016/j.coi.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boot RG, Blommaart EF, Swart E, Ghauharali-van der Vlugt K, Bijl N, Moe C, et al. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J Biol Chem. 2001;276:6770–8. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- 5.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, et al. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–6. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalaker A, Nkrumah L, Lee WK, Ramanathan M, Lane AP. Chitin stimulates expression of acidic mammalian chitinase and eotaxin-3 by human sinonasal epithelial cells in vitro. Am J Rhinol Allergy. 2009;23:8–14. doi: 10.2500/ajra.2009.23.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu Z, Zheng T, Homer RJ, Kim YK, Chen NY, Cohn L, et al. Acidic mammalian chitinase in asthmatic Th2 inflammation and IL-13 pathway activation. Science. 2004;304:1678–82. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 8.Ramanathan M, Jr, Lee WK, Lane AP. Increased expression of acidic mammalian chitinase in chronic rhinosinusitis with nasal polyps. Am J Rhinol. 2006;20:330–5. doi: 10.2500/ajr.2006.20.2869. [DOI] [PubMed] [Google Scholar]
- 9.Bierbaum S, Nickel R, Koch A, Lau S, Deichmann KA, Wahn U, et al. Polymorphisms and haplotypes of acid mammalian chitinase are associated with bronchial asthma. Am J Respir Crit Care Med. 2005;172:1505–9. doi: 10.1164/rccm.200506-890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsumoto T, Inoue H, Sato Y, Kita Y, Nakano T, Noda N, et al. Demethylallosamidin, a chitinase inhibitor, suppresses airway inflammation and hyperresponsiveness. Biochem Biophys Res Commun. 2009;390:103–8. doi: 10.1016/j.bbrc.2009.09.075. [DOI] [PubMed] [Google Scholar]
- 11.Yang CJ, Liu YK, Liu CL, Shen CN, Kuo ML, Su CC, et al. Inhibition of acidic mammalian chitinase by RNA interference suppresses ovalbumin-sensitized allergic asthma. Hum Gene Ther. 2009;20:1597–606. doi: 10.1089/hum.2008.092. [DOI] [PubMed] [Google Scholar]
- 12.Van Dyken SJ, Garcia D, Porter P, Huang X, Quinlan PJ, Blanc PD, et al. Fungal chitin from asthma-associated home environments induces eosinophilic lung infiltration. J Immunol. 2011;187:2261–7. doi: 10.4049/jimmunol.1100972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitz LJ, DeClercq C, Brooks J, Kuang W, Bates B, Demers D, et al. Acidic mammalian chitinase is not a critical target for allergic airway disease. Am J Respir Cell Mol Biol. 2012;46:71–9. doi: 10.1165/rcmb.2011-0095OC. [DOI] [PubMed] [Google Scholar]
- 14.Sherrill JD, Rothenberg ME. Genetic dissection of eosinophilic esophagitis provides insight into disease pathogenesis and treatment strategies. J Allergy Clin Immunol. 2011;128:23–32. doi: 10.1016/j.jaci.2011.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liacouras CA, Furuta GT, Hirano I, Atkins D, Attwood SE, Bonis PA, et al. Eosinophilic esophagitis: updated consensus recommendations for children and adults. J Allergy Clin Immunol. 2011;128:3–20. doi: 10.1016/j.jaci.2011.02.040. [DOI] [PubMed] [Google Scholar]
- 16.Aceves SS, Chen D, Newbury RO, Dohil R, Bastian JK, Broide DH. Mast cells infiltrate the esophageal smooth muscle in patients with eosinophilic esophagitis, express TGF-β1, and increase esophageal smooth muscle contraction. J Allergy Clin Immunol. 2010;126:1198–204. doi: 10.1016/j.jaci.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 17.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol. 2007;119:206–12. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 18.Aceves SS, Newbury RO, Chen D, Mueller J, Dohil R, Hoffman H, et al. Resolution of remodeling in eosinophilic esophagitis correlates with epithelial response to topical corticosteroids. 2010;65:109–16. doi: 10.1111/j.1398-9995.2009.02142.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Assa’ad AH, Gupta SK, Collins MH, Thomson M, Heath AT, Smith DA, et al. An antibody against IL-5 reduces numbers of esophageal intraepithelial eosinophils in children with eosinophilic esophagitis. Gastroenterology. 2011;141:1593–604. doi: 10.1053/j.gastro.2011.07.044. [DOI] [PubMed] [Google Scholar]
- 20.Rubinstein E, Cho JY, Rosenthal P, Chao J, Miller M, Pham A, et al. Siglec-F inhibition reduces esophageal eosinophilia and angiogenesis in a mouse model of eosinophilic esophagitis. J Pediatr Gastroenterol Nutr. 2011;53:409–16. doi: 10.1097/MPG.0b013e3182182ff8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakanishi E, Okamoto S, Matsuura H, Nagasawa H, Sakuda S. Allosamidin, a chitinase inhibitor produced by Streptomyces, acts as an inducer of chitinase production in its producing strain. Proc Japan Acad. 2001;77:79–82. [Google Scholar]
- 22.Song DJ, Cho JY, Miller M, Strangman W, Zhang M, Varki A, et al. Anti-Siglec-F antibody inhibits oral egg allergen induced intestinal eosinophilic inflammation in a mouse model. Clin Immunol. 2009;131:157–169. doi: 10.1016/j.clim.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho JY, Miller M, Baek KJ, Han JW, Nayar J, Lee SY, et al. Inhibition of airway remodeling in IL-5-deficient mice. J Clin Invest. 2004;113:551–60. doi: 10.1172/JCI19133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakuda S, Isogai A, Matsumoto S, Suzuki A, Koseki K. The structure of allosamidin, a novel insect chitinase inhibitor, produced by Streptomyces sp. Tetrahedron Lett. 1986;27:2475–8. [Google Scholar]
- 25.Berecibar A, Grandjean C, Siriwardena A. Synthesis and biological activity of natural aminocyclopentitol glycosidase inhibitors: mannostatins, trehazolin, allosamidins, and their analogues. Chem Rev. 1999;99:779–844. doi: 10.1021/cr980033l. [DOI] [PubMed] [Google Scholar]
- 26.Henrissat B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem J. 1991;280:309–16. doi: 10.1042/bj2800309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Scheltinga TAC, Armand S, Kalk KH, Isogai A, Henrissat B, Dijkstra BW. Stereochemistry of chitin hydrolysis by a plant chitinase/lysozyme and X-ray structure of a complex with allosamidin: evidence for substrate assisted catalysis. Biochemistry. 1995;34:15619–23. doi: 10.1021/bi00048a003. [DOI] [PubMed] [Google Scholar]
- 28.Monzingo AF, Marcotte EM, Hart PJ, Robertus JD. Chitinases, chitosanases, and lysozymes can be divided into procaryotic and eucaryotic families sharing a conserved core. Nat Struct Biol. 1996;3:133–40. doi: 10.1038/nsb0296-133. [DOI] [PubMed] [Google Scholar]
- 29.Rao FV, Houston DR, Boot RG, Aerts JM, Sakuda S, van Aalten DM. Crystal structures of allosamidin derivatives in complex with human macrophage chitinase. J Biol Chem. 2003;278:20110–6. doi: 10.1074/jbc.M300362200. [DOI] [PubMed] [Google Scholar]
- 30.Bussink AP, Speijer D, Aerts JM, Boot RG. Evolution of mammalian chitinase(-like) members of family 18 glycosyl hydrolases. Genetics. 2007;177:959–70. doi: 10.1534/genetics.107.075846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boot RG, Renkema GH, Verhoek M, Strijland A, Bliek J, de Meulemeester TM, et al. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J Biol Chem. 1998;273:25680–5. doi: 10.1074/jbc.273.40.25680. [DOI] [PubMed] [Google Scholar]
- 32.Boot RG, Bussink AP, Verhoek M, de Boer PA, Moorman AF, Aerts JM. Marked differences in tissue-specific expression of chitinases in mouse and man. J Histochem Cytochem. 2005;53:1283–92. doi: 10.1369/jhc.4A6547.2005. [DOI] [PubMed] [Google Scholar]