

Abstract
The total synthesis of the architecturally complex Daphniphyllum alkaloid (−)-calyciphylline N has been achieved. Highlights of the synthesis include a Et2AlCl promoted, highly stereoselective susbtrate controlled intramolecular Diels-Alder reaction, a transannular enolate alkylation, an effective Stille carbonylation/Nazarov cyclization sequence, and a high risk dia-stereoselective hydrogenation of a fully substituted conjugated diene ester.
The Daphniphyllum alkaloids comprise a large family of complex natural products including more than 180 known members,1 many with diverse biological activities, that have proven challenging as targets for total synthesis.2 We in particular became interested in the calyciphylline alkaloids, largely due to their unique frameworks and at the time, limited synthetic studies.3,4 Calyciphylline N [(−)-1, Figure 1], isolated in 2008 by Kobayashi and coworkers,5 was chosen as our initial target.
Figure 1.

(−)-Calyciphylline N and related congeners.6
While the biological activity of (−)-calyciphylline N (1) has not been investigated, we reasoned that a synthetic effort towards this alkaloid would not only unveil a wealth of interesting reactivity, but also permit access to other congeners of the family. Notable structural features of 1 include six contiguous stereogenic centers, three of which are quaternary bridgehead, a fused A ring dihydropyrrole, and a DEF decahydrocyclopentazulene ring system surrounding a central bicyclo[2.2.2]octane BC core.
Retrosynthetically (Figure 2), we envisioned that the dihydropyrrole A ring could arise via condensation of a primary amine with the carbonyl group in ring B, while the stereochemistry of the EF ring system could be installed by a challenging/critical, late stage α,β-reduction of an exceptionally hindered diene ester (2). The secondary hydroxyl in ring C could in turn be generated via a Tamao-Kumada7 oxidation of the siloxane ring, while construction of ring F would entail an aldol condensation, simplifying the structure to 3, the latter accessible from 4 via a cyclopentenone annulation involving a Stille carbonylation8/Nazarov cyclization9 sequence. Continuing with this analysis, tetracycle 4 could be accessed through elaboration of bicyclic ester 5, anticipated to be the product of an intramolecular Diels-Alder (IMDA) reaction.10 The requisite IMDA tri-ene, in turn, would arise via union of enantiomerically pure homoallylic alcohol 6 and known silyl acrylate 7.11
Figure 2.

Retrosynthetic Analysis.
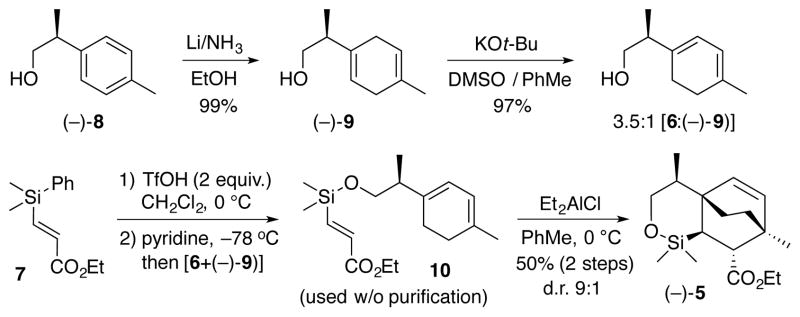
The synthesis of (−)-calyciphylline N (1) began with alcohol (−)-8 (Scheme 1), prepared in three steps from commercially available p-tolylacetic acid.12 Birch reduction13 readily furnished the desired cyclohexadiene; olefin isomerization with KOt-Bu in DMSO14 then provided an inseparable mixture (3.5:1) of the 1,3- and 1,4-dienes 6 and (−)-9, respectively, in excellent yield. Silylacrylate 7, obtained via oxidative hydrosilylation of ethyl acrylate with phenyldimethylsilane,11 was subsequently appended, employing a method introduced by Sieburth.15 To this end, treatment of 7 with TfOH at 0 °C, followed by sequential addition of pyridine and the mixture of the alcohols 6 and (−)-9 at −78 °C led to the requisite triene 10 for the IMDA reaction. However, due to the instability of 10 towards silica gel chromatography, the mixture was carried forward without purification. Interestingly, while the thermal Diels-Alder reaction led to a mixture of all possible diastereomers (as determined by 1H NMR), we were pleased to discover that the Et2AlCl promoted cyclization provided a 9:1 mixture of diastereomers in favor of the desired cycloadduct (−)-5.
Scheme 1.
One carbon homologation of (−)-5 to alcohol (−)-13 (Scheme 2) was next achieved by LiAlH4 reduction and conversion to the corresponding iodide (−)-11, followed by cyanide displacement and a two step reduction of the resulting nitrile. The overall yield for the five steps was 65%. Epoxidation of the olefin in (−)-13 with m-CPBAthen led to (−)-14 as a single diastereomer in 70% yield. Installation of the C1 ketone was next achieved in two steps and 67% yield via an acid promoted epoxide opening and oxidation of the resulting secondary alcohol, employing Dess-Martin periodinane (DMP),16 to deliver (+)-15. Reductive cleavage of the tetrahydropyran ring with SmI2 in a mixture of THF/MeOH then led to hydroxy ketone (+)-16 in 82% yield,17 the primary alcohol of which was protected as the TBS ether (TBSCl/imidazole in DMF) to provide (+)-17.
Scheme 2.

Elaboration of the requisite sidechain for eventual construction of ring D called for introduction of disubstitution α to the carbonyl in ketone (+)-17 (Scheme 3). Initially, (+)-17 proved unreactive towards standard acylating agents (EtOAc, Ac2O, 1-acetylimidazole, etc.), employing the lithium, potassium, or sodium enolates, with the exception of AcCl which led to complex mixtures of C- and O-acylated products. However, an LDA mediated aldol reaction with acetaldehyde, followed without purification by DMP oxidation of the β-hydroxy ketone, provided diketone (+)-18 in 91% yield for the 2 steps.18 Introduction of the allyl group via the Tsuji-Trost allylation19 then furnished (−)-19 as a single diastereomer (95%), which upon exposure to catalytic p-TsOH in MeOH cleanly led to the corresponding alcohol (−)-20, the latter converted to primary iodide (−)-21 in 97% yield. Pleasingly, transannular cyclization utilizing LDA delivered tetracycle (+)-4 as a crystalline solid (m.p. 123–125 °C), completing the construction of ring D. Interestingly, use of NaHMDS led only to elimination of the iodide. Single crystal X-ray analysis of (+)-4 confirmed the structure, as well as the relative and absolute configurations.
Scheme 3.

Before turning to elaboration of the eastern hemisphere, we investigated the proposed Tamao-Kumada6 oxidation of the siloxane ring in (+)-4. Unfortunately, the siloxane was found to be completely inert to the oxidation. Standard conditions (various fluoride sources, H2O2, and bicarbonate salts)7,20 led only to the recovery of starting material, while strongly basic conditions21 resulted in decomposition. Curiously, the use of TBAF (with or without oxidant) resulted in desilylation rather than oxidation.22 Attempts to transform the siloxane to a more reactive silane (e.g., silyl halide or hydride)23 prior to oxidation also proved unrewarding. Earlier studies, however, had demonstrated that a similar siloxane was a substrate for nucleophilic ring opening at the Si-O bond upon treatment with aryllithium reagents. Such reactivity was recently employed for the development of siloxanes as recoverable transfer agents in Pd-catalysed cross coupling reactions.24 We surmised that an arylsilane could be converted to the corresponding alcohol via the Fleming modification25 of the Tamao-Kumada oxidation.
With these earlier observations in mind, attempted siloxane opening in (+)-4 resulted in partial isomerization of the terminal alkene (not shown). We therefore functionalized the allyl group before moving forward (Scheme 4). Brown hydroboration (9-BBN) and oxidation (NaOH and H2O2)25 delivered the expected primary alcohol in 71% yield. Protection of the alcohol with TBSCl then furnished (+)-22 in 94% yield. Pleasingly, the siloxane could now be converted to arylsilane (+)-23 in excellent yield by treatment with 4-methoxyphenyllithium at room temperature. Selection of the methoxyphenyl substituent was in anticipation of greater reactivity towards the protodesilylation step of the Fleming-Tamao oxidation.26 Notably, both hindered carbonyls in (+)-23 remained completely inert to nucleophilic addition. Moving forward, rather than protect the newly generated primary alcohol, we introduced the requisite nitrogen with protection via treatment of (+)-23 with phthalimide under Mitsunobu conditions27 to provide (+)-24 in 99% yield.
Scheme 4.

Turning to construction of ring E via the proposed Stille carbonylation7/Nazarov8 cyclization sequence, exposure of (+)-24 to KHMDS in the presence of PhN(Tf)2 at −78 °C furnished vinyl triflate (+)-25, which underwent a highly efficient Stille carbonylation in DMF at 90 °C, employing only 1 atm. of CO, to provide dienone (+)-26 in 97% yield. Given that both Nazarov cyclizations and protodesilylations can be achieved with protic acid,8,24 we reasoned that both transformations could be accomplished in the same flask. Indeed, treatment of (+)-26 with HBF4•OEt2 at ambient temperature directly furnished silyl fluoride (+)-27 in 82% overall yield (Scheme 5). Under these conditions, the primary TBS group was also removed. We were also pleased to discover that treatment of (+)-27 with KF and m-CPBA in DMF resulted in the successful Fleming-Tamao oxidation to diol (+)-28 in 74% yield. Differentiation of the alcohols was then realized via chemoselective protection of the primary alcohol as the TES ether, followed by MOM protection of the secondary alcohol to afford protected diol (+)-30.
Scheme 5.

Next, oxidative removal of the TES ether with IBX (Scheme 6) directly provided aldehyde (+)-31 in excelent yield.28 The requisite aldol condensation to prepare (+)-32 was then achieved employing the conditions reported by Carreira and Weiss (Bn2NH2O2CCF3, PhH, 50 °C) in their synthesis of (+)-daphmanidin E.3
Scheme 6.

Turning to the critical 1,4-reduction of the conjugated diene in (+)-32, extensive experimentation on a less advanced aldehyde revealed a set of conditions involving the combination of ZnCl2, Ph2SiH2, and catalytic Pd(PPh3)429 as uniquely effective (see Supporting Information). Unfortunately, this reduction protocol when applied to (+)-32 resulted only in decomposition. Undeterred, aldehyde (+)-32 was oxidized to methyl ester (+)-2 (AcOH, NaCN, MeOH, then MnO2) via the method of Corey.30 A screen of cationic hydrogenation catalysts was next explored with the intent of directing the hydrogenation to the α,β-olefinic bond. Pleasingly, the BArF analog of the Crabtree catalyst,31 developed by Wuestenberg and Pfaltz,32 employing 900 psi of H2 pressure, proved effective in reducing (+)-2 to furnish a mixture (4:1) of diastereomers in 84% yield (Scheme 7). Detailed 2D NMR analysis revealed the major diastereomer to be (−)-34. This transformation, possibly directed by the C1 carbonyl in (+)-2, should prove useful in accessing natural congeners bearing the same mono-unsaturated DEF ring system (Figure 1).
Scheme 7.

Exposure of (−)-34 to hydrazine in EtOH led cleanly to removal of the phthalimide. Ring A construction involving imine formation was then readily achieved by heating the resulting amine with aq. NH4Cl (sat.) in EtOH at 70 °C.3 Completion of the total synthesis of (−)-calyciphylline N entailed treatment of (−)-34 with Ph2BBr33 to remove the MOM acetal. Totally synthetic (−)-calyciphylline N displayed spectral properties in excellent agreement with those derived from the natural product [i.e.,1H and 13C NMR (500 and 125 MHz, respectively), HRMS parent ion identification, and chiroptic properties].
In summary, the first total synthesis of a member of the calyciphylline alkaloids, (−)-calyciphylline N (1), has been achieved with a longest linear sequence of 37 steps from known alcohol (−)-8. Application of the strategies presented herein for the synthesis of other members of the Daphniphyllum alkaloids continues in our laboratory.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (National Cancer Institute) through CA-19033. We also thank Drs. George Furst, Rakesh Kohli, and Patrick Carroll for help in obtaining high resolution NMR, mass spectral analyses, and X-ray crystallographic data, respectively.
Footnotes
Experimental details, spectra, and X-ray crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kobayashi J, Kubota T. Nat Prod Rep. 2009;26:936. doi: 10.1039/b813006j. [DOI] [PubMed] [Google Scholar]
- 2.For recent synthetic studies, see: Coldham I, Watson L, Adams H, Martin NG. J Org Chem. 2011;76:2360. doi: 10.1021/jo2000868.Darses B, Michaelides IN, Sladojevich F, Ward JW, Rzepa PR, Dixon DJ. Org Lett. 2012;14:1684. doi: 10.1021/ol3002267.
- 3.For an elegant total synthesis of the closely related congener, (+)-daphmanidin E (Figure 1), see: Weiss ME, Carreira EM. Angew Chem Int Ed. 2011;50:11501. doi: 10.1002/anie.201104681.
- 4.For a very recent total synthesis of the Daphniphyllum alkaloid, daphenylline (Figure 1), see: Lu ZY, Li Y, Deng J, Li A. Nat Chem. 2013;5:679. doi: 10.1038/nchem.1694.
- 5.Yahata H, Kubota T, Kobayashi J. J Nat Prod. 2008;72:148. doi: 10.1021/np800515s. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kobayashi J, Ueno S, Morita H. J Org Chem. 2002;67:6546. doi: 10.1021/jo0258204. [DOI] [PubMed] [Google Scholar]; (b) Morita H, Ishioka N, Takatsu H, Iizuka T, Kobayashi J. J Nat Prod. 2006;69:418. doi: 10.1021/np0503799. [DOI] [PubMed] [Google Scholar]; (c) Saito S, Yahata H, Kubota T, Obara Y, Nakahata N, Kobayashi J. Tetrahedron. 2008;64:1901. [Google Scholar]; (d) Saito S, Kubota T, Fukushi E, Kawabata J, Zhang HP, Kobayashi J. Tetrahedron Lett. 2007;48:1587. [Google Scholar]; (e) Zhang H, Yang SP, Fan CQ, Ding J, Yue JM. J Nat Prod. 2006;69:553. doi: 10.1021/np050490e. [DOI] [PubMed] [Google Scholar]
- 7.Tamao K, Ishida N, Tanaka T, Kumada M. Organometallics. 1983;2:1694. [Google Scholar]
- 8.Merrifield JH, Godschalx JP, Stille JK. Organometallics. 1984;3:1108. [Google Scholar]
- 9.Nazarov IN, Zaretskaya II, Sorkina TI. Zh Obshch Khim. 1960;30:746. [Google Scholar]
- 10.Roush WR. J Am Chem Soc. 1978;100:3599.also see: Roush WR. In: Comp Org Synth. Trost BM, Fleming I, editors. Vol. 5. Pergamon Press; Oxford: 1991. p. 551.
- 11.Takeshita K, Seki Y, Kawamoto K, Murai S, Sonoda N. J Org Chem. 1987;52:4864. [Google Scholar]
- 12.(a) Mori K. Tetrahedron-Asymmetr. 2005;16:1721. [Google Scholar]; (b) Yadav JS, Basak AK, Srihari P. Tetrahedron Lett. 2007;48:2841. [Google Scholar]
- 13.Birch AJ. J Chem Soc. 1944:430. [Google Scholar]
- 14.Pearson DE, Buehler CA. Chem Rev. 1974;74:45. [Google Scholar]
- 15.Sieburth SM, Lang J. J Org Chem. 1999;64:1780. doi: 10.1021/jo990082d. [DOI] [PubMed] [Google Scholar]
- 16.Dess DB, Martin JC. J Am Chem Soc. 1991;113:7277. [Google Scholar]
- 17.Molander GA, Hahn G. J Org Chem. 1986;51:1135. [Google Scholar]
- 18.Smith AB, Levenberg PA. Synthesis. 1981:567. [Google Scholar]
- 19.(a) Trost BM, Fullerton TJ. J Am Chem Soc. 1973;95:292. [Google Scholar]; (b) Tsuji J, Takahash H, Morikawa M. Tetrahedron Lett. 1965:4387. [Google Scholar]
- 20.Tamao K. J Syn Org Chem Jpn. 1988;46:861. [Google Scholar]
- 21.Smitrovich JH, Woerpel KA. J Org Chem. 1996;61:6044. doi: 10.1021/jo991312r. [DOI] [PubMed] [Google Scholar]
- 22.A similar observation was noted in a recent total synthesis of Maoecrystal V: Lu P, Gu Z, Zakarian A. J Am Chem Soc. 2013;135:14552. doi: 10.1021/ja408231t.
- 23.Tamao K, Yamauchi T, Ito Y. Chem Lett. 1987:171. [Google Scholar]
- 24.Smith AB, Hoye AT, Martinez-Solorio D, Kim WS, Tong RBA. J Am Chem Soc. 2012;134:4533. doi: 10.1021/ja2120103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown HC, Knights EF, Scouten CG. J Am Chem Soc. 1974;96:7765. [Google Scholar]
- 26.Fleming I, Henning R, Plaut H. J Chem Soc Chem Commun. 1984:29. [Google Scholar]
- 27.(a) Mitsunobu O. Synthesis. 1981:1. [Google Scholar]; (b) Mitsunobu O, Yamada M. Bull Chem Soc Jpn. 1967;40:2380. [Google Scholar]
- 28.Wu YK, Huang JH, Shen X, Hu Q, Tang CJ, Li L. Org Lett. 2002;4:2141. doi: 10.1021/ol025946n. [DOI] [PubMed] [Google Scholar]
- 29.Keinan E, Greenspoon N. J Am Chem Soc. 1986;108:7314. [Google Scholar]
- 30.Corey EJ, Gilman NW, Ganem BE. J Am Chem Soc. 1968;90:5616. [Google Scholar]
- 31.Crabtree R. Acc Chem Res. 1979;12:331. [Google Scholar]
- 32.Wuestenberg B, Pfaltz A. Adv Synth Catal. 2008;350:174. [Google Scholar]
- 33.Guindon Y, Yoakim C, Morton HE. J Org Chem. 1984;49:3912. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.